
Identification, Classification of the MIKC-Type MADS-Box Gene Family, and Expression Analysis of Female and Male Flower Buds in Walnut (Juglans regia, Juglandaceae)


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Identification of MIKC-Type MADS-Box Genes in Walnut
2.3. Phylogenetic Relationship Analysis of MIKC-Type MADS-Box Genes in Walnut
2.4. Exon–Intron Structural and Conserved Motif Analysis
2.5. Chromosome Localization, Duplication Events, and Divergence Time Estimation with Other Plants
2.6. Functional Annotation Analysis
2.7. Cis-Acting Elements and Protein Interaction Network Analysis
2.8. Subcellular Localization
2.9. Genes Expression and Quantitative Real-Time PCR
3. Results
3.1. Characterization and Distribution of MIKC-Type MADS-Box Genes in Walnut
3.2. Phylogenetic Analysis of MIKC-Type MADS-Box Gene Family Members
3.3. Analysis of Conserved Motifs and Gene Structure in Walnut
3.4. Chromosomal Locations, Gene Duplications, and Synteny Relationship of MIKC-Type MADS-Box Gene Family in Walnut
3.5. Cis-Regulatory Motif Analysis of MIKC-Type MADS-Box Genes
3.6. Transient Expression Analysis
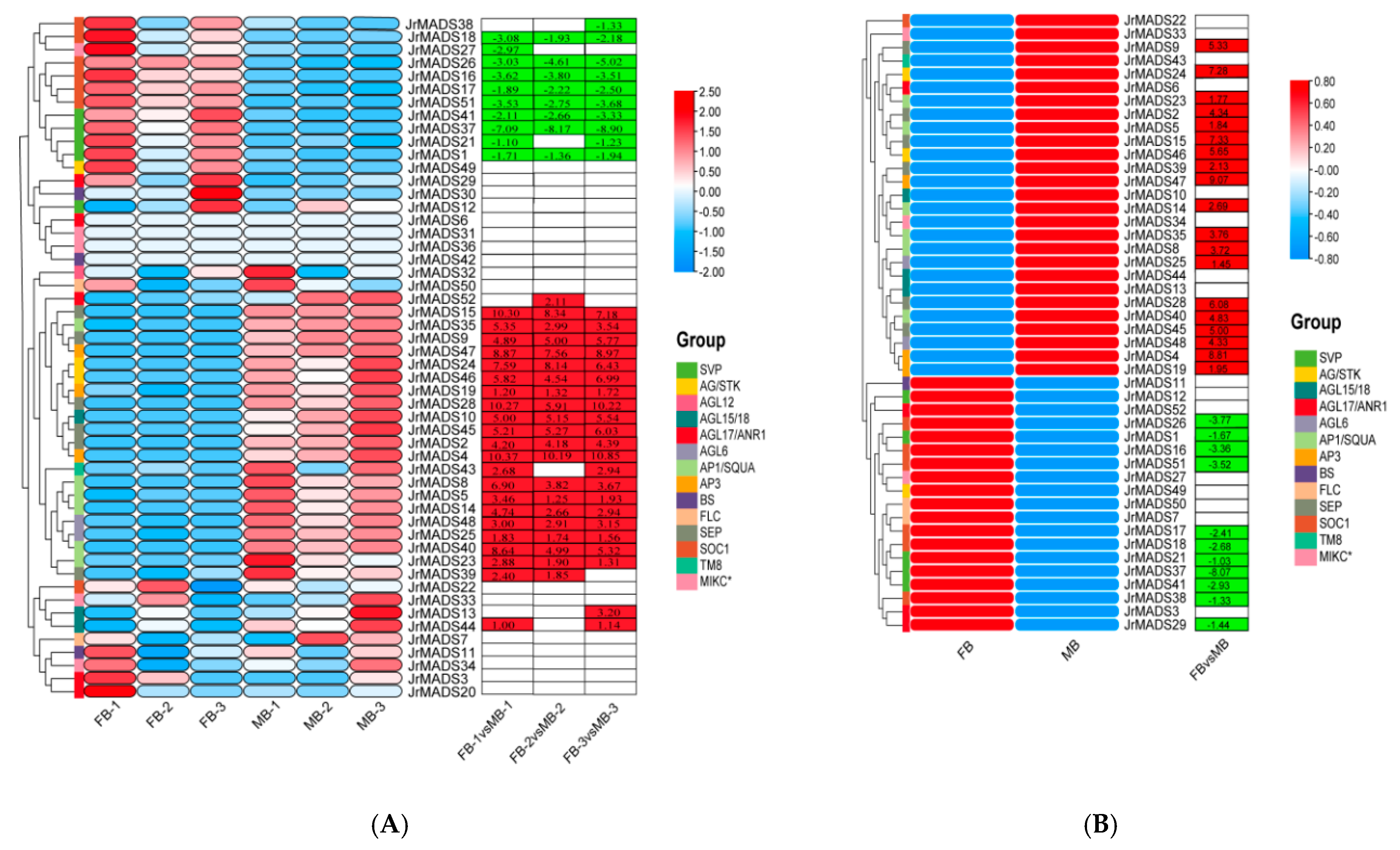
3.7. Analysis of the Expression Patterns of MIKC-Type MADS-Box Genes
3.8. Functional Annotation Analysis
3.9. Regulatory Relationships of Genes Involved in Flower Development
3.10. qRT-PCR to Verify the Expression Pattern of JrMADS Genes and Gibberellin Biosynthetic Enzymes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HMM | Hidden Markov model |
| NCBI | National Center for Biotechnology Information |
| CDD | Conserved Domain Database |
| PI | Theoretical isoelectric point |
| MW | Molecular weight |
| NJ | Neighbor joining |
| CDS | Coding sequence |
| DEGs | Differential expression genes |
| GO | Gene Ontology |
References
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, E.R.; Liljegren, S.J.; Pelaz, S.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Vergara-Silva, F.; Yanofsky, M.F. MADS-box gene evolution beyond flowers: Expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. Cell Mol. Biol. 2000, 24, 457–466. [Google Scholar] [CrossRef]
- Yu, H.; Goh, C.J. Identification and characterization of three orchid MADS-box genes of the AP1/AGL9 subfamily during floral transition. Plant Physiol. 2000, 123, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, L.; Li, Y.; Jia, C.; Zhang, J.; Miao, H.; Hu, W.; Wang, Z.; Xu, B.; Jin, Z. Role for the banana AGAMOUS-like gene MaMADS7 in regulation of fruit ripening and quality. Physiol. Plant. 2015, 155, 217–231. [Google Scholar] [CrossRef]
- Theißen, G.; Melzer, R.; Rümpler, F. MADS-domain transcription factors and the floral quartet model of flower development: Linking plant development and evolution. Development 2016, 143, 3259–3271. [Google Scholar] [CrossRef]
