
Transcriptome Analysis Reveals Molecular Mechanisms of Melanin Synthesis in Auricularia heimuer Under Different Fermentation Times
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Auricularia Heimuer Strain
2.2. Physiological Assay
2.3. Transcriptome Analysis
2.4. Real-Time-Quantitative PCR (RT-qPCR) Analysis
2.5. Statistical Analysis
3. Results
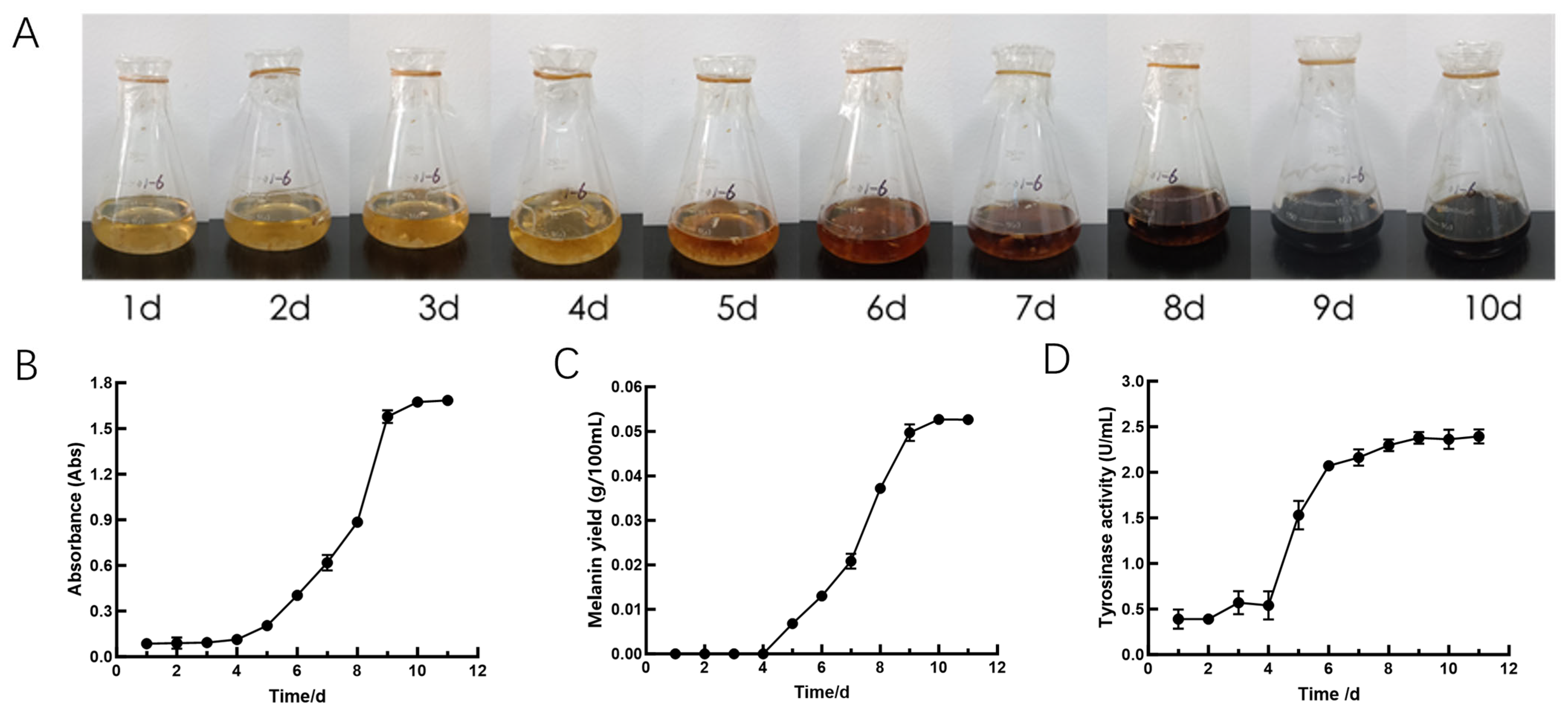
3.1. Physiological Indicators
3.2. RNA-Seq Data Quality Statistics
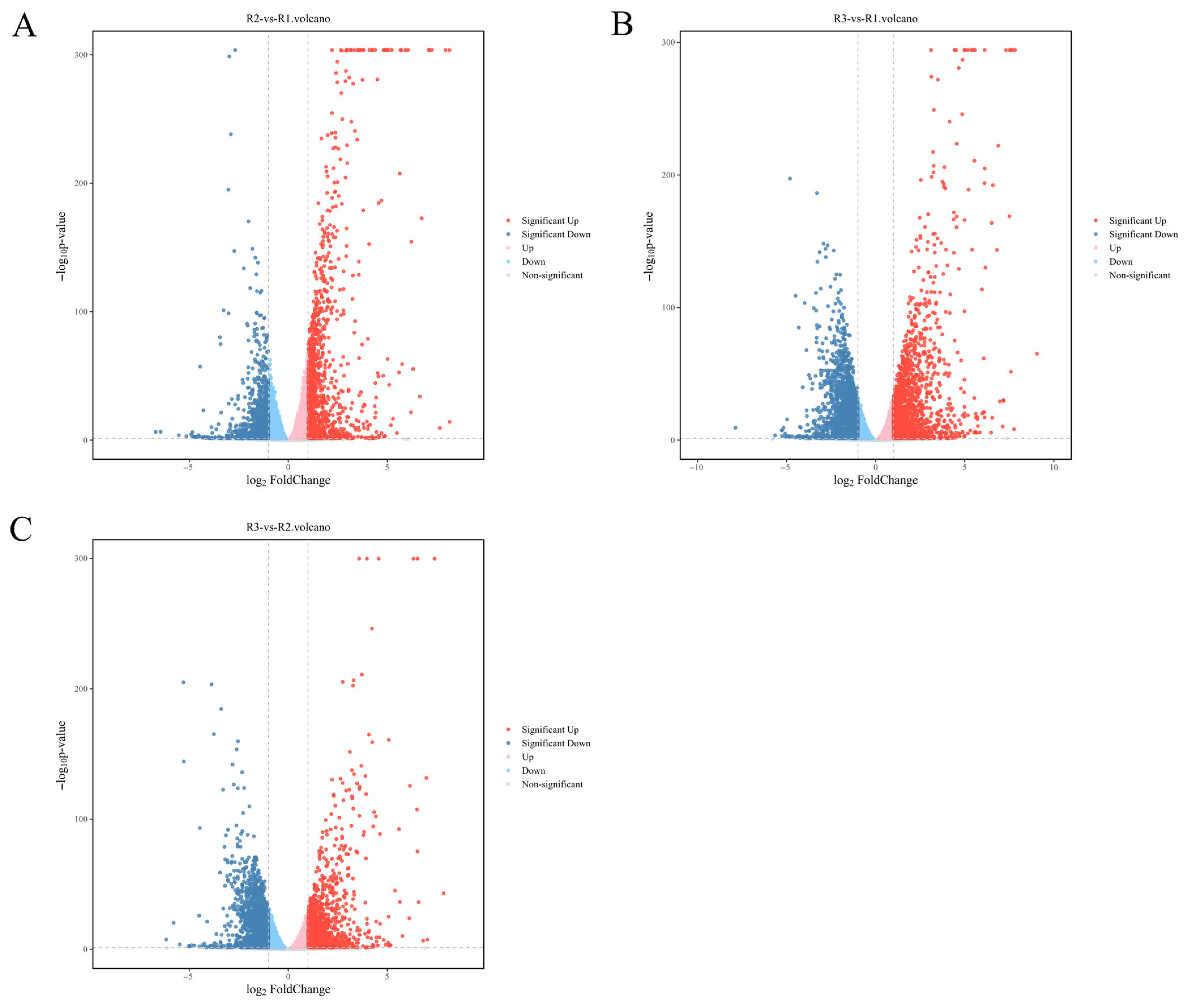
3.3. DEGs Analysis
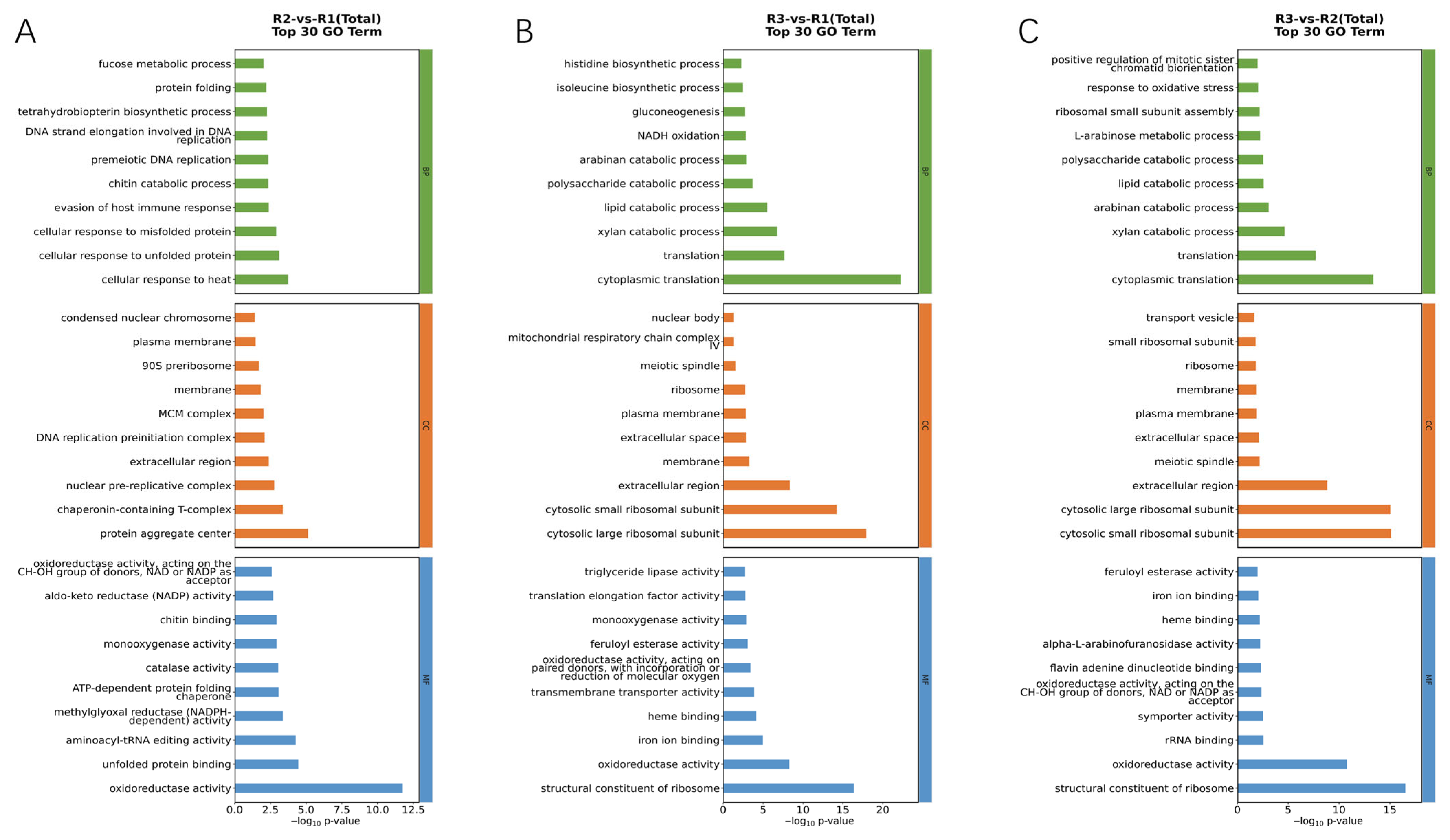
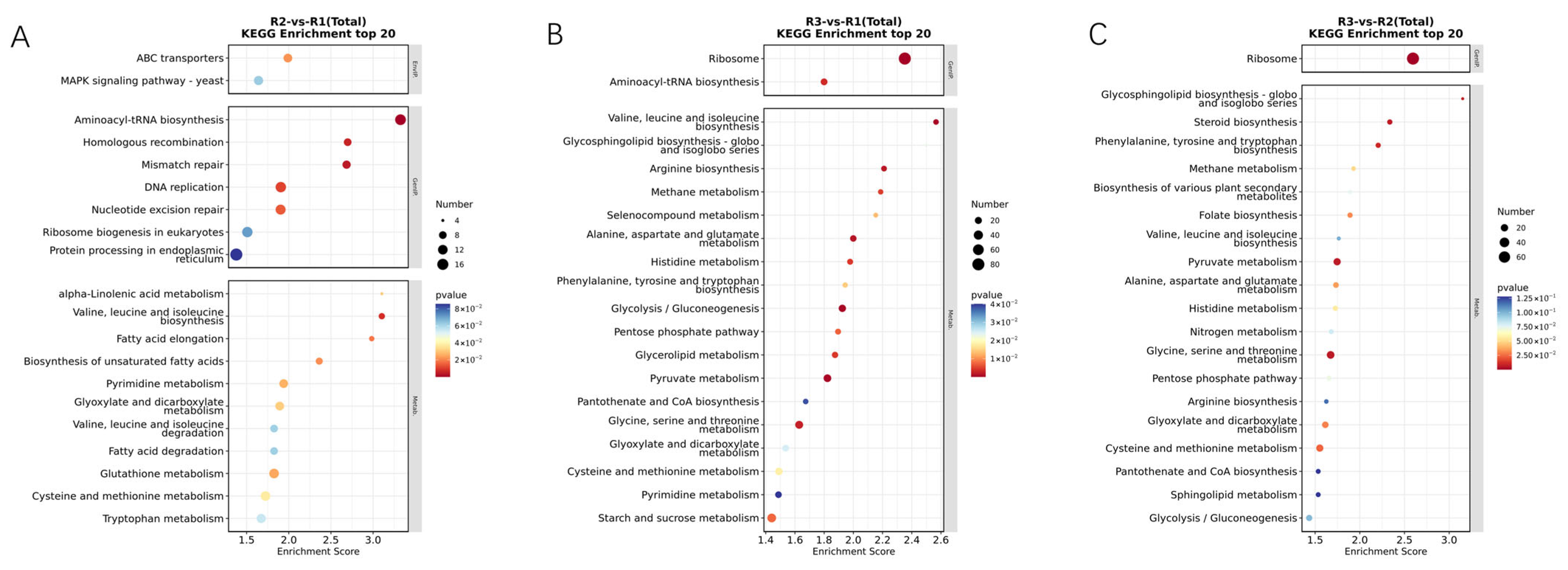
3.4. GO and KEGG Classification
3.5. Transcriptomic Responses to Melanin Synthesis
3.5.1. Analysis of Transcription Factor-Related Genes
3.5.2. Analysis of Glycosidase-Related Genes
3.5.3. Analysis of Metabolism-Related Genes
3.5.4. Analysis of Genes Related to Growth and Development
3.5.5. Analysis of Nitrogen Utilization-Related Genes
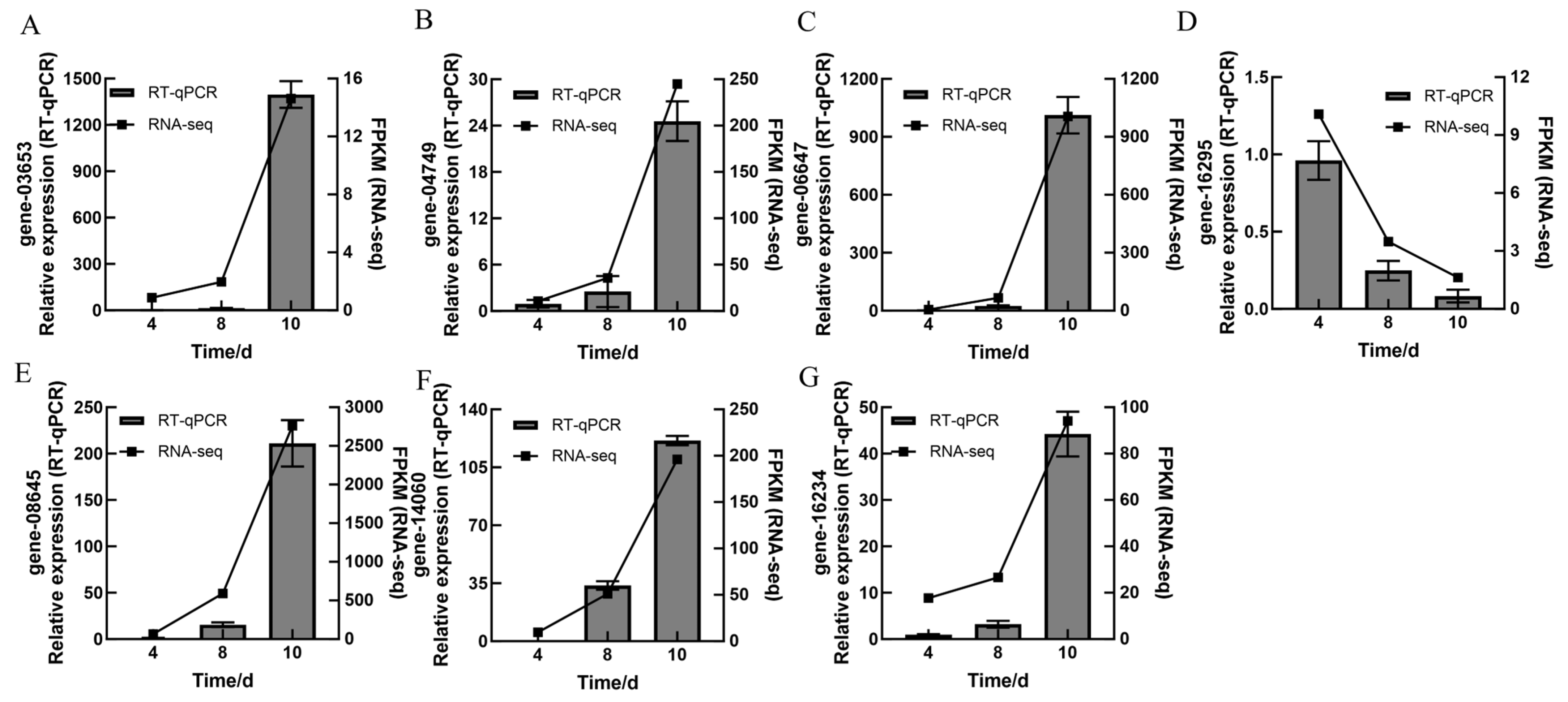
3.6. RT-qPCR Verification of DEGs
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dai, Y.; Li, X.; Song, B.; Sun, L.; Yang, C.; Zhang, X.; Wang, Y.; Zhang, Z.; Fu, Y.; Li, Y. Genomic Analyses Provide Insights Into the Evolutionary History and Genetic Diversity of Auricularia Species. Front. Microbiol. 2019, 10, 2255. [Google Scholar] [CrossRef]
- Chen, N.; Zhang, H.; Zong, X.; Li, S.; Wang, J.; Wang, Y.; Jin, M. Polysaccharides from Auricularia auricula: Preparation, structural features and biological activities. Carbohydr. Polym. 2020, 247, 116750. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, L.; Xue, B.; Zhao, D.; Zhang, Y.; Yan, X. A New Lectin from Auricularia auricula Inhibited the Proliferation of Lung Cancer Cells and Improved Pulmonary Flora. BioMed Res. Int. 2021, 2021, 5597135. