
Comparative Proteomic Analysis of Flammulina filiformis Reveals Substrate-Specific Enzymatic Strategies for Lignocellulose Degradation


Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strain
2.2. Reagents and Apparatus
2.3. Culture Conditions
2.4. Secreted Protein Preparation
2.5. LC-MS/MS Data Acquisition
2.6. Sequence Database Search and Data Analysis
2.7. Statistical Analysis
3. Results
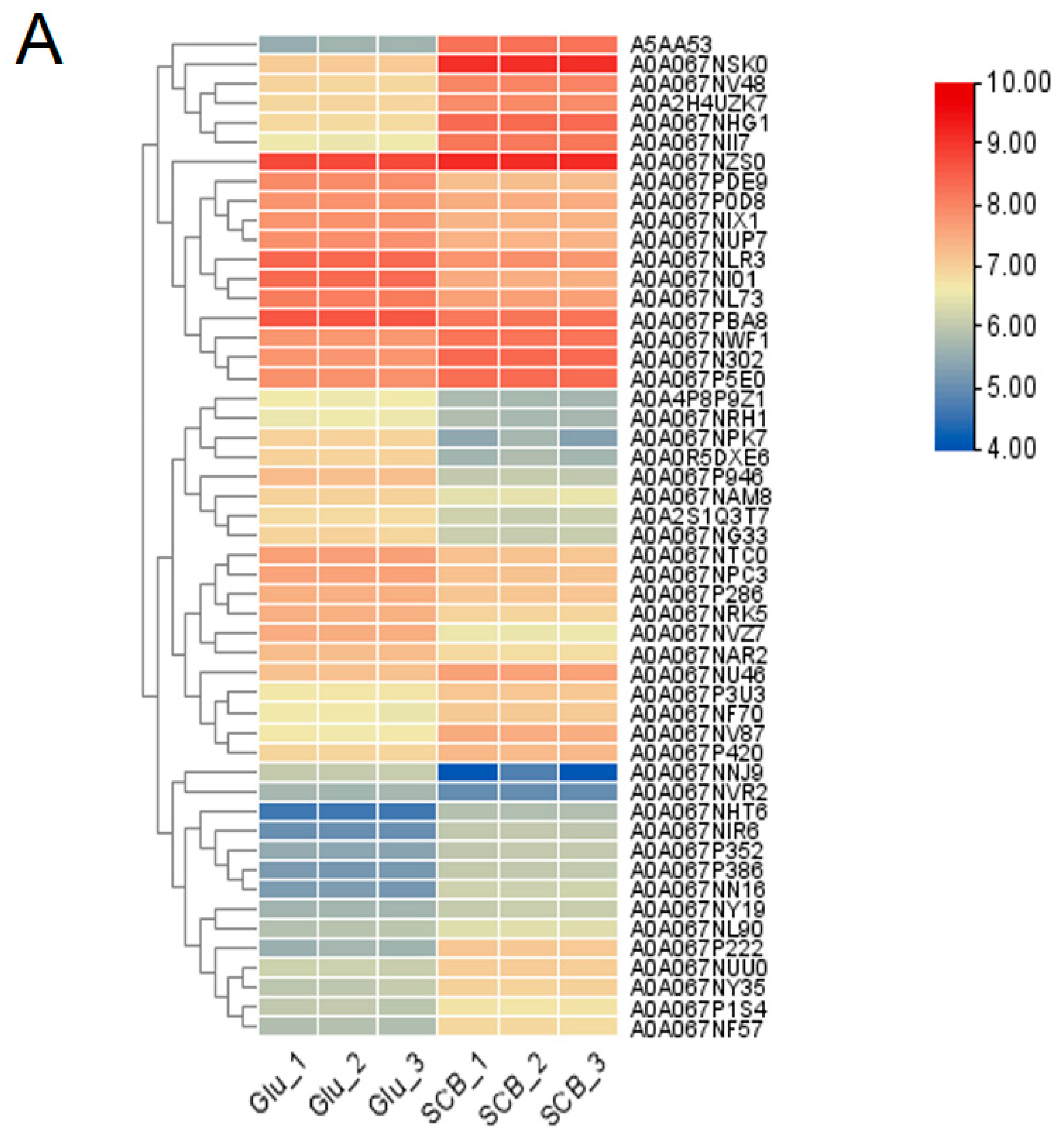
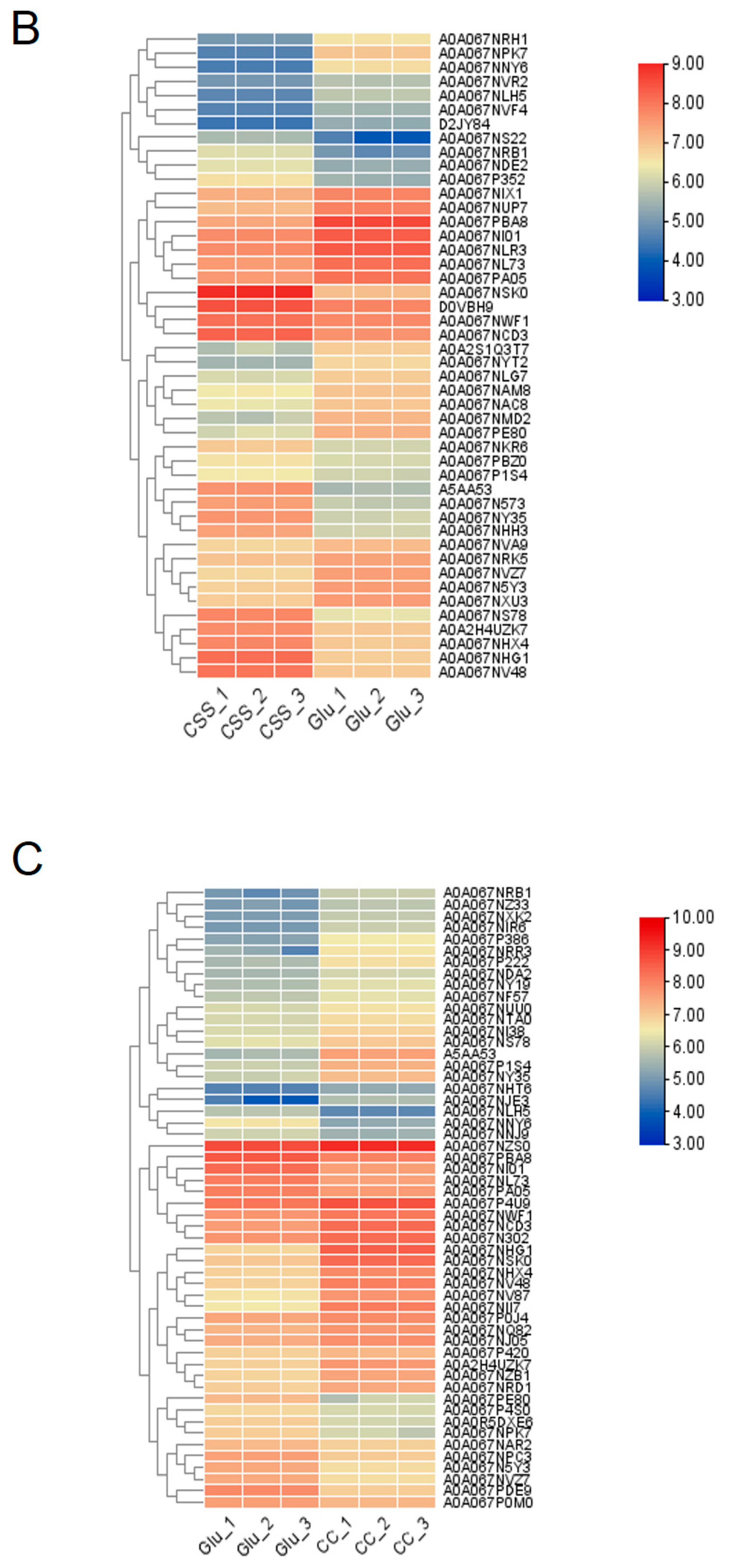
3.1. Differential Protein Expression Profiles
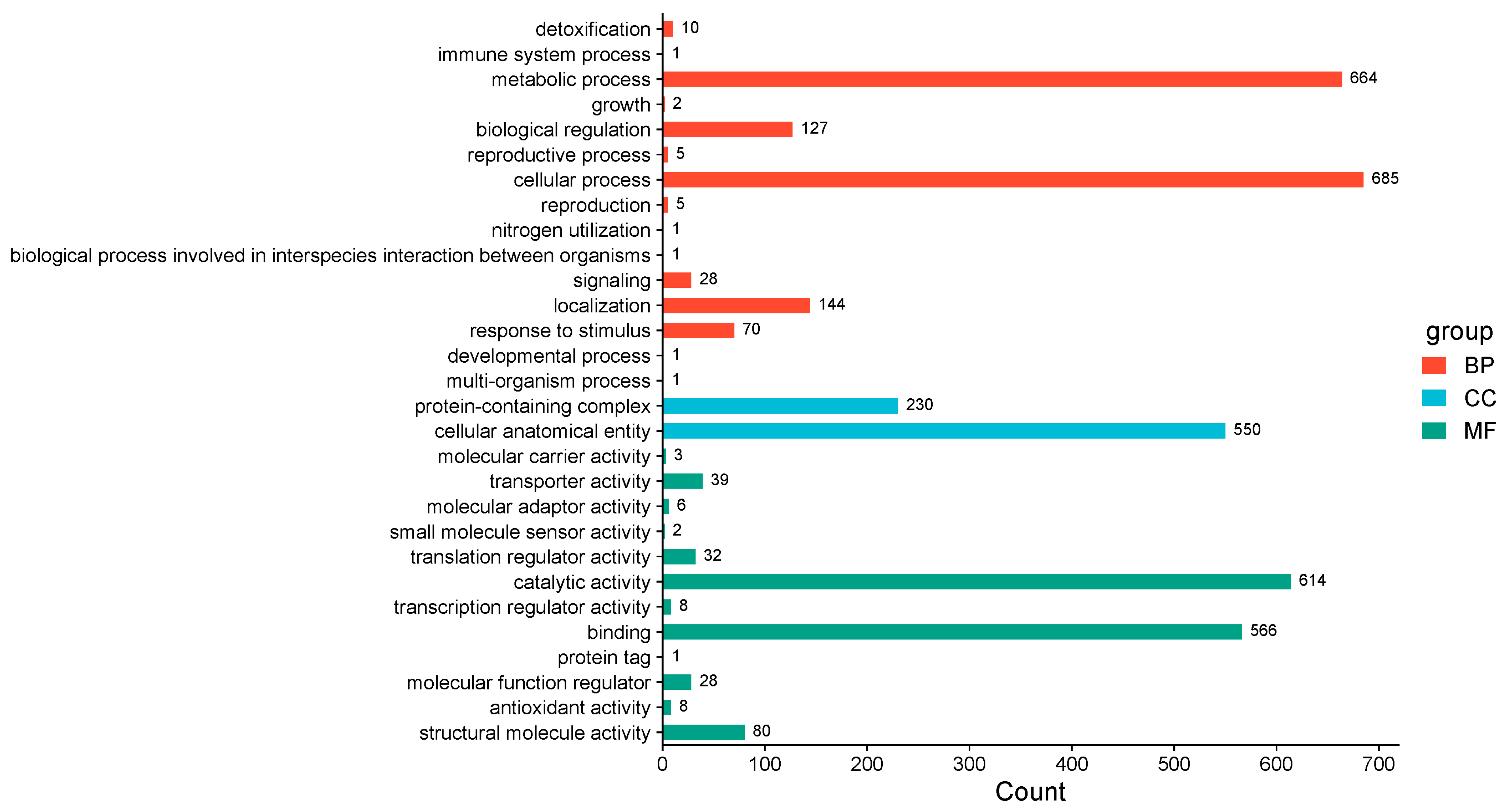
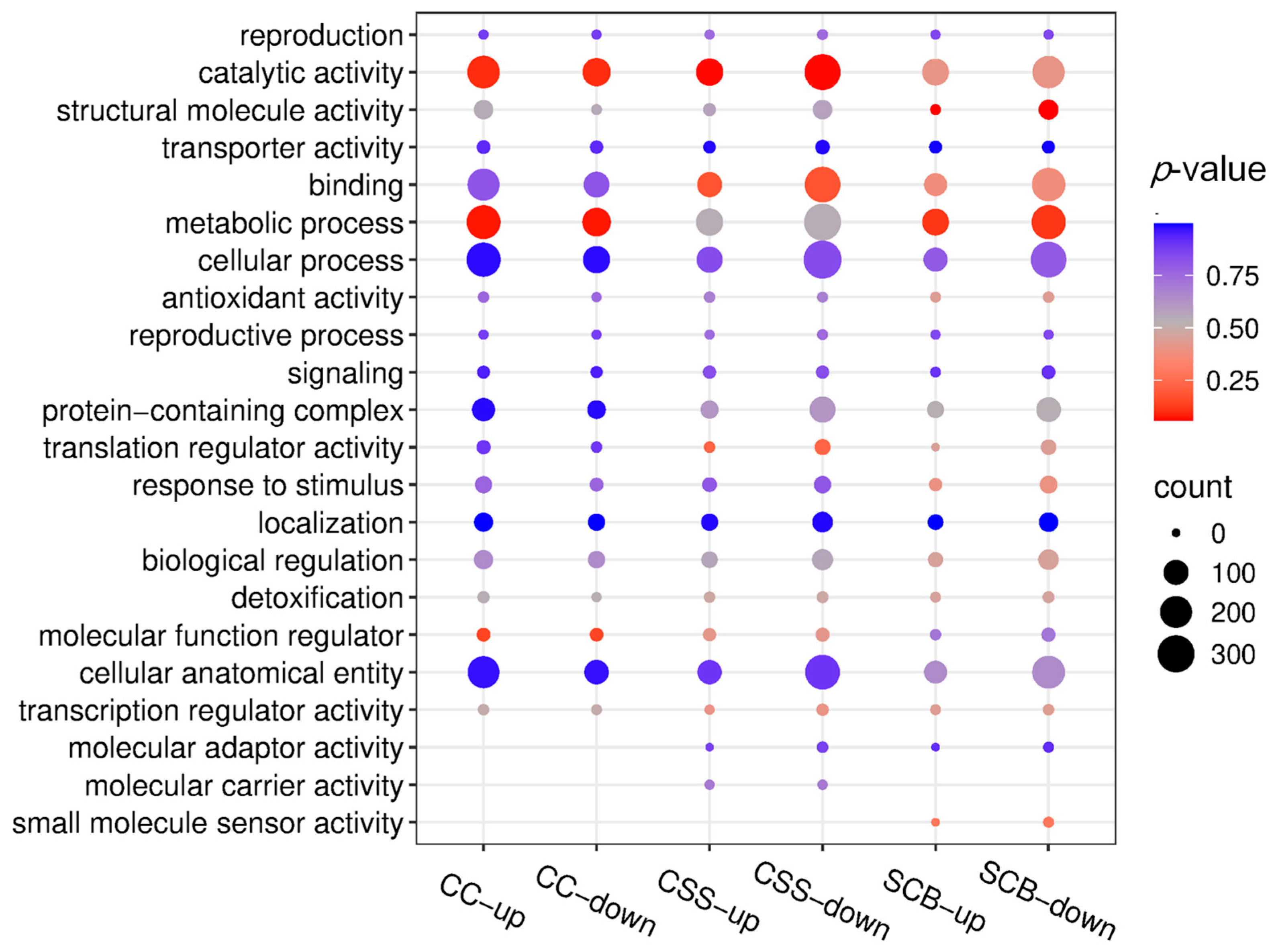
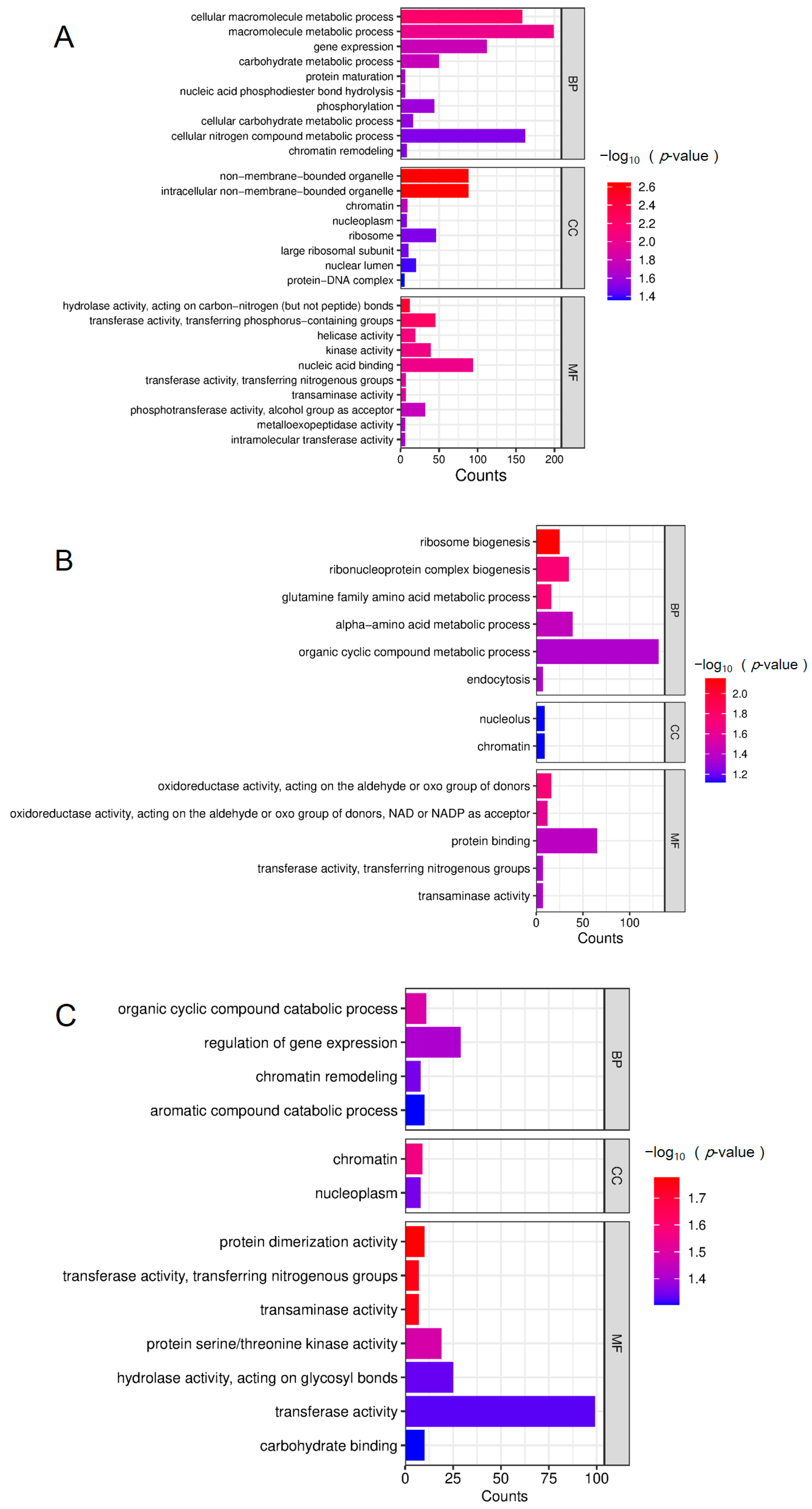
3.2. GO Functional Classification of Differential Proteins
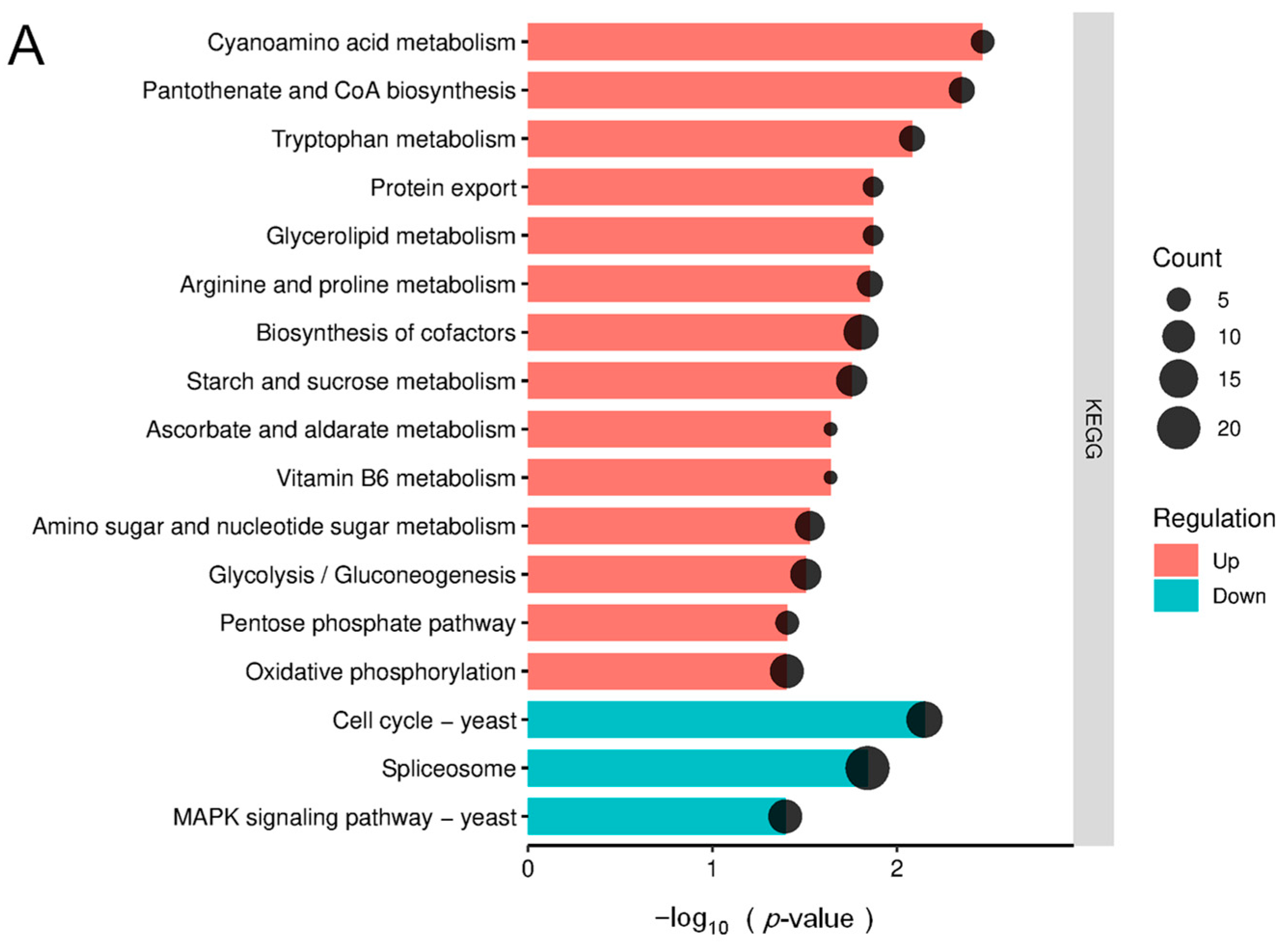
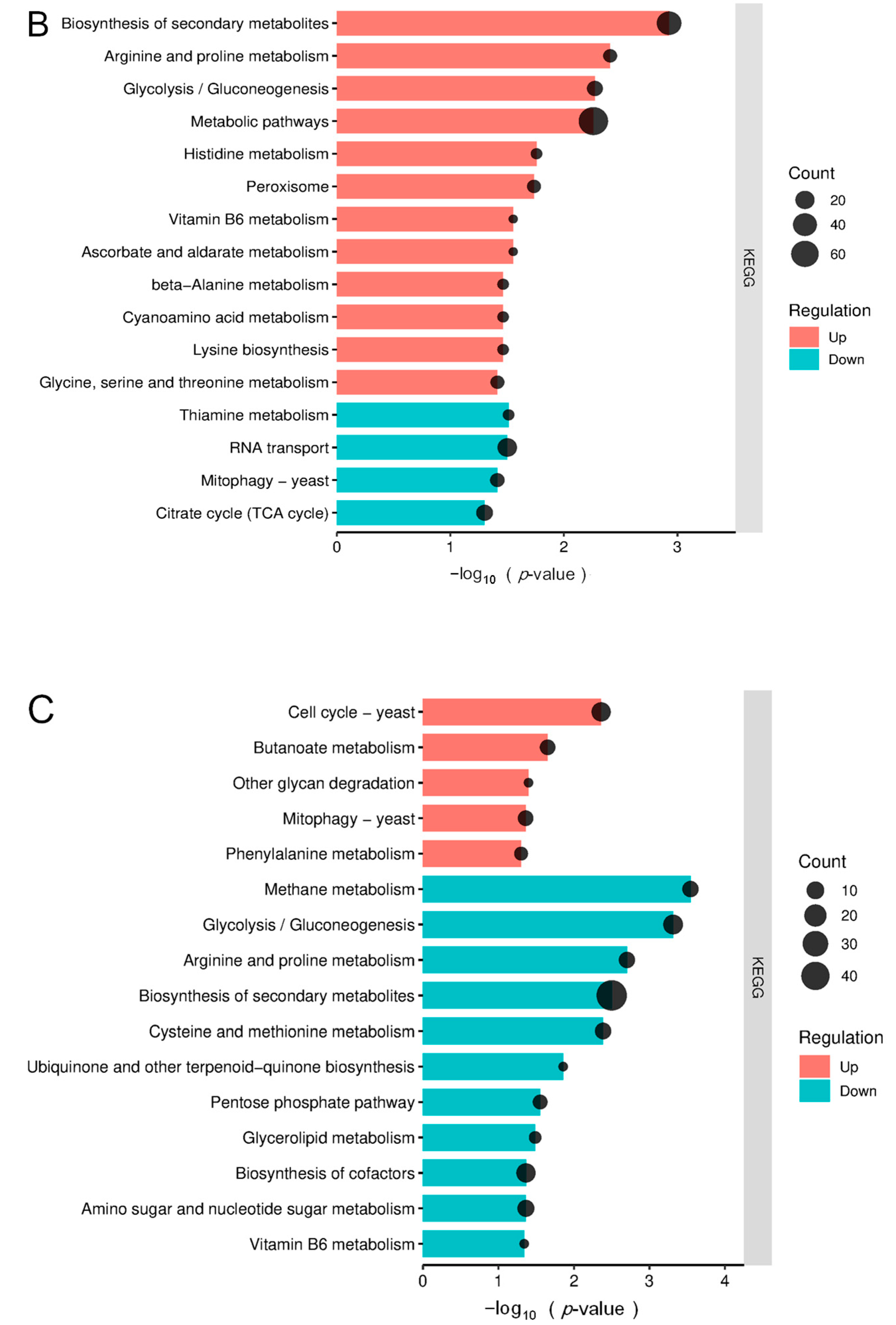
3.3. KEGG Pathway Enrichment Analysis of Differentially Expressed Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cheng, D.Y.; Liang, Y.Z. Notes on the nomenclature of five important edible fungi in China. Mycosystema 2018, 37, 1572–1577. [Google Scholar]
- Tang, C.; Hoo, P.C.-X.; Tan, L.T.-H.; Pusparajah, P.; Khan, T.M.; Lee, L.-H.; Goh, B.-H.; Chan, K.-G. Golden Needle Mushroom: A Culinary Medicine with Evidenced-Based Biological Activities and Health Promoting Properties. Front. Pharmacol. 2016, 7, 474. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, L.-W.; Yang, Z.-L.; Bau, T.; Li, T.-H.; Dai, Y.-C. Resource diversity of Chinese macrofungi: Edible, medicinal and poisonous species. Fungal Divers. 2019, 98, 1–76. [Google Scholar] [CrossRef]
- Zhao, Q.C. Fat Reduction Function of Flammulina Velutipe Dietary Fiber Analyzed Byintestinal Flora-Metabolomics-Proteomics. Master’s Thesis, Henan University, Kaifeng, China, 2023. [Google Scholar]
- Huang, Q.; Jia, Y.; Wan, Y.; Li, H.; Jiang, R. Market Survey and Risk Assessment for Trace Metals in Edible Fungi and the Substrate Role in Accumulation of Heavy Metals. J. Food Sci. 2015, 80, H1612–H1618. [Google Scholar] [CrossRef]
- Li, Z.; Han, J.; Xie, H.; Hu, Q.; Gong, Z.; Zou, Y. Analysis on nutrient changes in the mushroom substrate and the spent mushroom substrate of Flammulina velutipes. Acta Edulis Fungi 2021, 28, 47–54. [Google Scholar] [CrossRef]
- Chen, J.M.; Yu, W.W.; Wu, H.; Lai, F.R. Analysis of the nutritional components of corn cobs. Mod. Food Sci. Technol. 2012, 28, 1073–1075. [Google Scholar]
- Xie, C.; Yan, L.; Gong, W.; Zhu, Z.; Tan, S.; Chen, D.; Hu, Z.; Peng, Y. Effects of Different Substrates on Lignocellulosic Enzyme Expression, Enzyme Activity, Substrate Utilization and Biological Efficiency of Pleurotus eryngii. Cell. Physiol. Biochem. 2016, 39, 1479–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gao, N.; Kong, X.; Han, H.; Wu, D. Research Progress on the Cultivation of Edible Mushroom Substrates Using Cotton Waste. North. Hortic. 2024, 19, 126–132. [Google Scholar]