- Yu, L.H.; Wu, J.; Zhang, Z.S.; Miao, Z.Q.; Zhao, P.X.; Wang, Z.; Xiang, C.B. Arabidopsis MADS-Box Transcription Factor AGL21 Acts as Environmental Surveillance of Seed Germination by Regulating ABI5 Expression. Mol. Plant 2017, 10, 834–845. [Google Scholar] [CrossRef]
- Callens, C.; Tucker, M.R.; Zhang, D.; Wilson, Z.A. Dissecting the role of MADS-box genes in monocot floral development and diversity. J. Exp. Bot. 2018, 69, 2435–2459. [Google Scholar] [CrossRef]
- Yin, W.; Yu, X.; Chen, G.; Tang, B.; Wang, Y.; Liao, C.; Zhang, Y.; Hu, Z. Suppression of SlMBP15 Inhibits Plant Vegetative Growth and Delays Fruit Ripening in Tomato. Front. Plant Sci. 2018, 9, 938. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Ribas de Pouplana, L.; Martínez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Münster, T.; Theissen, G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Liu, J.; Fu, X.; Dong, Y.; Lu, J.; Ren, M.; Zhou, N.; Wang, C. MIKC(C)-type MADS-box genes in Rosa chinensis: The remarkable expansion of ABCDE model genes and their roles in floral organogenesis. Hortic. Res. 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Theissen, G.; Kim, J.T.; Saedler, H. Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 1996, 43, 484–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jack, T. Defining subdomains of the K domain important for protein-protein interactions of plant MADS proteins. Plant Mol. Biol. 2004, 55, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Melzer, R.; Verelst, W.; Theissen, G. The class E floral homeotic protein SEPALLATA3 is sufficient to loop DNA in ‘floral quartet’-like complexes in vitro. Nucleic Acids Res. 2009, 37, 144–157. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef]
- Kwantes, M.; Liebsch, D.; Verelst, W. How MIKC* MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol. Biol. Evol. 2012, 29, 293–302. [Google Scholar] [CrossRef]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenetics Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Schilling, S.; Kennedy, A.; Pan, S.; Jermiin, L.S.; Melzer, R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar] [CrossRef]
- Samach, A.; Onouchi, H.; Gold, S.E.; Ditta, G.S.; Schwarz-Sommer, Z.; Yanofsky, M.F.; Coupland, G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 2000, 288, 1613–1616. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Shen, L.; Wu, Y.; Chen, H.; Robertson, M.; Helliwell, C.A.; Ito, T.; Meyerowitz, E.; Yu, H. A repressor complex governs the integration of flowering signals in Arabidopsis. Dev. Cell 2008, 15, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.M.; Ung, N.; Lal, S.; Courtier, J. Specification of reproductive meristems requires the combined function of SHOOT MERISTEMLESS and floral integrators FLOWERING LOCUS T and FD during Arabidopsis inflorescence development. J. Exp. Bot. 2011, 62, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Kosaka, S.; Shibuta, M.; Nagata, K.; Uemura, T.; Nakano, A.; Kaya, H. Transient activity of the florigen complex during the floral transition in Arabidopsis thaliana. Development 2019, 146, dev171504. