
Comparative Analysis of Rhizosphere Microbiomes in Different Blueberry Cultivars


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Preparation
2.2. Extraction of Rhizosphere Soil DNA and Sequencing of 16S rRNA, ITS
2.3. Bioinformatics Analyses
3. Results
3.1. Diversity and Structure of the Rhizosphere Microbiome in Different Blueberry
3.2. Bacterial and Fungal Community Diversity in Rhizosphere Soil of Different Blueberry Varieties
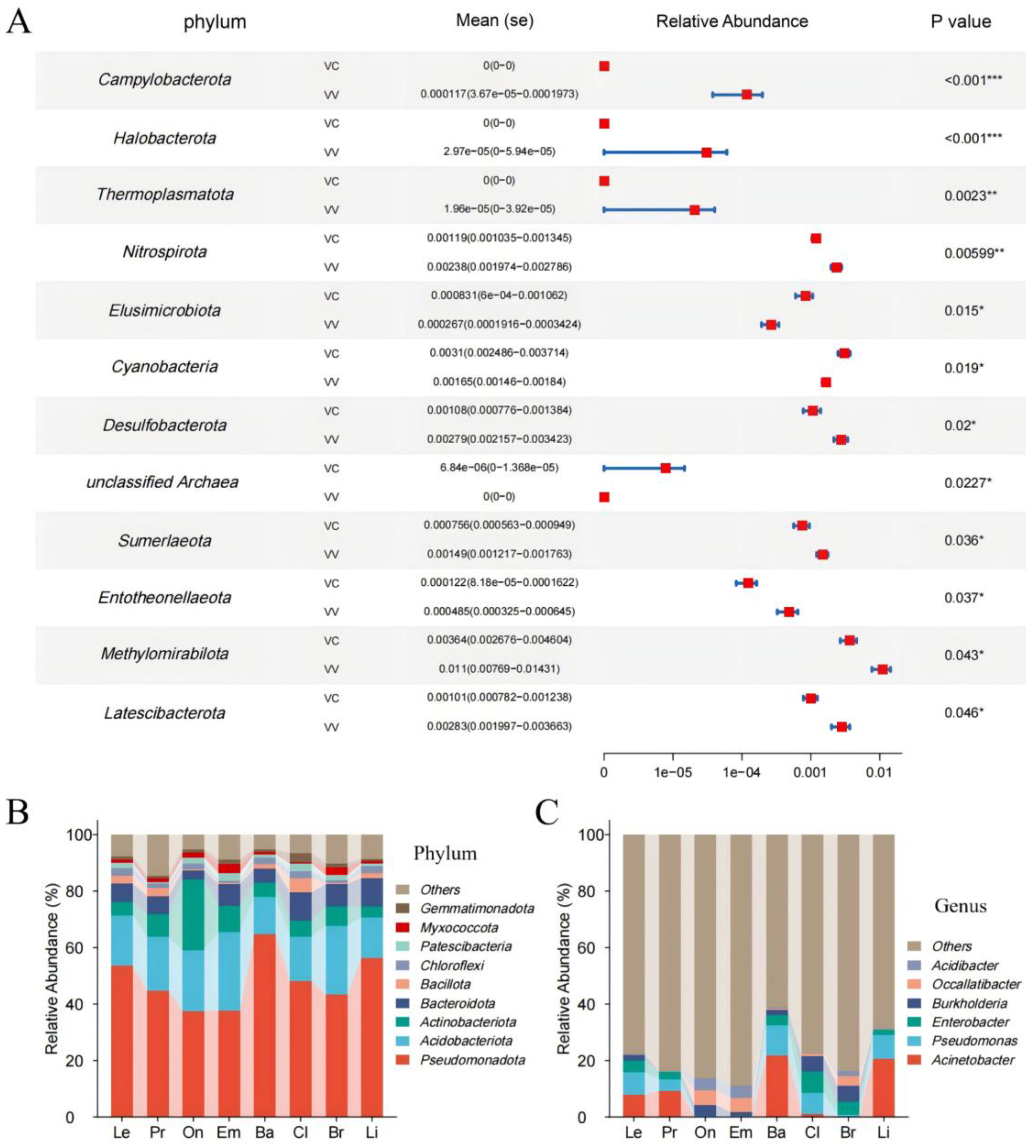
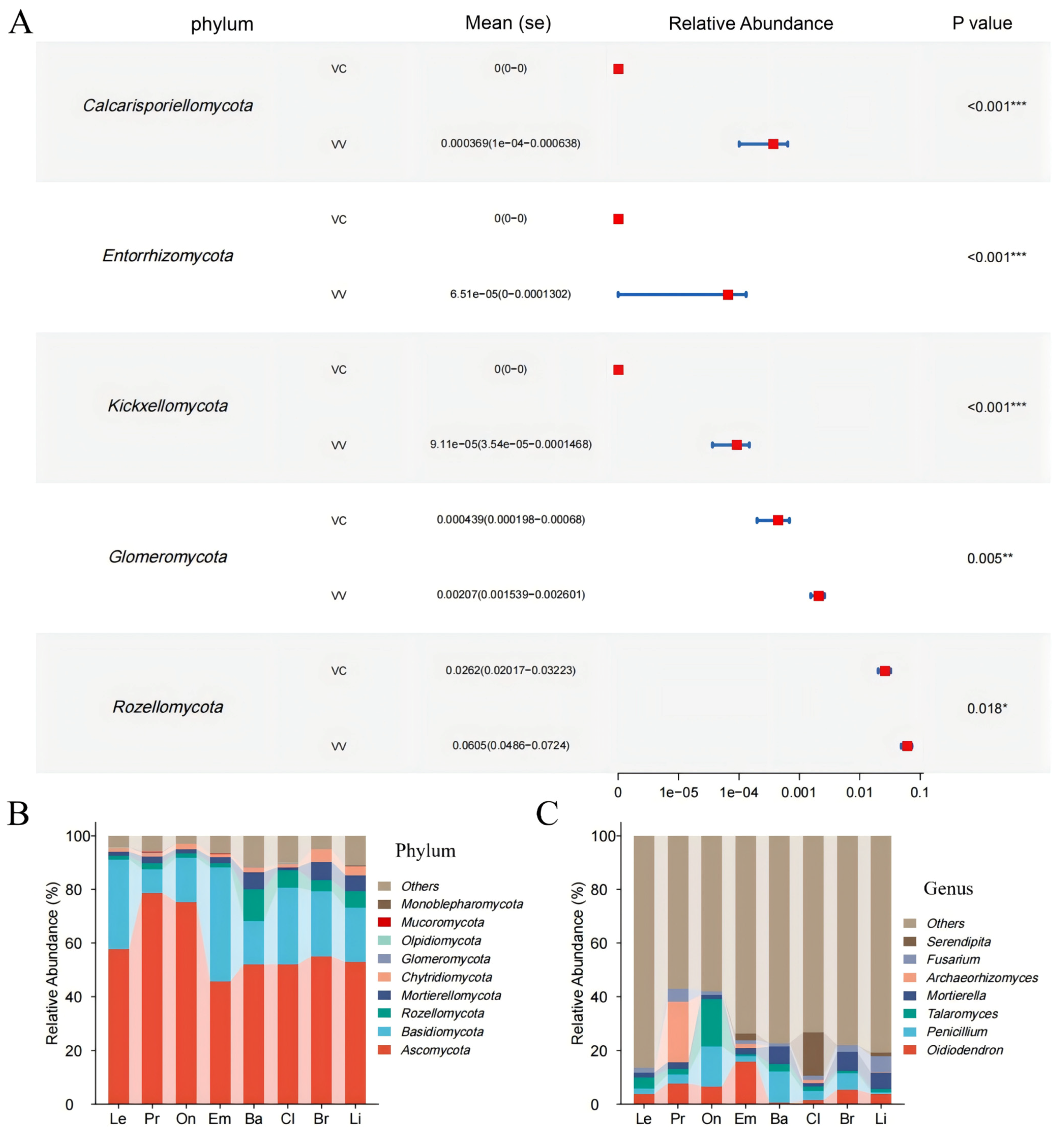
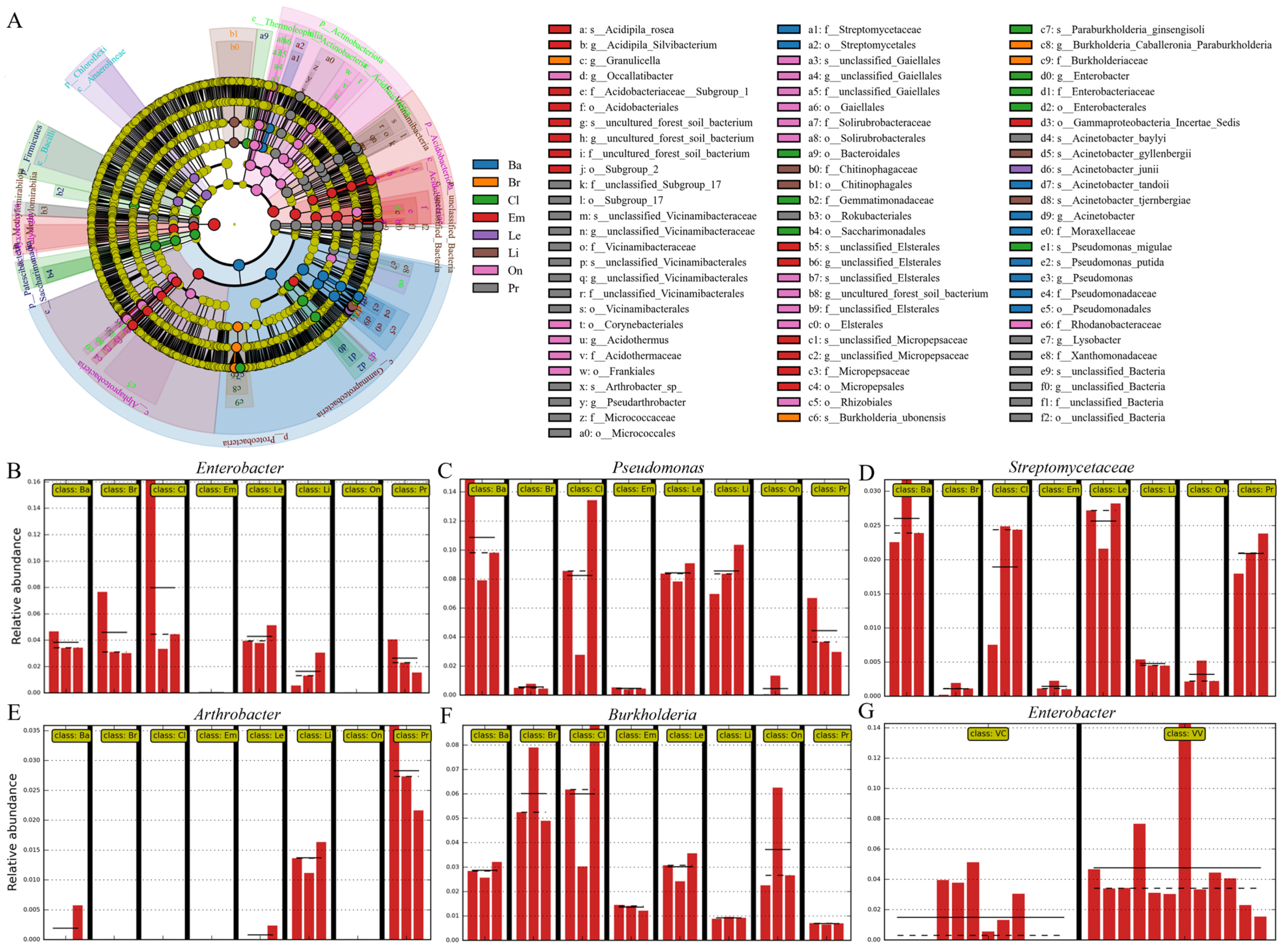
3.3. Bacterial and Fungal Composition Diversity in Different Blueberry Rhizospheres
3.4. Differences Between Rhizobiomes of Blueberry Cultivars
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PGPR | plant growth-promoting rhizobacteria |
| OTUs | operational taxonomic units |
| ASVs | amplicon sequence variants |
| ITS | internal transcribed spacer |
| ERM | ericoid mycorrhizal |
References
- Miller, K.; Feucht, W.; Schmid, M. Bioactive Compounds of Strawberry and Blueberry and Their Potential Health Effects Based on Human Intervention Studies: A Brief Overview. Nutrients 2019, 11, 1510. [Google Scholar] [CrossRef] [PubMed]
- Conner, C.J.; Bocsanczy, A.M.; Spakes-Richter, B.; Norman, D.J. Bacterial Wilt Resistance in Blueberry Species. Int. J. Fruit Sci. 2022, 22, 852–859. [Google Scholar] [CrossRef]
- Singh, U.B.; Malviya, D.; Wasiullah, H.; Singh, S.; Pradhan, J.K.; Singh, B.P.; Roy, M.; Imram, M.; Pathak, N.; Baisyal, B.M.; et al. Bio-protective microbial agents from rhizosphere eco-systems trigger plant defense responses provide protection against sheath blight disease in rice (Oryza sativa L.). Microbiol. Res. 2016, 192, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; He, Y.; Chen, Y. Rhizosphere microbiome regulation: Unlocking the potential for plant growth. Curr. Res. Microbiol. Sci. 2025, 8, 100322. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.; Kruijt, M.; Bruijn, I.D.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Petersen, C.; Hamerich, I.K.; Adair, K.L.; Griem-Krey, H.; Oliva, M.T.; Hoeppner, M.P.; Bohannan, B.J.M.; Schulenburg, H. Host and microbiome jointly contribute to environmental adaptation. ISME J. 2023, 17, 1953–1965. [Google Scholar] [CrossRef]