- Torgbo, S.; Quan, V.M.; Sukyai, P. Cellulosic value-added products from sugarcane bagasse. Cellulose 2021, 28, 5219–5240. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Y.; Lu, D.; Yang, X.; Lu, H. Application and Research Progress of Edible Fungi Cultivation with Sugarcane Bagasse. Chin. Agric. Sci. Bull. 2016, 32, 44–48. [Google Scholar]
- Huang, Q.; Wang, Q.; Zhang, L.; Kang, P.; Liang, L. Amino Acid Composition and Proteins Nutritional Evaluation of Different Kinds of Colored Pleurotus Cultivated with Bagasse. North. Hortic. 2019, 10, 127–133. [Google Scholar]
- Rezaeian, S.; Pourianfar, H.R. A Comparative Study on Bioconversion of Different Agro Wastes by Wild and Cultivated Strains of Flammulina velutipes. Waste Biomass Valorization 2017, 8, 2631–2642. [Google Scholar] [CrossRef]
- Kumar, P.; Barrett, D.M.; Delwiche, M.J.; Stroeve, P. Methods for Pretreatment of Lignocellulosic Biomass for Efficient Hydrolysis and Biofuel Production. Ind. Eng. Chem. Res. 2009, 48, 3713–3729. [Google Scholar] [CrossRef]
- Tian, C.; Ma, Y. Progress in lignocellulose deconstruction by fungi. Chin. J. Biotechnol. 2010, 26, 1333–1339. [Google Scholar]
- Chen, Y.; Zhang, H.Q.; Zhao, Y.; Gao, P.J.; Wang, L.S. Biosynthesis of Natural Crystal Cellulose and Its Decrystallization. Prog. Biochem. Biophys. 2016, 43, 747–757. [Google Scholar] [CrossRef]
- Chen, S.; Ge, W.; Buswell, J.A. Molecular cloning of a new laccase from the edible straw mushroom Volvariella volvacea: Possible involvement in fruit body development. FEMS Microbiol. Lett. 2004, 230, 171–176. [Google Scholar] [CrossRef]
- Park, Y.-J.; Lee, C.-S.; Kong, W.-S. Genomic Insights into the Fungal Lignocellulolytic Machinery of Flammulina rossica. Microorganisms 2019, 7, 421. [Google Scholar] [CrossRef]
- Park, Y.J.; Kong, W.S. Genome-Wide Comparison of Carbohydrate-Active Enzymes (CAZymes) Repertoire of Flammulina ononidis. Mycobiology 2018, 46, 349–360. [Google Scholar] [CrossRef]
- Zhou, Z.; Ju, X.; Chen, J.; Wang, R.; Zhong, Y.; Li, L. Charge-oriented strategies of tunable substrate affinity based on cellulase and biomass for improving in situ saccharification: A review. Bioresour. Technol. 2021, 319, 124159. [Google Scholar] [CrossRef]
- Zhou, S. The Mechanism of Substrate Degradation, Carbohydrateand Triterpene Metabolism of Ganoderma lucidum. Ph.D. Thesis, Huazhong University Of Science And Technology, Wuhan, China, 2019. [Google Scholar]
- Ge, Y.-j.; Shang, X.-d.; Tan, Q. Restricting nitrogen content in culture medium promotes Lentinula edodes to secrete lignin peroxidase. Acta Edulis Fungi 2022, 29, 25–34. [Google Scholar] [CrossRef]
- Benoit, I.; Culleton, H.; Zhou, M.; DiFalco, M.; Aguilar-Osorio, G.; Battaglia, E.; Bouzid, O.; Brouwer, C.P.J.M.; El-Bushari, H.B.O.; Coutinho, P.M.; et al. Closely related fungi employ diverse enzymatic strategies to degrade plant biomass. Biotechnol. Biofuels 2015, 8, 107. [Google Scholar] [CrossRef]