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, P.; Guo, D.; Wang, N.; Lin, H.; Wang, X.; Niu, L. Transcriptional repressor AGL79 positively regulates flowering time in Arabidopsis. J. Plant Physiol. 2023, 285, 153985. [Google Scholar] [CrossRef]
- Xing, M.; Li, H.; Liu, G.; Zhu, B.; Zhu, H.; Grierson, D.; Luo, Y.; Fu, D. A MADS-box transcription factor, SlMADS1, interacts with SlMACROCALYX to regulate tomato sepal growth. Plant Sci. 2022, 322, 111366. [Google Scholar] [CrossRef]
- Zhao, T.J.; Gai, J.Y. Discovery of new male-sterile cytoplasm sources and development of a new cytoplasmic-nuclear male-sterile line NJCMS3A in soybean. Euphytica 2006, 152, 387–396. [Google Scholar] [CrossRef]
- Wang, J.; Sun, W.; Wang, L.; Liu, X.; Xu, Y.; Sabir, I.A.; Jiu, S.; Wang, S.; Zhang, C. FRUITFULL is involved in double fruit formation at high temperature in sweet cherry. Environ. Exp. Bot. 2022, 201, 104986. [Google Scholar] [CrossRef]
- Xu, H.X.; Meng, D.; Yang, Q.; Chen, T.; Qi, M.; Li, X.Y.; Ge, H.; Chen, J.W. Sorbitol induces flower bud formation via the MADS-box transcription factor EjCAL in loquat. J. Integr. Plant Biol. 2023, 65, 1241–1261. [Google Scholar] [CrossRef]
- Xia, Y.; Xue, B.; Shi, M.; Zhan, F.; Wu, D.; Jing, D.; Wang, S.; Guo, Q.; Liang, G.; He, Q. Comparative transcriptome analysis of flower bud transition and functional characterization of EjAGL17 involved in regulating floral initiation in loquat. PLoS ONE 2020, 15, e0239382. [Google Scholar] [CrossRef]
- Zhang, S.; Yao, J.; Wang, L.; Wu, N.; van Nocker, S.; Li, Z.; Gao, M.; Wang, X. Role of grapevine SEPALLATA-related MADS-box gene VvMADS39 in flower and ovule development. Plant J. Cell Mol. Biol. 2022, 111, 1565–1579. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Zhang, X.; Cai, K.; Li, Y.; Wang, Q.; Qu, G.; Han, R.; Zhao, X. Genome-wide identification and expression analysis of the MADS-box gene family during female and male flower development in Juglans mandshurica. Front. Plant Sci. 2022, 13, 1020706. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, L.; Liu, R.; Cao, S.; Lu, Z. Comparative Proteomic Analysis of Floral Buds before and after Opening in Walnut (Juglans regia L.). Int. J. Mol. Sci. 2024, 25, 7878. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Wu, G.Q.; Li, Z.Q.; Cao, H.; Wang, J.L. Genome-wide identification and expression analysis of the WRKY genes in sugar beet (Beta vulgaris L.) under alkaline stress. PeerJ 2019, 7, e7817. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef]