- Philippot, L.; Chenu, C.; Kappler, A.; Rillig, M.C.; Fierer, N. The interplay between microbial communities and soil properties. Nat. Rev. Microbiol. 2024, 22, 226–239. [Google Scholar] [CrossRef]
- Fan, X.; Ge, A.H.; Qi, S.; Guan, Y.; Wang, R.; Yu, N.; Wang, E. Root exudates and microbial metabolites: Signals and nutrients in plant-microbe interactions. Sci. China Life Sci. 2025. [Google Scholar] [CrossRef]
- Thepbandit, W.; Athinuwat, D. Rhizosphere Microorganisms Supply Availability of Soil Nutrients and Induce Plant Defense. Microorganisms 2024, 12, 558. [Google Scholar] [CrossRef]
- Al-qaysi, S.A.A.S.; Alwan, A.H. Molecular Identification of Rhizosphere Trichoderma spp. and Their Antagonistic Impact Against Some Plant Pathogenic Fungi. Baghdad Sci. J. 2016, 13, 53–65. [Google Scholar] [CrossRef]
- Chepsergon, J.; Moleleki, L.N. Rhizosphere bacterial interactions and impact on plant health. Curr. Opin. Microbiol. 2023, 73, 102297. [Google Scholar] [CrossRef] [PubMed]
- Ge, A.H.; Wang, E. Exploring the plant microbiome: A pathway to climate-smart crops. Cell 2025, 188, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Si, P.; Shao, W.; Yu, H.; Yang, X.; Gao, D.; Qiao, X.; Wang, Z.; Wu, G. Rhizosphere Microenvironments of Eight Common Deciduous Fruit Trees Were Shaped by Microbes in Northern China. Front. Microbiol. 2018, 9, 3147. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xue, R.; Wang, D.; Hu, Y.; Gu, K.; Yang, L.; Zhao, J.; Guan, S.; Su, J.; Jiang, Y. Variations in different preceding crops on the soil environment, bacterial community richness and diversity of tobacco-planting soil. Front. Microbiol. 2024, 15, 1389751. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Q.; Du, W.; Ai, F.; Yin, Y.; Ji, R.; Guo, H. Microbial communities in the rhizosphere of different willow genotypes affect phytoremediation potential in Cd contaminated soil. Sci. Total Environ. 2021, 769, 145224. [Google Scholar] [CrossRef]