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, X.; Liu, Y.; Chen, W.; Wang, C. Studies on the biosynthetic pathways of melanin in Auricularia auricula. J. Basic Microbiol. 2022, 62, 843–856. [Google Scholar] [CrossRef]
- Zong, X.; Zhang, H.; Zhu, L.; Deehan, E.C.; Fu, J.; Wang, Y.; Jin, M. Auricularia auricula polysaccharides attenuate obesity in mice through gut commensal Papillibacter cinnamivorans. J. Adv. Res. 2023, 52, 203–218. [Google Scholar] [CrossRef]
- Shi, Q.; Yang, Z.; Fan, R.; Chu, J.; Fang, C.; Zhang, Y.; Shi, W.; Zhang, Y. Isolation, Characterization, and Antioxidant Activity of Melanin from Auricularia auricula (Agaricomycetes). Int. J. Med. Mushrooms 2023, 25, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Chen, S.; Huang, Q.; Tan, J.; Zeng, J.; Yao, J.; Feng, T.; Wang, G.; Zhang, Y. The lipid lowering and antioxidative stress potential of polysaccharide from Auricularia auricula prepared by enzymatic method. Int. J. Biol. Macromol. 2021, 187, 651–663. [Google Scholar] [CrossRef]
- Han, Q.; Li, H.; Zhao, F.; Gao, J.; Liu, X.; Ma, B. Auricularia auricula Peptides Nutritional Supplementation Delays H2O2-Induced Senescence of HepG2 Cells by Modulation of MAPK/NF-κB Signaling Pathways. Nutrients 2023, 15, 3731. [Google Scholar] [CrossRef]
- Cordero, R.J.B.; Casadevall, A. Melanin. Curr. Biol. 2020, 30, R142–R143. [Google Scholar] [CrossRef]
- Montefiori, D.C.; Zhou, J.Y. Selective antiviral activity of synthetic soluble L-tyrosine and L-dopa melanins against human immunodeficiency virus in vitro. Antivir. Res. 1991, 15, 11–25. [Google Scholar] [CrossRef]
