
Genome-Wide Identification of SNP and SSR Markers from Cymbidium goeringii and C. faberi for Their Potential Application in Breeding

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
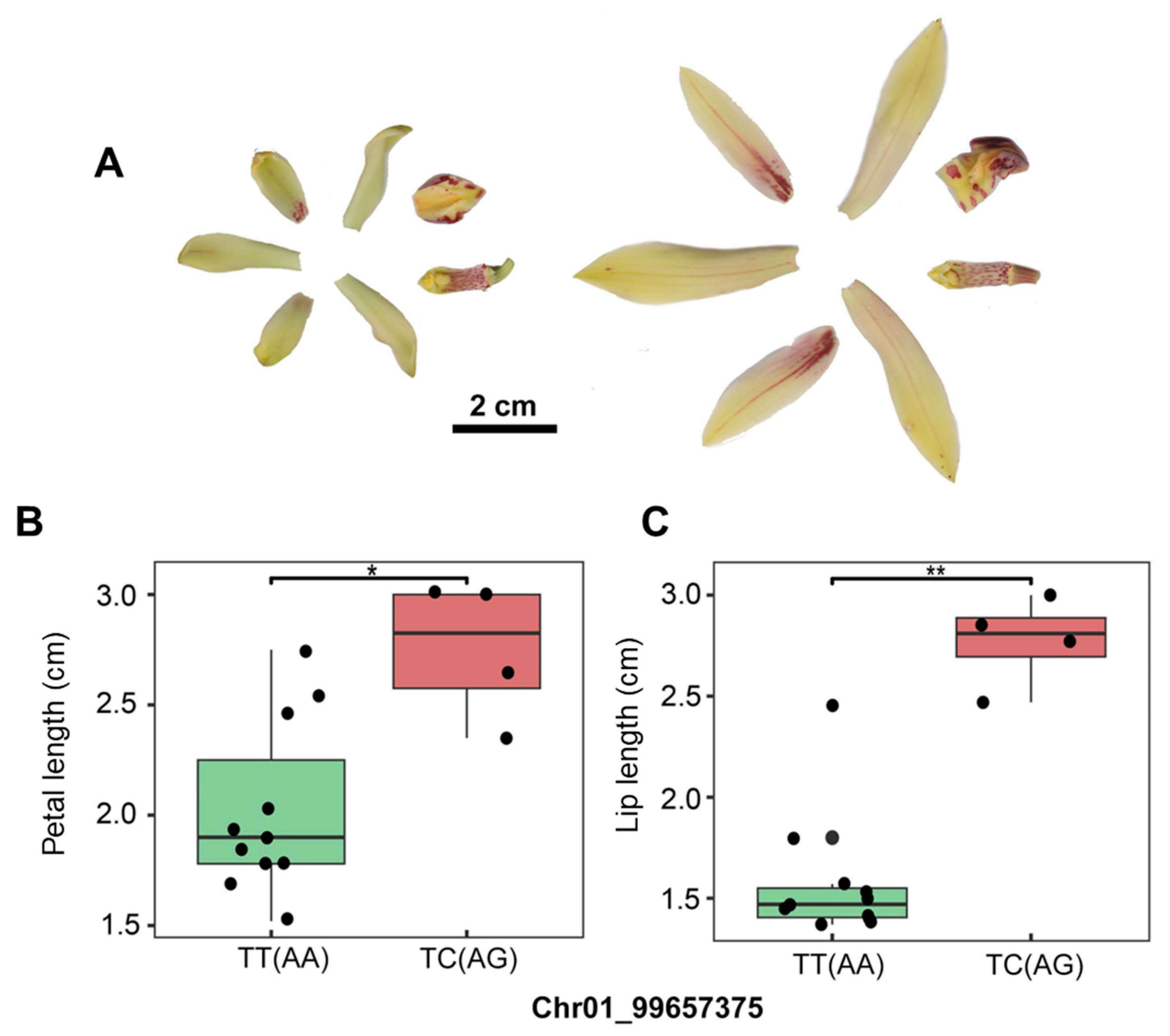
2.2. Measurement of Petal and Lip Length
2.3. DNA Isolation and Quality Control
2.4. RAD Library Preparation, Sequencing, Assembly
2.5. SSR Marker Identification and Genomic Distribution Analysis
2.6. SNP and InDel Detection Pipeline