- Zhang, Z.; Ge, S.; Fan, L.C.; Guo, S.; Hu, Q.; Ahammed, G.J.; Yan, P.; Zhang, L.P.; Li, Z.Z.; Zhang, J.Y.; et al. Diversity in rhizospheric microbial communities in tea varieties at different locations and tapping potential beneficial microorganisms. Front. Microbiol. 2022, 13, 1027444. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Shen, Z.; Wang, J.; Chen, Y.; Wang, D.; Liu, G.; Han, M. Comparison and interpretation of characteristics of Rhizosphere microbiomes of three blueberry varieties. BMC Microbiol. 2021, 21, 30. [Google Scholar] [CrossRef]
- Xun, W.; Ren, Y.; Yan, H.; Ma, A.; Liu, Z.; Wang, L.; Zhang, N.; Xu, Z.; Miao, Y.; Feng, H.; et al. Sustained Inhibition of Maize Seed-Borne Fusarium Using a Bacillus-Dominated Rhizospheric Stable Core Microbiota with Unique Cooperative Patterns. Adv. Sci. 2023, 10, 2205215. [Google Scholar] [CrossRef]
- Hong, S.; Yuan, X.; Yang, J.; Yang, Y.; Jv, H.; Li, R.; Jia, Z.; Ruan, Y. Selection of rhizosphere communities of diverse rotation crops reveals unique core microbiome associated with reduced banana Fusarium wilt disease. New Phytol. 2023, 238, 2194–2209. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, T.; Huang, Q.; Guo, H.; Zhang, H.; Xu, Q.; Shen, Q.; Ling, N. Core species impact plant health by enhancing soil microbial cooperation and network complexity during community coalescence. Soil Biol. Biochem. 2024, 188, 109231. [Google Scholar] [CrossRef]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.M.; Zhou, X.; Zhang, A.M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Z.D.; Wang, E.T.; Sun, L.Q.; Li, Y. The Effect of Climate Variables, Soil Characteristics, and Peanut Cultivars on the Rhizobial Bacteria Community. Microorganisms 2025, 13, 926. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mavrodi, O.V.; Hou, J.; Blackmon, C.; Babiker, E.M.; Mavrodi, D.V. Comparative Analysis of Rhizosphere Microbiomes of Southern Highbush Blueberry (Vaccinium corymbosum L.), Darrow’s Blueberry (V. darrowii Camp), and Rabbiteye Blueberry (V. virgatum Aiton). Front. Microbiol. 2020, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Wu, Y.; Yang, H.; Wang, S.; Wu, W.; Lyu, L.; Li, W. Long-term cultivation drives dynamic changes in the rhizosphere microbial community of blueberry. Front. Plant Sci. 2022, 13, 96275. [Google Scholar] [CrossRef]