- Huang, Z.; Duan, W.; Song, X.; Tang, J.; Wu, P.; Zhang, B.; Hou, X. Retention, Molecular Evolution, and Expression Divergence of the Auxin/Indole Acetic Acid and Auxin Response Factor Gene Families in Brassica Rapa Shed Light on Their Evolution Patterns in Plants. Genome Biol. Evol. 2015, 8, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Quan, S.; Zhang, Z.; Kang, C.; Liu, J.; Niu, J. Genome-wide Identification, Characterization and Expression profile of TALE gene family in (Juglans regia L.). Sci. Hortic. 2022, 297, 110945. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Ji, F.; Qiu, J.; Song, X.; Bu, D.; Pan, G.; Ma, Q.; Chen, J.; Huang, R.; et al. A high-quality walnut genome assembly reveals extensive gene expression divergences after whole-genome duplication. Plant Biotechnol. J. 2020, 18, 1848–1850. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Shiu, S.H.; Karlowski, W.M.; Pan, R.; Tzeng, Y.H.; Mayer, K.F.; Li, W.H. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell 2004, 16, 1220–1234. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Parenicová, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef]
- Ma, J.; Yang, Y.; Luo, W.; Yang, C.; Ding, P.; Liu, Y.; Qiao, L.; Chang, Z.; Geng, H.; Wang, P.; et al. Genome-wide identification and analysis of the MADS-box gene family in bread wheat (Triticum aestivum L.). PLoS ONE 2017, 12, e0181443. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Islam, T. Genome-wide analysis and expression profiling of glyoxalase gene families in soybean (Glycine max) indicate their development and abiotic stress specific response. BMC Plant Biol. 2016, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Hu, Z.; Guo, X.; Tian, S.; Chen, G. Genome-Wide Analysis of the MADS-Box Transcription Factor Family in Solanum lycopersicum. Int. J. Mol. Sci. 2019, 20, 2961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fatima, M.; Zhou, P.; Ma, Q.; Ming, R. Analysis of MADS-box genes revealed modified flowering gene network and diurnal expression in pineapple. BMC Genom. 2020, 21, 8. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Zhang, J.; Miao, H.; Wang, J.; Gao, P.; Hu, W.; Jia, C.; Wang, Z.; Xu, B.; et al. Genome-wide analysis of banana MADS-box family closely related to fruit development and ripening. Sci. Rep. 2017, 7, 3467. [Google Scholar] [CrossRef]
- Grimplet, J.; Martínez-Zapater, J.M.; Carmona, M.J. Structural and functional annotation of the MADS-box transcription factor family in grapevine. BMC Genom. 2016, 17, 80. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Feng, C.; Liu, M.; Wang, J.; Hu, Y. Genome-wide identification, characterization of the MADS-box gene family in Chinese jujube and their involvement in flower development. Sci. Rep. 2017, 7, 1025. [Google Scholar] [CrossRef]
- Tian, Y.; Dong, Q.; Ji, Z.; Chi, F.; Cong, P.; Zhou, Z. Genome-wide identification and analysis of the MADS-box gene family in apple. Gene 2015, 555, 277–290. [Google Scholar] [CrossRef]