- An, Q.; Wu, W.X.; Wu, W.B.; Dai, D.Y. Effects of carbon and nitrogen sources on lignocellulose decomposition enzyme activities in Flammulina velutipes. Mycosystema 2015, 34, 761–771. [Google Scholar]
- Xiao, Q.; Yu, H.; Zhang, J.; Li, F.; Li, C.; Zhang, X.; Ma, F. The potential of cottonseed hull as biorefinery substrate after biopretreatment by Pleurotus ostreatus and the mechanism analysis based on comparative proteomics. Ind. Crops Prod. 2019, 130, 151–161. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, C.; Zhou, Y.; Zheng, S.; Hu, Q.; Zou, Y. Label-free comparative proteomic analysis of Pleurotus eryngii grown on sawdust, bagasse, and peanut shell substrates. J. Proteom. 2024, 294, 105074. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fueyo, E.; Ruiz-Dueñas, F.J.; López-Lucendo, M.F.; Pérez-Boada, M.; Rencoret, J.; Gutiérrez, A.; Pisabarro, A.G.; Ramírez, L.; Martínez, A.T. A secretomic view of woody and nonwoody lignocellulose degradation by Pleurotus ostreatus. Biotechnol. Biofuels 2016, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, R.; Huang, C.; Xie, H.; Gao, X.; Yao, Q.; Yang, P.; Li, J.; Gong, Z. Effects of Different Carbon and Nitrogen Ratios on Yield, Nutritional Value, and Amino Acid Contents of Flammulina velutipes. Life 2024, 14, 598. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, D.; Wang, X.; Li, T.; Wang, B. Targeted metabolomic analysis of active substances in Auricularia heimuer cultivated on different substrates. Mycosystema 2024, 43, 79–91. [Google Scholar]
- Angelini, R.; Cona, A.; Tavladoraki, P. Determination of Copper Amine Oxidase Activity in Plant Tissues. Methods Mol. Biol. 2018, 1694, 129–139. [Google Scholar] [CrossRef]
- Planas-Portell, J.; Gallart, M.; Tiburcio, A.F.; Altabella, T. Copper-containing amine oxidases contribute to terminal polyamine oxidation in peroxisomes and apoplast of Arabidopsis thaliana. BMC Plant Biol. 2013, 13, 109. [Google Scholar] [CrossRef]
- Xu, X.; Gu, L.; He, P.; Zhou, R. Characterization of five putative aspartate aminotransferase genes in the N2-fixing heterocystous cyanobacterium Anabaena sp. strain PCC 7120. Microbiology 2015, 161, 1219–1230. [Google Scholar] [CrossRef]
- Wei, Z.X. Identification of Drought Resistance Function of Aspartatetransaminase Gene VfAAT from Vicia faba L. Master’s Thesis, Yangtze University, Jingzhou, China, 2023. [Google Scholar]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Liu, A. Omics Analysis of Fruiting Body Development and Changesof Antioxidant Activity in Flammulina filiformis. Master’ Thesis, Shandong Agricultural University, Taian, China, 2023. [Google Scholar]
- Xia, L.; Li, L.; Zhang, X.; Zhang, L. Identification of Phosphoglucomutase and UDP-Glucose Pyrophosphorylase Involved in Biosynthesis of Polysaccharide from Wolfiporia cocos. Chem. Bioeng. 2024, 41, 44–52. [Google Scholar]