- Zhang, W.B.; Luo, Q.Q.; Zhu, Y.; Ma, J.; Cao, L.; Yang, M.; He, X.L. Microbial diversity in two traditional bacterial douchi from Gansu province in northwest China using illumina sequencing. PLoS ONE 2018, 13, e0197527. [Google Scholar] [CrossRef]
- Orgiazzi, A.; Lumini, E.; Nilsson, R.H.; Girlanda, M.; Vizzini, A.; Bonfante, P.; Bianciotto, V. Unravelling Soil Fungal Communities from Different Mediterranean Land-Use Backgrounds. PLoS ONE 2012, 7, e34847. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput comm unity sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.; McMurdie, P.; Rosen, M.A.W.; Johnson, A.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Dong, L.; Xu, J.; Feng, G.; Li, X.; Chen, S. Soil bacterial and fungal community dynamics in relation to Panax notoginseng death rate in a continuous cropping system. Sci. Rep. 2016, 6, 31802. [Google Scholar] [CrossRef]
- Gao, Z.; Han, M.; Hu, Y.; Li, Z.; Liu, C.; Wang, X.; Tian, Q.; Jiao, W.; Hu, J.; Liu, L.; et al. Effects of Continuous Cropping of Sweet Potato on the Fungal Community Structure in Rhizospheric Soil. Front. Microbiol. 2019, 10, 2269. [Google Scholar] [CrossRef] [PubMed]
- Yurgel, S.N.; Douglas, G.M.; Comeau, A.M.; Mammoliti, M.; Dusault, A.; Percival, D.; Langille, M.G.I. Variation in bacterial and eukaryotic communities associated with natural and managed wild blueberry habitats. Phytobiomes 2017, 1, 102–113. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Y.; Wang, P.; Wang, C.; Ma, J.; Yang, X.; Ma, D.; Li, M. Study on the diversity of fungal and bacterial communities in continuous cropping fields of Chinese chives (Allium tuberosum). BioMed Res. Int. 2020, 2020, e3589758. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Sun, L.; Qiu, C.; Ding, Y.; Gu, H.; Wang, L.; Wang, Z.; Ding, Z. Organic mulching positively regulates the soil microbial communities and ecosystem functions in tea plantation. BMC Microbiol. 2020, 20, 103–112. [Google Scholar] [CrossRef]