2.7. Validation of SSR Polymorphisms
2.8. SNP Validation and Genotyping
3. Results
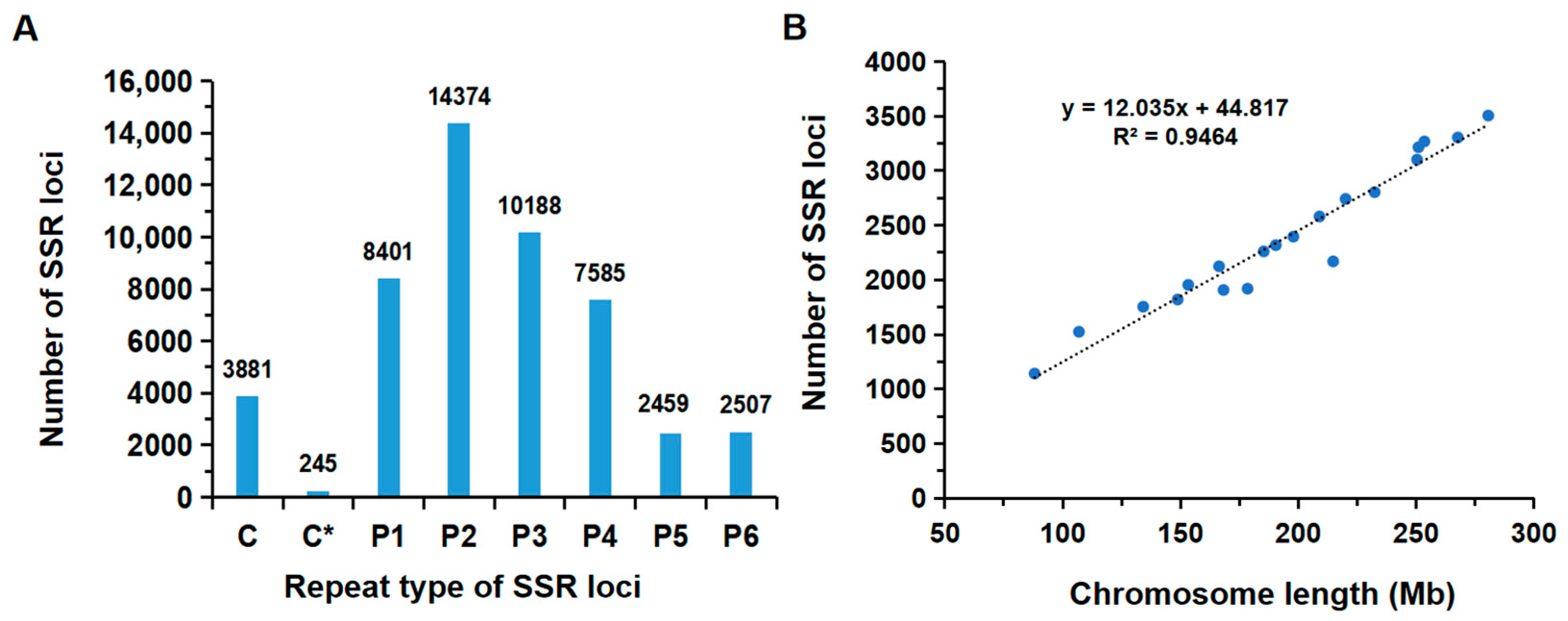
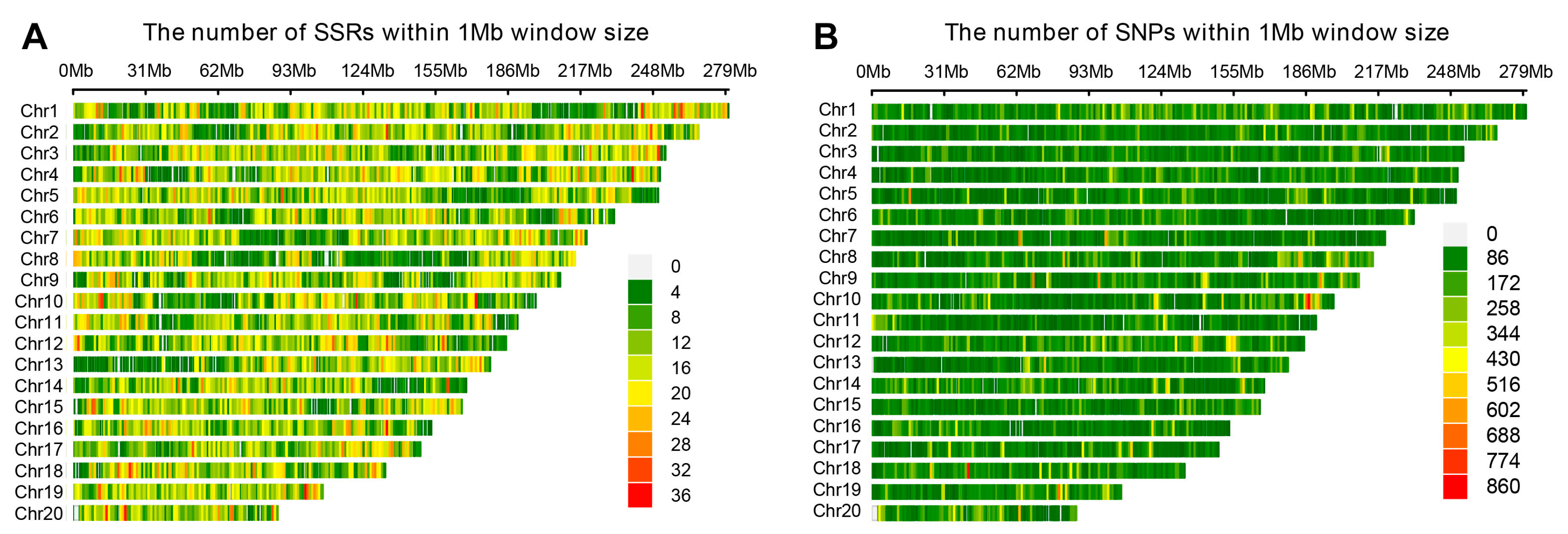
3.1. Genomic Distribution and Characteristics of SSR Markers
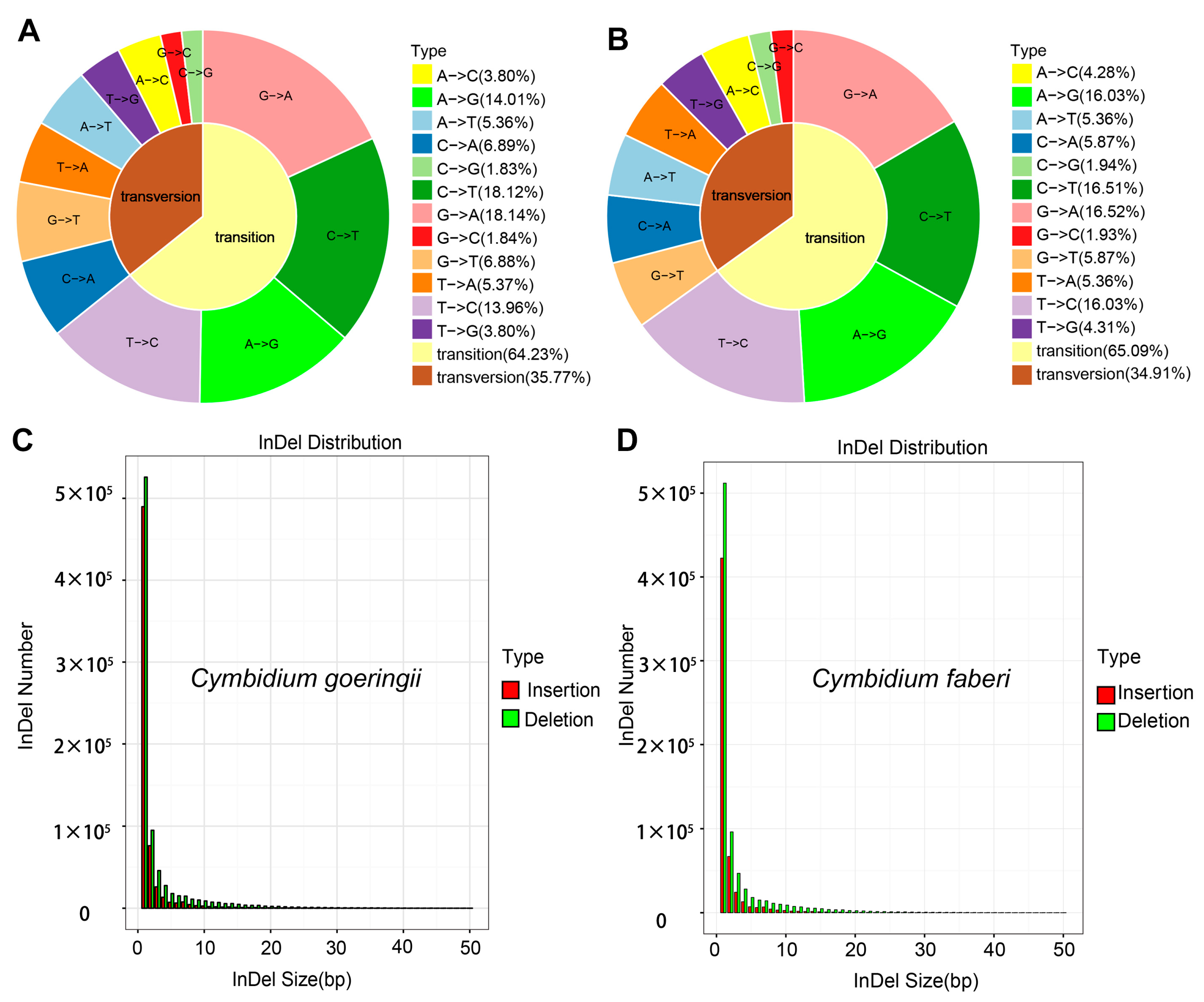
3.2. Identification and Characteristics of SNPs and InDels in C. goeringii and C. faberi
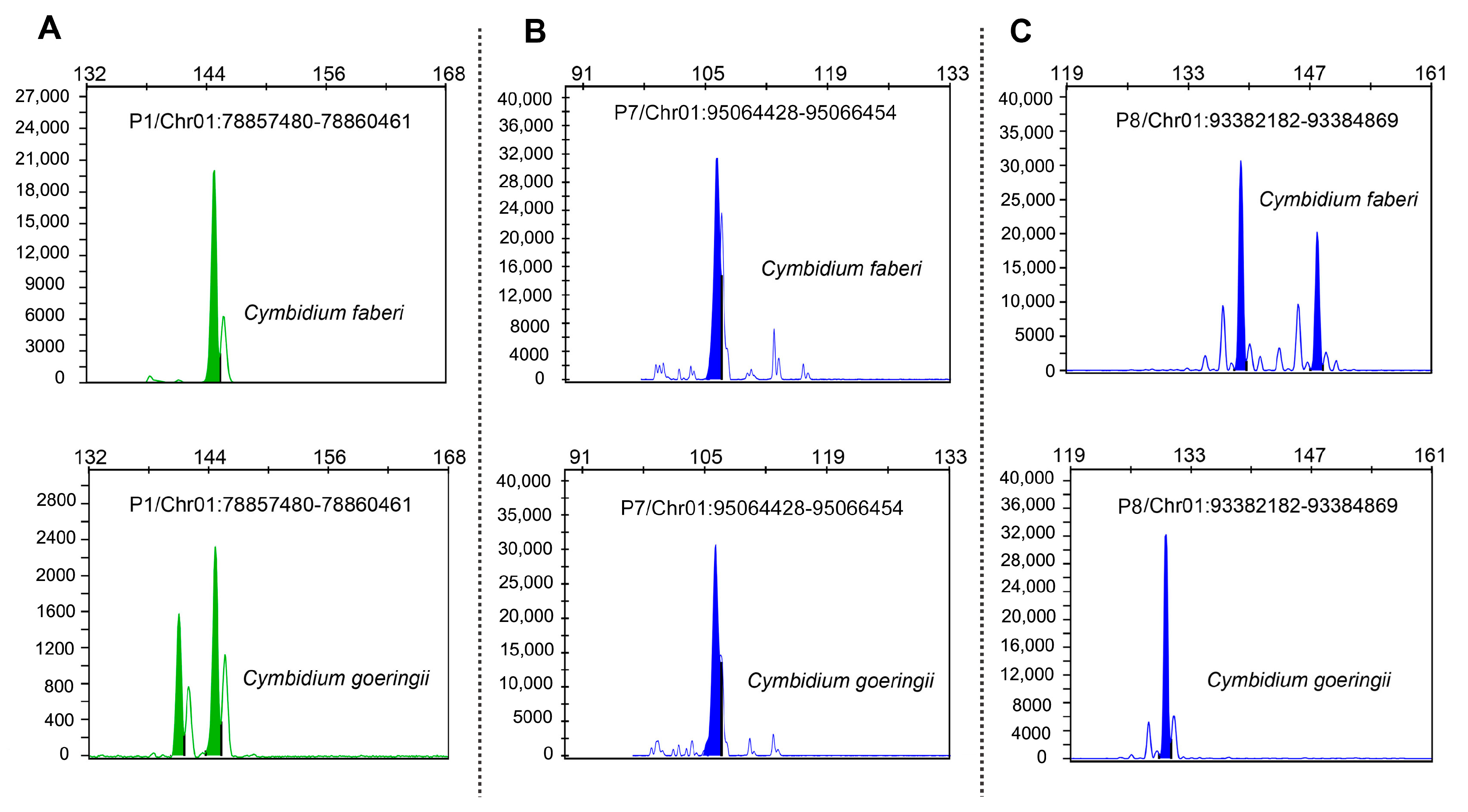
3.3. SSR Marker Validation and Polymorphism Assessment