- Leseberg, C.H.; Li, A.; Kang, H.; Duvall, M.; Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 2006, 378, 84–94. [Google Scholar] [CrossRef]
- Marrano, A.; Britton, M.; Zaini, P.A.; Zimin, A.V.; Workman, R.E.; Puiu, D.; Bianco, L.; Pierro, E.A.D.; Allen, B.J.; Chakraborty, S.; et al. High-quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. GigaScience 2020, 9, giaa050. [Google Scholar] [CrossRef]
- Jain, R.; Jenkins, J.; Shu, S.; Chern, M.; Martin, J.A.; Copetti, D.; Duong, P.Q.; Pham, N.T.; Kudrna, D.A.; Talag, J.; et al. Genome sequence of the model rice variety KitaakeX. BMC Genom. 2019, 20, 905. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.J.N.; Coito, J.L.; Faísca-Silva, D.; Cunha, J.; Costa, M.M.R.; Amâncio, S.; Rocheta, M. Portuguese wild grapevine genome re-sequencing (Vitis vinifera sylvestris). Sci. Rep. 2020, 10, 18993. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Bai, Y.; Sun, H.; Wang, N.; Ma, Y.; Li, M.; Wang, X.; Jiao, C.; Legall, N.; Mao, L.; et al. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 2017, 8, 249. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, B.T.; Denkena, J.; Colomé-Tatché, M.; Shahryary, Y.; Hazarika, R.; Grimwood, J.; Mamidi, S.; Jenkins, J.; Grabowski, P.P.; Sreedasyam, A.; et al. A genome assembly and the somatic genetic and epigenetic mutation rate in a wild long-lived perennial Populus trichocarpa. Genome Biol. 2020, 21, 259. [Google Scholar] [CrossRef]
- Díaz-Riquelme, J.; Lijavetzky, D.; Martínez-Zapater, J.M.; Carmona, M.J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009, 149, 354–369. [Google Scholar] [CrossRef]
- Daminato, M.; Masiero, S.; Resentini, F.; Lovisetto, A.; Casadoro, G. Characterization of TM8, a MADS-box gene expressed in tomato flowers. BMC Plant Biol. 2014, 14, 319. [Google Scholar] [CrossRef]
- Coenen, H.; Viaene, T.; Vandenbussche, M.; Geuten, K. TM8 represses developmental timing in Nicotiana benthamiana and has functionally diversified in angiosperms. BMC Plant Biol. 2018, 18, 129. [Google Scholar] [CrossRef]
- Theissen, G.; Strater, T.; Fischer, A.; Saedler, H. Structural characterization, chromosomal localization and phylogenetic evaluation of two pairs of AGAMOUS-like MADS-box genes from maize. Gene 1995, 156, 155–166. [Google Scholar] [CrossRef]
- Xu, G.; Guo, C.; Shan, H.; Kong, H. Divergence of duplicate genes in exon-intron structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1187–1192. [Google Scholar] [CrossRef]