- Pascoe, M.J.; Maillard, J.Y. The role of melanin in Aspergillus tolerance to biocides and photosensitizers. Lett. Appl. Microbiol. 2021, 72, 375–381. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, P.; Dai, X.; Yao, X.; Zhou, S.; Ma, Q.; Liu, J.; Tian, S.; Zhu, J.; Zhang, J.; et al. Extraction, physicochemical properties, and antioxidant activity of natural melanin from Auricularia heimuer fermentation. Front. Nutr. 2023, 10, 1131542. [Google Scholar] [CrossRef]
- Yin, C.M.; Yao, F.; Wu, W.; Fan, X.Z.; Chen, Z.; Ma, K.; Shi, D.F.; Gao, H. Physicochemical Properties and Antioxidant Activity of Natural Melanin Extracted from the Wild Wood Ear Mushroom, Auricularia auricula (Agaricomycetes). Int. J. Med. Mushrooms 2022, 24, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Chen, X.D.; Yu, L.S. Biosynthesis, function and applications of melanin. Biot. Resour. 2020, 42, 652–659. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, X.; Chen, W.; Zhang, L.; Zhu, H. Production of natural edible melanin by Auricularia auricula and its physicochemical properties. Food Chem. 2016, 196, 486–492. [Google Scholar] [CrossRef]
- Wang, J.; Ma, Z.; Wang, C.; Chen, W. Melanin in Auricularia auricula: Biosynthesis, production, physicochemical characterization, biological functions, and applications. Food Sci. Biotechnol. 2024, 33, 1751–1758. [Google Scholar] [CrossRef]
- Fan, X. The study of melanin property and color evaluation of Auricularia cornea and the expression of its key enzymes gene for pigment synthesis. Master’s Thesis, Jilin Agricultural University, Jilin, China, 2019. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; Yan, D.; Gao, Q.; Fan, Y.; Yu, Z.; Liu, Y. Analyses of melanin synthesis pathway and the related genes in Lentinula edodes based on whole genome sequence comparison. Mycosystema 2023, 42, 1114–1128. [Google Scholar] [CrossRef]