- Qiong, W. Analysis of Triterpenoid and Polysaccharide Highyield Mechanism in Ganoderma lucidum based on Omics Technology. Ph.D. Thesis, Jiangnan University, Wuxi, China, 2020. [Google Scholar]
- Xu, Y.L.; Yuan, H.; Li, N.; Xiao, J.H.; Xu, J.-W. Increased production and anti-senescence activity of exopolysaccharides in Ganoderma lingzhi by co-overexpression of β-1,3-glucan synthase and UDP-glucose pyrophosphorylase. Int. J. Biol. Macromol. 2023, 253, 126778. [Google Scholar] [CrossRef]
- Liu, J.-h.; Shang, X.-d.; Liu, J.-y.; Tan, Q. Changes in trehalose content, enzyme activity and gene expression related to trehalose metabolism in Flammulina velutipes under heat shock. Microbiology 2016, 162, 1274–1285. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, S.-t.; Xu, Z.-h.; Shi, J.-s. Degradation of hemicellulose by microbial enzymes and its application in brewing industry. Food Ferment. Ind. 2020, 46, 255–262. [Google Scholar] [CrossRef]
- Zhu, N.; Yang, J.; Ji, L.; Liu, J.; Yang, Y.; Yuan, H. Metagenomic and metaproteomic analyses of a corn stover-adapted microbial consortium EMSD5 reveal its taxonomic and enzymatic basis for degrading lignocellulose. Biotechnol. Biofuels 2016, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Malgas, S.; Thoresen, M.; van Dyk, J.S.; Pletschke, B.I. Time dependence of enzyme synergism during the degradation of model and natural lignocellulosic substrates. Enzym. Microb. Technol. 2017, 103, 1–11. [Google Scholar] [CrossRef]
- Yuan, L.; Qin, X. ATP synthase beta subunit function and disease. Chin. J. Clin. Pharmacol. Ther. 2018, 23, 464–470. [Google Scholar]
- Gao, X.; Meng, H.; Li, R.; Li, X. Advances in Cellulose Degradation by Glycoside Hydrolase Family 7 Proteins. J. Mirobiology 2020, 40, 113–117. [Google Scholar]
- Bernardi, A.V.; de Gouvêa, P.F.; Gerolamo, L.E.; Yonamine, D.K.; de Lourdes de Lima Balico, L.; Uyemura, S.A.; Dinamarco, T.M. Functional characterization of GH7 endo-1,4-β-glucanase from Aspergillus fumigatus and its potential industrial application. Protein Expr. Purif. 2018, 150, 1–11. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Z.; Zheng, C.; Wang, X.; Wu, Y.; Li, H.; Zhang, G. Status and Progress on Functions of Plant Enolase Gene ENO2. J. Plant Genet. Resour. 2018, 19, 1030–1037. [Google Scholar]
- Van Dyk, J.S.; Pletschke, B.I. A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes—Factors affecting enzymes, conversion and synergy. Biotechnol. Adv. 2012, 30, 1458–1480. [Google Scholar] [CrossRef]
- Yu, H.-W.; Im, J.-H.; Kong, W.-S.; Park, Y.-J. Comparative Analysis of Carbohydrate Active Enzymes in the Flammulina velutipes var. lupinicola Genome. Microorganisms 2021, 9, 20. [Google Scholar] [CrossRef]