- Tyagi, S.; Singh, K.; Upadhyay, S.K. Molecular characterization revealed the role of catalases under abiotic and arsenic stress in bread wheat (Triticum aestivum L.). J. Hazard. Mater. 2021, 403, 123585. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. Int. J. Exp. Plant Biol. 2014, 217–218, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, R.; Chen, T.; Qin, D.; An, X. Comprehensive genome-wide characterization of the MIKC-type MADS-box family members and the dynamic expression profiling throughout the development of floral buds in Populus tomentosa. Ind. Crops Prod. 2024, 222, 119968. [Google Scholar] [CrossRef]
- Cheng, Y.; Cheng, L.; Hu, G.; Guo, X.; Lan, Y. Auxin and CmAP1 regulate the reproductive development of axillary buds in Chinese chestnut (Castanea mollissima). Plant Cell Rep. 2023, 42, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Wang, Y.; Zhu, J.; Zhang, Y.; Hou, H. Genomic Organization, Phylogenetic Comparison, and Differential Expression of the Nuclear Factor-Y Gene Family in Apple (Malus domestica). Plants 2020, 10, 16. [Google Scholar] [CrossRef]
- Nie, P.; Xu, G.; Yu, B.; Lyu, D.; Xue, X.; Qin, S. Genome-Wide Identification and Expression Profiling Reveal the Potential Functions of the SWEET Gene Family during the Sink Organ Development Period in Apple (Malus × domestica Borkh.). Agronomy 2022, 12, 1747. [Google Scholar] [CrossRef]
- Rosso, L.; Cantamessa, S.; Bergante, S.; Biselli, C.; Fricano, A.; Chiarabaglio, P.M.; Gennaro, M.; Nervo, G.; Secchi, F.; Carra, A. Responses to Drought Stress in Poplar: What Do We Know and What Can We Learn? Life 2023, 13, 533. [Google Scholar] [CrossRef]
- Yu, H.; Ito, T.; Zhao, Y.; Peng, J.; Kumar, P.; Meyerowitz, E.M. Floral homeotic genes are targets of gibberellin signaling in flower development. Proc. Natl. Acad. Sci. USA 2004, 101, 7827–7832. [Google Scholar] [CrossRef]
- Lai, Y.S.; Zhang, W.; Zhang, X.; Shen, D.; Wang, H.; Qiu, Y.; Song, J.; Li, X. Integrative Analysis of Transcriptomic and Methylomic Data in Photoperiod-Dependent Regulation of Cucumber Sex Expression. G3 2018, 8, 3981–3991. [Google Scholar] [CrossRef]
- Wu, R.M.; Walton, E.F.; Richardson, A.C.; Wood, M.; Hellens, R.P.; Varkonyi-Gasic, E. Conservation and divergence of four kiwifruit SVP-like MADS-box genes suggest distinct roles in kiwifruit bud dormancy and flowering. J. Exp. Bot. 2012, 63, 797–807. [Google Scholar] [CrossRef]
- Ito, T.; Ng, K.H.; Lim, T.S.; Yu, H.; Meyerowitz, E.M. The homeotic protein AGAMOUS controls late stamen development by regulating a jasmonate biosynthetic gene in Arabidopsis. Plant Cell 2007, 19, 3516–3529. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, X.; Su, X.; Rao, P.; Gao, K.; Lei, B.; An, X. Identification and expression analysis of APETALA1 homologues in poplar. Acta Physiol. Plant. 2015, 37, 50. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.; Muiño, J.M.; Blanvillain, R.; Busscher, M.; Busscher-Lange, J.; Dinh, Q.D.; Liu, S.; Westphal, A.H.; Boeren, S.; et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. USA 2012, 109, 1560–1565. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Protein | Chromosome | Protein Length (aa) | MW (kDa) | PI | Subcellular Location |