- Wang, G.; Li, D.; Zhu, B.; Ma, H.; Mu, Y.; Lv, F.; Jiang, M. Cloning and bioinformatics analysis of dopachrome tautomerase gene from Auricularia auricula. Hubei Agric. Sci. 2022, 61, 153–156. [Google Scholar] [CrossRef]
- He, M.; Wang, T.; Tang, C.; Xiao, M.; Pu, X.; Qi, J.; Li, Y.; Li, X. Metabolomics and transcriptomics reveal the effects of different fermentation times on antioxidant activities of Ophiocordyceps sinensis. J. Fungi. 2025, 11, 51. [Google Scholar] [CrossRef]
- Qiu, Z.; Gao, Y.; Wang, S.; Wang, J.; Wang, X.; Cai, N.; Zhao, J.; Li, T.; Li, H.; Li, T.; et al. Mechanism Underlying Light Intensity-Induced Melanin Synthesis of Auricularia heimuer Revealed by Transcriptome Analysis. Cells 2022, 12, 56. [Google Scholar] [CrossRef]
- Yao, X.G.; Guo, Y.; Han, C.; Tian, S.; Zhu, J.N.; Zhou, S.Y.; Liu, J.N.; Dai, X.D.; Zhang, P.Q.; Ma, Q.F.; et al. Optimization of fermentation medium formula for increasing melanin yield and physicochemical properties of melanin of Auricularia heimuer. Mycosystema 2024, 43, 241–254. [Google Scholar] [CrossRef]
- Fu, R.; Sun, W.; Liu, B.; Sun, J.; Wu, Q.; Liu, X.; Xiang, M. Genome and transcriptome reveal lithophilic adaptation of Cladophialophora brunneola, a new rock-inhabiting fungus. Mycology 2024, 14, 326–343. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Chu, T.; Shang, J.J.; Guan, W.; Yang, R.H.; Bao, D.P.; Tang, L.H. Transcriptome analysis of Lentinula edodes during brown mycelium-film formation. Mycosystema 2022, 41, 260–273. [Google Scholar] [CrossRef]
- Omura, T. Forty years of cytochrome P450. Biochem. Biophys. Res. Commun. 1999, 266, 690–698. [Google Scholar] [CrossRef]