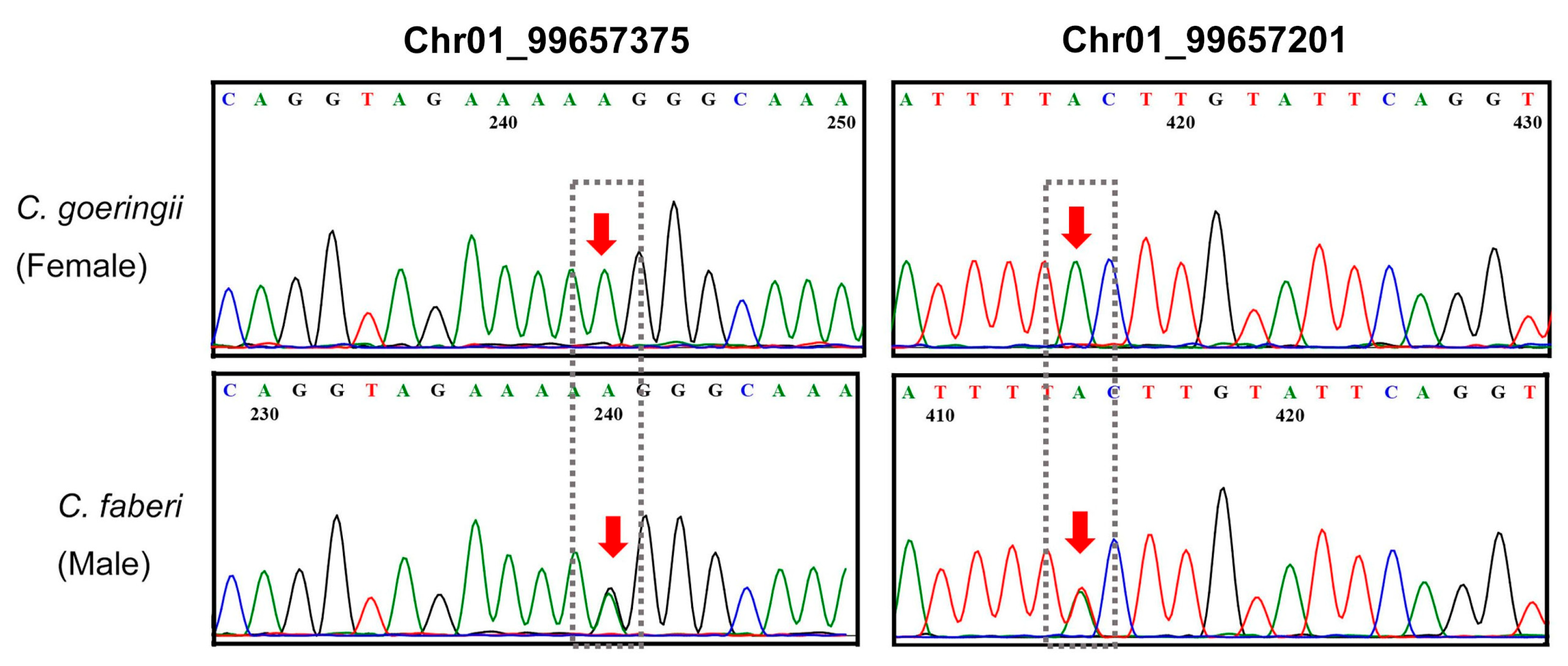
3.4. SNP Marker Validation and Polymorphism Analysis
3.5. Applications of SSR-Base Hybrid Identification
3.6. Applications of SNP-Assisted Trait Prediction in Cymbidium Hybrids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. Genetic interactions among floral homeotic genes of Arabidopsis. Development 1991, 112, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Palomino, M.; Theißen, G. Conserved