|---|---|---|---|---|---|---|---|
| JrMADS1 | gene25279 | XP_018806751.1 | Chr11 | 228 | 25.67 | 6.13 | nucleus |
| JrMADS2 | gene25477 | XP_018807034.1 | Chr16 | 252 | 28.64 | 7.67 | nucleus |
| JrMADS3 | gene27721 | XP_018810079.1 | Chr13 | 270 | 31.36 | 9.30 | nucleus |
| JrMADS4 | gene28276 | XP_018810897.1 | Chr08 | 225 | 25.86 | 9.33 | nucleus |
| JrMADS5 | gene29720 | XP_018813004.1 | Chr02 | 248 | 28.72 | 8.73 | nucleus |
| JrMADS6 | gene29996 | XP_018813442.1 | Chr08 | 219 | 25.29 | 8.97 | nucleus |
| JrMADS7 | gene31372 | XP_018815444.1 | Chr10 | 203 | 23.78 | 5.96 | nucleus |
| JrMADS8 | gene32213 | XP_018816775.1 | Chr01 | 255 | 28.98 | 9.14 | nucleus |
| JrMADS9 | gene32212 | XP_018816776.1 | Chr01 | 245 | 28.13 | 8.68 | nucleus |
| JrMADS10 | gene3078 | XP_018817259.1 | Chr05 | 255 | 29.03 | 6.21 | nucleus |
| JrMADS11 | gene32624 | XP_018817273.1 | Chr13 | 231 | 27.05 | 6.01 | nucleus |
| JrMADS12 | gene33316 | XP_018818329.1 | Chr13 | 223 | 25.51 | 6.65 | nucleus |
| JrMADS13 | gene33390 | XP_018818423.1 | Chr03 | 248 | 28.43 | 7.03 | nucleus |
| JrMADS14 | gene33459 | XP_018818517.1 | Chr16 | 246 | 28.34 | 8.50 | nucleus |
| JrMADS15 | gene33549 | XP_018818571.1 | Chr10 | 245 | 28.09 | 9.05 | nucleus |
| JrMADS16 | gene34631 | XP_018820266.1 | Chr02 | 205 | 23.74 | 9.79 | nucleus |
| JrMADS17 | gene34707 | XP_018820385.1 | Chr07 | 232 | 26.74 | 9.00 | nucleus |
| JrMADS18 | gene34785 | XP_018820468.2 | Chr09 | 226 | 25.93 | 9.36 | nucleus |
| JrMADS19 | gene35264 | XP_018821032.1 | Chr11 | 271 | 30.97 | 9.29 | nucleus |
| JrMADS20 | gene39166 | XP_018826847.1 | Chr16 | 231 | 26.71 | 8.91 | nucleus |
| JrMADS21 | gene39189 | XP_018826905.1 | Chr16 | 221 | 25.25 | 6.61 | nucleus |
| JrMADS22 | gene40183 | XP_018828400.1 | Chr10 | 221 | 25.60 | 9.20 | nucleus |
| JrMADS23 | gene3840 | XP_018828587.1 | Chr15 | 248 | 28.73 | 9.50 | nucleus |
| JrMADS24 | gene41314 | XP_018830028.1 | Chr03 | 242 | 27.84 | 9.45 | nucleus |
| JrMADS25 | gene41827 | XP_018830769.1 | Chr01 | 247 | 28.73 | 9.30 | nucleus |
| JrMADS26 | gene41823 | XP_018830813.1 | Chr01 | 207 | 23.85 | 8.70 | nucleus |
| JrMADS27 | gene4912 | XP_018833820.2 | Chr06 | 346 | 38.65 | 5.87 | nucleus |
| JrMADS28 | gene6737 | XP_018836792.1 | Chr13 | 245 | 28.24 | 8.33 | nucleus |
| JrMADS29 | gene6905 | XP_018837041.1 | Chr11 | 226 | 26.01 | 8.69 | nucleus |
| JrMADS30 | gene7096 | XP_018837323.1 | Chr03 | 275 | 32.26 | 6.84 | mitochondrion |
| JrMADS31 | gene8020 | XP_018838782.1 | Chr10 | 366 | 41.12 | 5.46 | nucleus |
| JrMADS32 | gene9597 | XP_018840982.1 | Chr01 | 208 | 23.66 | 8.27 | nucleus |
| JrMADS33 | gene12230 | XP_018845246.2 | Chr16 | 391 | 44.34 | 6.92 | nucleus |
| JrMADS34 | gene12804 | XP_018846114.1 | Chr13 | 355 | 40.60 | 6.36 | nucleus |