- Chen, H.L.; Zhang, Q.Y.; Sun, K. Laccase-Mediated Oxidative Coupling of Phenolic Compounds in vivo: From Fundamentals to Multifunctional Applications in Green Synthesis. Biotechnol. Bull. 2020, 36, 193–204. [Google Scholar] [CrossRef]
- Bok, J.W.; Balajee, S.A.; Marr, K.A.; Andes, D.; Nielsen, K.F.; Frisvad, J.C.; Keller, N.P. LaeA, a regulator of morphogenetic fungal virulence factors. Eukaryot. Cell 2005, 4, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, M.; Li, L.; Si, J.; Song, B.; Zhou, C.; Yu, M.; Wang, X.; Zhang, Y.; Ding, G.; et al. Overexpression of the Global Regulator LaeA in Chaetomium globosum Leads to the Biosynthesis of Chaetoglobosin Z. J. Nat. Prod. 2016, 79, 2487–2494. [Google Scholar] [CrossRef]
- Luo, Q.; Li, N.; Xu, J.W. A methyltransferase LaeA regulates ganoderic acid biosynthesis in Ganoderma lingzhi. Front. Microbiol. 2022, 13, 1025983. [Google Scholar] [CrossRef]
- Rispail, N.; Soanes, D.M.; Ant, C.; Czajkowski, R.; Grünler, A.; Huguet, R.; Perez-Nadales, E.; Poli, A.; Sartorel, E.; Valiante, V.; et al. Comparative genomics of MAP kinase and calcium-calcineurin signalling components in plant and human pathogenic fungi. Fungal Genet. Biol. 2009, 46, 287–298. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef]
- Lu, Z.M.; Zhang, R.T.; Huang, X.B.; Cao, X.T.; Shen, X.Y.; Fan, L.; Hou, C.L. Optimisation of hypocrellin production in Shiraia-like fungi via genetic modification involving a transcription factor gene and a putative monooxygenase gene. Mycology 2023, 15, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, M.H.; Li, X.Y.; Li, Q.R.; Peng, L.T. Cloning and expression analysis of the PacC gene of the postharvest pathogen Penicillium italicum in citrus. J. Food Sci. Technol. 2022, 43, 145–152. [Google Scholar] [CrossRef]