- Do, T.; Huynh, V.; Le, L.; Nguyen, T.; Nguyen-Pham, A.; Bui-Thi, M.; Chau-Thi, A.; Tran, S.; Nguyen, V.; Ho-Huynh, T. Microbial diversity analysis using 16S rRNA gene amplicon sequencing of rhizosphere soils from double-cropping rice and rice-shrimp farming systems in Soc Trang, Vietnam. Microbiol. Resour. Announc. 2021, 10, e0059521. [Google Scholar] [CrossRef]
- Zhao, Y.; Fu, W.; Hu, C.; Chen, G.; Xiao, Z.; Chen, Y.; Wang, Z.; Cheng, H. Variation of rhizosphere micro bial community in continuous mono-maize seed production. Sci. Rep. 2021, 11, e1544. [Google Scholar] [CrossRef]
- Saad, N.; Alcala-briseno, R.I.; Polston, J.E.; Olmstead, J.W.; Varsani, A.; Harmon, P.F. Blueberry red ringspot virus genomes from Florida inferred through analysis of blueberry root transcriptomes. Sci. Rep. 2020, 10, e12043. [Google Scholar] [CrossRef]
- Wang, M.; Sun, D.; Xu, Z. Effects of Spent Mushroom Substrate Treated with Plant Growth-Promoting Rhizobacteria on Blueberry Growth and Soil Quality. Microorganisms 2025, 13, 932. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Sui, J.; Liu, Z.; Li, Q.; Ji, C.; Song, X.; Hu, Y.; Wang, C.; Sa, R.; et al. Isolation and characterization of phosphofungi, and screening of their plant growth-promoting activities. AMB Express 2018, 8, 63–71. [Google Scholar] [CrossRef]
- Gómez-Godínez, L.J.; Cisneros-Saguilán, P.; Toscano-Santiago, D.D.; Santiago-López, Y.E.; Fonseca-Pérez, S.N.; Ruiz-Rivas, M.; Aguirre-Noyola, J.L.; García, G. Cultivable and Non-Cultivable Approach to Bacteria from Undisturbed Soil with Plant Growth-Promoting Capacity. Microorganisms 2025, 13, 909. [Google Scholar] [CrossRef]
- Negrușier, C.; Colișar, A.; Rózsa, S.; Chiș, M.S.; Sîngeorzan, S.-M.; Borsai, O.; Negrean, O.-R. Bilberries vs. Blueberries: A Comprehensive Review. Horticulturae 2024, 10, 1343. [Google Scholar] [CrossRef]