| JrMADS35 | gene13376 | XP_018846950.1 | Chr10 | 249 | 28.53 | 9.78 | nucleus |
| JrMADS36 | gene15119 | XP_018849786.1 | Chr16 | 337 | 38.14 | 5.47 | nucleus |
| JrMADS37 | gene16258 | XP_018851438.1 | Chr11 | 281 | 32.36 | 9.63 | nucleus |
| JrMADS38 | gene16410 | XP_018851691.1 | Chr12 | 218 | 25.21 | 9.42 | nucleus |
| JrMADS39 | gene16863 | XP_018852400.1 | Chr13 | 245 | 28.35 | 8.67 | nucleus |
| JrMADS40 | gene16864 | XP_018852402.2 | Chr13 | 259 | 30.01 | 9.22 | nucleus |
| JrMADS41 | gene22497 | XP_018852859.2 | Chr08 | 229 | 25.79 | 5.92 | nucleus |
| JrMADS42 | gene22569 | XP_018858716.2 | Chr16 | 244 | 28.59 | 7.07 | nucleus |
| JrMADS43 | gene371 | XP_035541113.1 | Chr02 | 204 | 23.74 | 10.11 | nucleus |
| JrMADS44 | gene11730 | XP_035541415.1 | Chr14 | 255 | 29.07 | 6.04 | nucleus |
| JrMADS45 | gene29718 | XP_035543716.1 | Chr02 | 246 | 27.67 | 6.35 | chloroplast |
| JrMADS46 | gene1703 | XP_035544964.1 | Chr04 | 246 | 28.10 | 9.42 | nucleus |
| JrMADS47 | gene28585 | XP_035546734.1 | Chr06 | 213 | 24.75 | 7.76 | nucleus |
| JrMADS48 | gene34708 | XP_035547562.1 | Chr07 | 260 | 29.67 | 8.84 | nucleus |
| JrMADS49 | gene9586 | XP_035547898.1 | Chr01 | 256 | 29.24 | 9.39 | nucleus |
| JrMADS50 | gene8967 | XP_035550066.1 | Chr01 | 212 | 24.83 | 6.13 | chloroplast |
| JrMADS51 | gene40184 | XP_035550324.1 | Chr10 | 208 | 24.01 | 9.10 | nucleus |
| JrMADS52 | gene8205 | XP_035551623.1 | Chr11 | 246 | 28.45 | 9.30 | cytoplasm and nucleus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, C.; Fesobi, O.P.; Zhang, Z.; Yuan, X.; Zhao, H.; Quan, S.; Niu, J. Identification, Classification of the MIKC-Type MADS-Box Gene Family, and Expression Analysis of Female and Male Flower Buds in Walnut (Juglans regia, Juglandaceae). Horticulturae 2025, 11, 787. https://doi.org/10.3390/horticulturae11070787
Guo C, Fesobi OP, Zhang Z, Yuan X, Zhao H, Quan S, Niu J. Identification, Classification of the MIKC-Type MADS-Box Gene Family, and Expression Analysis of Female and Male Flower Buds in Walnut (Juglans regia, Juglandaceae). Horticulturae. 2025; 11(7):787. https://doi.org/10.3390/horticulturae11070787
Chicago/Turabian StyleGuo, Caihua, Olumide Phillip Fesobi, Zhongrong Zhang, Xing Yuan, Haochang Zhao, Shaowen Quan, and Jianxin Niu. 2025. "Identification, Classification of the MIKC-Type MADS-Box Gene Family, and Expression Analysis of Female and Male Flower Buds in Walnut (Juglans regia, Juglandaceae)" Horticulturae 11, no. 7: 787. https://doi.org/10.3390/horticulturae11070787
APA StyleGuo, C., Fesobi, O. P., Zhang, Z., Yuan, X., Zhao, H., Quan, S., & Niu, J. (2025). Identification, Classification of the MIKC-Type MADS-Box Gene Family, and Expression Analysis of Female and Male Flower Buds in Walnut (Juglans regia, Juglandaceae). Horticulturae, 11(7), 787. https://doi.org/10.3390/horticulturae11070787