- Schmitz, E.; Leontakianakou, S.; Norlander, S.; Nordberg Karlsson, E.; Adlercreutz, P. Lignocellulose degradation for the bioeconomy: The potential of enzyme synergies between xylanases, ferulic acid esterase and laccase for the production of arabinoxylo-oligosaccharides. Bioresour. Technol. 2022, 343, 126114. [Google Scholar] [CrossRef]
- Han, M.-L.; Bian, L.-S.; Jiang, H.-H.; An, Q. Effects of different carbon and nitrogen sources on lignocellulolytic enzyme activities of Pleurotus ostreatus. Mycosystema 2020, 39, 1538–1550. [Google Scholar] [CrossRef]
- Park, Y.-J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.-S.; Park, H.-R.; Yoon, D.-E.; Nam, J.-Y.; et al. Whole Genome and Global Gene Expression Analyses of the Model Mushroom Flammulina velutipes Reveal a High Capacity for Lignocellulose Degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Huang, T.; Zheng, T.; Gong, M.; Guo, T.; Li, F.; Wang, W.; Li, Z.; Tang, L. Advances in low temperature stimulation for the fruiting body formation of edible fungi. Microbiol. China 2023, 50, 5518–5533. [Google Scholar]
- Li, Y.; Zhao, L.; Gu, Z.; Li, Y.; Shi, G.; Ding, Z. Heterologous expression and characterization of the key enzymes involved in sugar donor synthesis of polysaccharide in Ganoderma lucidum. Microbiol. China 2019, 46, 3233–3247. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | CAZy Family | Protein Description | Signalp | Protein Abundance | |||
|---|---|---|---|---|---|---|---|
| Glucose | Sugarcane Bagasse | Corn Cobs | Cotton Seed Shells | ||||
| A0A067N481 | GH5 | Glycoside hydrolase family 5 protein | N | ||||
| A0A067N5C6 | GT2 | Chitin synthase | N | 1,674,382.92233333 | 1,553,722.012 | 761,452.014833333 | 1,626,613.11766667 |
| A0A067N7N9 | AA1 | Fet3 ferroxidase | Y | 1,923,473.892 | 1,569,898.35733333 | 814,216.989533333 | 1,917,835.20633333 |
| A0A067N9N5 | GT35 | Alpha-1,4 glucan phosphorylase | N | 5,045,299.339 | 2,661,980.72466667 | 3,300,366.021 | 2,613,296.99 |
| A0A067N9V0 | GH3 | Glycoside hydrolase family 3 protein | N | 73,966,935.1533333 | 65,251,685.5266667 | 42,499,127.7966667 | 125,931,711.033333 |
| A0A067NB35 | GH35 | Glycoside hydrolase family 35 protein | N | 44,803,576.2366667 | 54,079,091.2 | 40,553,862.6666667 | 62,096,653.43 |
| A0A067NCX6 | GH35 | Beta-galactosidase | N | 26,270,162.46 | 12,170,355.1233333 | 9,266,884.40666667 | 4,327,699.927 |
| A0A067NDH9 | GH16 | Glycoside hydrolase family 16 protein | N | 705,834.522833333 | 589,586.945666667 | 724,761.576 | 1,811,456.84333333 |
| A0A067NE50 | PL26 | Autophagy-related protein 27 | Y | 37,554,727.2766667 | 27,057,562.07 | 13,562,714.79 | 13,853,933.0233333 |
| A0A067NG33 | GT3 | Glycogen [starch] synthase | N | 8,748,214.16666667 | 1,328,355.905 | 2,525,576.068 | 1,244,528.46533333 |
| A0A067NG43 | GH179 | GFO_IDH_MocA domain-containing protein | N | 687,454.406266667 | 1,619,932.57866667 | 807,516.327666667 | 1,428,833.614 |