- Zhang, S.J. Functional studies of the thiol-disulfide reductase Trx in Magnaporthe oryzae and a unique secreted protein in Fusarium graminearum. Unpublished doctoral dissertation, Northwest A&F University, Yangling, China, 2015. [Google Scholar]
- Sun, K.; Li, Y.; Gai, Y.; Wang, J.; Jian, Y.; Liu, X.; Wu, L.; Shim, W.B.; Lee, Y.W.; Ma, Z.; et al. HapX-mediated H2B deub1 and SreA-mediated H2A.Z deposition coordinate in fungal iron resistance. Nucleic Acids Res. 2023, 51, 10238–10260. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Shen, D.; Wang, J.; Dong, Y.; Zhang, M.; Tang, Z.; Xia, Q.; Nyawira, K.T.; Jing, M.; Dou, D.; Xia, A. The glycoside hydrolase 18 family chitinases are associated with development and virulence in the mosquito pathogen Pythium guiyangense. Fungal Genet. Biol. 2009, 135, 103290. [Google Scholar] [CrossRef]
- Jiang, X.Y.; Han, W.; Liu, Y.F.; Tang, C.H.; Feng, J.; Zhang, J.S. Identification of key factors affecting liquid fermentation of Ganoderma lucidum for triterpenes: A review. Microbiol. Bull. 2023, 50, 2155–2172. [Google Scholar] [CrossRef]
- Li, X.W.; Liu, Z.Y.; Xu, Y.J.; Zhu, J.B.; Wu, Y.M. Explore of Molecular Mechanism on Fungal Elicitors Regulating Shikonin Synthesis. Chin. Agric. Sci. Bull. 2024, 26, 78–88. [Google Scholar] [CrossRef]
- Hu, Y.S.; Lv, A.; Zhang, J.Y.; LEI, Y.; Wang, L.; Lv, Y.Y. Research progresses of aflatoxin biosynthetic regulation by LaeA. J. Henan Univ. Technol. 2018, 39, 127–132. [Google Scholar] [CrossRef]
- Wang, G.; Sun, P.; Gong, Z.; Gu, L.; Lou, Y.; Fang, W.; Zhang, L.; Su, L.; Yang, T.; Wang, B.; et al. Srk1 kinase, a SR protein-specific kinase, is important for sexual reproduction, plant infection and pre-mRNA processing in Fusarium graminearum. Environ. Microbiol. 2018, 20, 3261–3277. [Google Scholar] [CrossRef]