- Van Deynze, A.; Zamora, P.; Delaux, P.M.; Heitmann, C.; Jayaraman, D.; Rajasekar, S.; Graham, D.; Maeda, J.; Gibson, D.; Schwartz, K.D.; et al. Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota. PLoS Biol. 2018, 16, e2006352. [Google Scholar] [CrossRef] [PubMed]
- Koyama, R.; Aparecido Ribeiro Júnior, W.; Mariani Zeffa, D.; Tadeu Faria, R.; Mitsuharu Saito, H.; Simões Azeredo Gonçalves, L.; Ruffo Roberto, S. Association of Indolebutyric Acid with Azospirillum brasilense in the Rooting of Herbaceous Blueberry Cuttings. Horticulturae 2019, 5, 68. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Vaccinium Species | Cultivars | Vaccinium Genotypes (Breeding Selections) | Pedigree | Blueberry Type |
|---|---|---|---|---|---|
| VV | V. virgatum (2n = 6× = 72) | Pr | Premier | Tifblue/Woodard | Rabbiteye |
| Ba | Baldwin | Callaway/Tifblue | |||
| Cl | Climax | Black Giant/Myers | |||
| Br | Brightwell | Tifblue/Menditoo | |||
| VC | V. corymbosum (2n = 4× = 48) | Le | Legacy | Elizabeth/US 75 | Highbush |
| Li | Liberty | Elliott/Duke | |||
| Em | Emerald | Florida 4B/Avonblue | |||
| On | O’Neal | US 11/G-144 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, L.; Zhao, Q.; Deng, J.; Cui, L.; Zhang, T.; Yang, Q.; Zhao, S. Comparative Analysis of Rhizosphere Microbiomes in Different Blueberry Cultivars. Horticulturae 2025, 11, 696. https://doi.org/10.3390/horticulturae11060696
Xiao L, Zhao Q, Deng J, Cui L, Zhang T, Yang Q, Zhao S. Comparative Analysis of Rhizosphere Microbiomes in Different Blueberry Cultivars. Horticulturae. 2025; 11(6):696. https://doi.org/10.3390/horticulturae11060696
Chicago/Turabian StyleXiao, Lifeng, Qiuyue Zhao, Jie Deng, Lingyan Cui, Tingting Zhang, Qin Yang, and Sifeng Zhao. 2025. "Comparative Analysis of Rhizosphere Microbiomes in Different Blueberry Cultivars" Horticulturae 11, no. 6: 696. https://doi.org/10.3390/horticulturae11060696
APA StyleXiao, L., Zhao, Q., Deng, J., Cui, L., Zhang, T., Yang, Q., & Zhao, S. (2025). Comparative Analysis of Rhizosphere Microbiomes in Different Blueberry Cultivars. Horticulturae, 11(6), 696. https://doi.org/10.3390/horticulturae11060696