| A0A067NG45 | GT69 | Glycosyltransferase family 69 protein | Y | 4,409,595.69266667 | 4,622,098.876 | 2,747,698.281 | 2,672,916.48933333 |
| A0A067NHG4 | GH51 | Non-reducing end alpha-L-arabinofuranosidase | N | 1,153,395.48766667 | 3,097,140.86366667 | 3,939,891.408 | 10,696,011.0986667 |
| A0A067NHT6 | GH5 | Glycoside hydrolase family 5 protein | Y | 43,335.89372 | 761,726.745733333 | 219,575.423666667 | 43,335.89372 |
| A0A067NHZ2 | AA8 | GMC_OxRdtase_N domain-containing protein | N | 545,214.741766667 | 91,463,449.0566667 | 6,676,565.561 | 2,890,166.06066667 |
| A0A067NI47 | GH30 | Glycoside hydrolase family 30 protein | N | 2,387,567.25 | 215,776.937866667 | 36,963.9312 | 36,963.9312 |
| A0A067NIA1 | CE15 | Carbohydrate esterase family 15 protein | Y | 339,189.8876 | 115,408,491.666667 | 41,968,924.2733333 | 8,447,431.35666667 |
| A0A067NJ89 | GH5 | Glycoside hydrolase family 5 protein | N | 284,593.773466667 | 326,019.267 | 115,600.020666667 | 21,217.80416 |
| A0A067NJZ2 | GH115 | Glycoside hydrolase family 115 protein | Y | 1,930,693.30433333 | 4,595,319.543 | 1,213,666.526 | 369,670.60656 |
| A0A067NKG1 | GH63 | Glyco_hydro_63 domain-containing protein | N | 2,747,527.69166667 | 983,334.406933333 | 1,246,596.6286 | 1,117,502.767 |
| A0A067NKR6 | GH3 | Glycoside hydrolase family 3 protein | N | 1,172,579.32166667 | 4,990,872.88133333 | 2,641,664.84333333 | 7,850,058.13933333 |
| A0A067NL12 | CE4 | Chitin deacetylase | Y | 2,532,754.30033333 | 924,863.084666667 | 1,155,317.05666667 | 2,255,575.432 |
| A0A067NL90 | AA3 | GMC_OxRdtase_N domain-containing protein | Y | 882,786.094366667 | 2,610,349.02066667 | 808,885.0888 | 3,630,119.53566667 |
| A0A067NLS2 | GH38 | Alpha-mannosidase | N | 7,706,159.83633333 | 12,219,068.8266667 | 1,657,370.85133333 | 1,078,749.07483333 |
| A0A067NNJ9 | GH13 | 1,4-alpha-glucan-branching enzyme | N | 1,242,357.576 | 29,290.3292333333 | 302,425.2643 | 109,230.33996 |
| A0A067NNT3 | GT15 | Glycosyltransferase family 15 protein | N | 1,208,833.28266667 | 1,664,963.70766667 | 552,614.189366667 | 210,154.392233333 |
| A0A067NP57 | GH74 | Glycoside hydrolase family 74 protein | Y | 388,005.1119 | 47,425,233.0866667 | 7,718,332.32966667 | 22,058,456.8833333 |
| A0A067NPC3 | GH20 | Beta-hexosaminidase | Y | 40,254,045.4666667 | 14,772,757.8533333 | 9,881,569.315 | 24,482,395.38 |
| A0A067NQN1 | GH74 | Glycoside hydrolase family 74 protein | Y | 463,429.509266667 | 58,716,335.94 | 10,340,246.84 | 25,877,955.2 |
| A0A067NRB1 | GH3 | Beta-glucosidase | N | 82,215.74544 | 2,068,114.20433333 | 1,022,417.6928 | 1,592,823.36066667 |
| A0A067NRS6 | AA4 | FAD-binding PCMH-type domain-containing protein | N | 12,488,633.6083333 | 9,745,718.80066667 | 5,172,592.48033333 | 40,775,298.46 |
| A0A067NSK0 | GH7 | Glucanase | Y | 11,217,736.1 | 1,268,879,946 | 230,125,783.033333 | 923,739,948.666667 |