- Zang, H.; Shackelford, R.; Bewley, A.; Beeser, A.E. Mutational Analyses of the Cysteine-Rich Domain of Yvh1, a Protein Required for Translational Competency in Yeast. Biology 2022, 11, 1246. [Google Scholar] [CrossRef] [PubMed]
- Han, T.L.; Cannon, R.D.; Gallo, S.M.; Villas-Bôas, S.G. A metabolomic study of the effect of Candida albicans glutamate dehydrogenase deletion on growth and morphogenesis. NPJ Biofilms Microbiomes 2019, 5, 13. [Google Scholar] [CrossRef]
- Pan, Y.Y.; Liu, G. Research advances on molecular regulation of filamentous fungal secondary metabolism in China. Hereditas 2018, 40, 874–887. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Annotation | Primer Sequence (5’→3′) |
|---|---|---|
| Reference gene | 18S rRNA | F: CTGGCTCTGTCAGTGTAG R: TCCGATAACGAACGAGAC |
| gene-03653 | aes1 | F: TCACTGGCGTTGATCCTG R: TGAACAGCGCCCATTCCGTTGTGTT |
| gene-04749 | CYP075 | F: CCATTGGCAAGCACACAG R: TATTCCCGAAGAACTCCG |
| gene-16234 | srk1 | F: GGCTTCCCGCCTTTCTAC R: GCTGCTCAGCGATCACCT |
| gene-16295 | yvh1 | F: ACCGCTACCTCTGAAACA R: TAAGTCGGCGTTCGTTCGAGAAGTG |
| gene-14060 | ustYa | F: ATGACCATCCCTCGCGCCGTTGCTT R: AAGCAACGGCGCGAGGGATGGTCAT |
| gene-06647 | mdlA | F: CGCACCAGGGAACGGATA R: AGGGAGTGGGAGGCAAGC |
| gene-08645 | adh-1 | F: GACCTGCACGCCATGAAA R: CAAGCAAGGCACGAACCA |
| Sample | Raw Reads (M) | Raw Bases (G) | Clean Reads (M) | Clean Bases (G) | Valid Bases (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| R1_1 | 47.55 | 7.08 | 47.16 | 7.02 | 99.18 | 96.3 |
| R1_2 | 45.66 | 6.79 | 45.26 | 6.73 | 99.13 | 96.63 |
| R1_3 | 46.7 | 6.96 | 46.36 | 6.91 | 99.26 | 95.61 |