| A0A067NTR2 | GH13 | Glycoside hydrolase family 13 protein | Y | 285,318.224566667 | 502,909.6734 | 212,830.326166667 | 186,036.48156 |
| A0A067NTS1 | GH72 | 1,3-beta-glucanosyltransferase | N | 54,130,175.98 | 4,963,599.49233333 | 11,825,230.4266667 | 105,578,133.73 |
| A0A067NU72 | GH47 | alpha-1,2-Mannosidase | N | 1,769,389.19833333 | 280,684.350966667 | 372,926.358866667 | 49,006.6803 |
| A0A067NUA1 | GH3 | Glycoside hydrolase family 3 protein | N | 795,122.5487 | 127,320.676066667 | 929,881.6146 | 517,719.2041 |
| A0A067NUJ5 | GT20 | Glycosyltransferase family 20 protein | N | 4,084,710.286 | 810,559.760733333 | 548,366.213233333 | 561,918.323533333 |
| A0A067NUM9 | GH43 | Glycoside hydrolase family 43 protein | N | 1,511,309.05833333 | 59,552,658.17 | 56,268,782.9433333 | 76,230,697.6633333 |
| A0A067NVF4 | GH47 | alpha-1,2-Mannosidase | Y | 328,118.7679 | 632,440.375066667 | 251,327.522866667 | 46,763.0276 |
| A0A067NVK8 | GH179 | GFO_IDH_MocA domain-containing protein | N | 972,190.939133333 | 796,142.437633333 | 834,063.037033333 | 2,027,235.743 |
| A0A067NW26 | GH27 | Alpha-galactosidase | N | 231,569.9718 | 2,884,487.07166667 | 35,724.60152 | 35,724.60152 |
| A0A067NXJ5 | GT69 | Glycosyltransferase family 69 protein | N | 579,444.1575 | 772,410.943 | 1,040,558.4363 | 615,105.0206 |
| A0A067NXX6 | GH5 | Glycoside hydrolase family 5 protein | N | 142,572.07057 | 425,257.856066667 | 308,028.0275 | 537,522.797633333 |
| A0A067NYC9 | AA9 | Glycoside hydrolase family 61 protein | N | 32,413.33185 | 191,864.378366667 | 98,566.1137166667 | 516,223.5153 |
| A0A067NYD8 | GT39 | Dolichyl-phosphate-mannose--protein mannosyltransferase | N | 184,289.4021 | 256,503.235966667 | 45,563.4125 | 45,563.4125 |
| A0A067NYI6 | AA9 | Glycoside hydrolase family 61 protein | Y | 111,182.064 | 1,637,476.95666667 | 595,988.317233333 | 111,182.064 |
| A0A067P039 | GH3 | Beta-glucosidase | N | 496,367.998333333 | 5,285,172.74033333 | 2,872,587.55566667 | 1,756,140.254 |
| A0A067P0D5 | GT2 | Chitin synthase | N | 115,901.747933333 | 156,000.033433333 | 20,483.6242 | 20,483.6242 |
| A0A067P120 | GH2 | Glycoside hydrolase family 2 protein | N | 616,712.845333333 | 687,912.198833333 | 213,204.624633333 | 126,479.038473333 |
| A0A067P222 | GH3 | Beta-glucosidase | N | 463,925.3207 | 12,174,613.98 | 5,160,192.30733333 | 3,392,101.14166667 |
| A0A067P2G6 | GH74 | Glycoside hydrolase family 74 protein | N | 1,029,203.9254 | 92,302,563.21 | 12,674,182.64 | 29,404,276.5166667 |
| A0A067P3G0 | GH43 | Glycoside hydrolase family 43 protein | Y | 2,249,040.07066667 | 1,483,016.085 | 334,123.3293 | 61,306.25716 |
| A0A067P3W7 | GH10 | Beta-xylanase | Y | 1,473,446.39433333 | 196,961,002.933333 | 25,016,172.1333333 | 24,481,648.0066667 |
| A0A067P3Z6 | GH5 | Glycoside hydrolase family 5 protein | Y | 22,239,740.3866667 | 12,050,158.5566667 | 14,542,128.2233333 | 26,057,009.0033333 |
| A0A067P4G8 | PL8 | Polysaccharide lyase family 8 protein | Y | 11,472,925.3766667 | 3,982,520.96933333 | 1,567,346.429 | 536,265.1585 |
| A0A067P5W8 | GT24 | Glycosyltransferase family 24 protein | Y | 48,176.3044 | 544,094.1723 | 253,305.0977 | 348,904.692666667 |
| A0A067P7X8 | GH105 | Glycoside hydrolase family 105 protein | Y | 29,812,627.2533333 | 25,171,134.76 | 12,159,050.3666667 | 21,464,407.3833333 |
| A0A067P8J0 | GT48 | 1,3-beta-glucan synthase | N | 279,285.116566667 | 1,133,638.01133333 | 590,600.269866667 | 151,762.7827 |
| A0A067P9S7 | GT4 | Glycosyltransferase family 4 protein | N | 33,378,855.0866667 | 13,535,715.7066667 | 27,846,050.02 | 10,315,850.4533333 |
| A0A067PAK9 | GH43 | Glycoside hydrolase family 43 protein | N | 14,733.40841 | 543,993.021933333 | 85,749.15857 | 14,733.40841 |
| A0A067PCV4 | GH13 | Alpha-amylase | Y | 217,508.8117 | 148,575.9674 | 178,161.0273 | 157,544.848753333 |
| A0A067PE74 | GH31 | Glycoside hydrolase family 31 protein | Y | 260,600.315933333 | 23,874.92206 | 90,987.84162 | 23,874.92206 |
| A0A0F6R9C3 | AA1 | Laccase A | Y | 4,092,911.83733333 | 11,578,570.09 | 12,466,733.3766667 | 6,823,822.44233333 |
| A0A2S1Q3T7 | GT20 | Alpha, alpha-trehalose-phosphate synthase (UDP-forming) | N | 7,161,878.82433333 | 1,439,578.51166667 | 2,626,963.73133333 | 643,211.974533333 |
| A0A482GMN1 | AA2 | Peroxidase | Y | 30,817,611.0333333 | 13,001,488.19 | 28,196,940.2466667 | 23,719,528.7833333 |
| A0A482GQ69 | GH37 | Trehalase | N | 1,444,615.75266667 | 218,731.31909 | 51,030.5576433333 | 465,147.892066667 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Han, J.; Xie, H.; Sun, Y.; Li, F.; Gong, Z.; Zou, Y. Comparative Proteomic Analysis of Flammulina filiformis Reveals Substrate-Specific Enzymatic Strategies for Lignocellulose Degradation. Horticulturae 2025, 11, 912. https://doi.org/10.3390/horticulturae11080912
Li W, Han J, Xie H, Sun Y, Li F, Gong Z, Zou Y. Comparative Proteomic Analysis of Flammulina filiformis Reveals Substrate-Specific Enzymatic Strategies for Lignocellulose Degradation. Horticulturae. 2025; 11(8):912. https://doi.org/10.3390/horticulturae11080912
Chicago/Turabian StyleLi, Weihang, Jiandong Han, Hongyan Xie, Yi Sun, Feng Li, Zhiyuan Gong, and Yajie Zou. 2025. "Comparative Proteomic Analysis of Flammulina filiformis Reveals Substrate-Specific Enzymatic Strategies for Lignocellulose Degradation" Horticulturae 11, no. 8: 912. https://doi.org/10.3390/horticulturae11080912
APA StyleLi, W., Han, J., Xie, H., Sun, Y., Li, F., Gong, Z., & Zou, Y. (2025). Comparative Proteomic Analysis of Flammulina filiformis Reveals Substrate-Specific Enzymatic Strategies for Lignocellulose Degradation. Horticulturae, 11(8), 912. https://doi.org/10.3390/horticulturae11080912