| R2_1 | 47.07 | 7.00 | 46.67 | 6.94 | 99.14 | 96.10 |
| R2_2 | 47.20 | 7.03 | 46.82 | 6.97 | 99.20 | 95.06 |
| R2_3 | 47.65 | 7.09 | 47.24 | 7.03 | 99.14 | 96.35 |
| R3_1 | 46.39 | 6.91 | 46.08 | 6.87 | 99.32 | 95.73 |
| R3_2 | 47.26 | 7.04 | 46.91 | 6.99 | 99.26 | 96.04 |
| R3_3 | 47.13 | 7.03 | 46.88 | 7.00 | 99.47 | 95.66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, X.; Zhang, J.; Sheng, C.; Guo, Y.; Han, C.; Tian, S.; Zhu, J.; Zhang, X.; Zhou, S.; Liu, J.; et al. Transcriptome Analysis Reveals Molecular Mechanisms of Melanin Synthesis in Auricularia heimuer Under Different Fermentation Times. Horticulturae 2025, 11, 817. https://doi.org/10.3390/horticulturae11070817
Yao X, Zhang J, Sheng C, Guo Y, Han C, Tian S, Zhu J, Zhang X, Zhou S, Liu J, et al. Transcriptome Analysis Reveals Molecular Mechanisms of Melanin Synthesis in Auricularia heimuer Under Different Fermentation Times. Horticulturae. 2025; 11(7):817. https://doi.org/10.3390/horticulturae11070817
Chicago/Turabian StyleYao, Xiuge, Jiechi Zhang, Chunge Sheng, Yan Guo, Chuang Han, Shuang Tian, Jianan Zhu, Xiaojia Zhang, Shuyang Zhou, Jianing Liu, and et al. 2025. "Transcriptome Analysis Reveals Molecular Mechanisms of Melanin Synthesis in Auricularia heimuer Under Different Fermentation Times" Horticulturae 11, no. 7: 817. https://doi.org/10.3390/horticulturae11070817
APA StyleYao, X., Zhang, J., Sheng, C., Guo, Y., Han, C., Tian, S., Zhu, J., Zhang, X., Zhou, S., Liu, J., Dai, X., Zhang, P., Yin, B., & Ma, Y. (2025). Transcriptome Analysis Reveals Molecular Mechanisms of Melanin Synthesis in Auricularia heimuer Under Different Fermentation Times. Horticulturae, 11(7), 817. https://doi.org/10.3390/horticulturae11070817