differential expression of paralogous DEFICIENS-and GLOBOSA-like MADS-box genes in the flowers of Orchidaceae: Refining the ‘orchid code’. Plant J. 2011, 66, 1008. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-Y.; Kao, N.-H.; Li, J.-Y.; Hsu, W.-H.; Liang, Y.-L.; Wu, J.-W.; Yang, C.-H. Characterization of the possible roles for B class MADS box genes in regulation of perianth formation in orchid. Plant Physiol. 2010, 152, 837–853. [Google Scholar] [CrossRef]
- Yang, F.; Guo, Y.; Li, J.; Lu, C.; Wei, Y.; Gao, J.; Xie, Q.; Jin, J.; Zhu, G. Genome-wide association analysis identified molecular markers and candidate genes for flower traits in Chinese orchid (Cymbidium sinense). Hortic. Res. 2023, 10, uhad206. [Google Scholar] [CrossRef]
- Shi, Y.; Zhu, H.; Zhang, J.; Bao, M.; Zhang, J. Development and validation of molecular markers for double flower of Prunus mume. Sci. Hortic. 2023, 310, 111761. [Google Scholar] [CrossRef]
- Tan, C.; Zhang, H.; Chen, H.; Guan, M.; Zhu, Z.; Cao, X.; Ge, X.; Zhu, B.; Chen, D. First report on development of genome-wide microsatellite markers for stock (Matthiola incana L.). Plants 2023, 12, 748. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef] [PubMed]
- García, C.; Guichoux, E.; Hampe, A. A comparative analysis between SNPs and SSRs to investigate genetic variation in a juniper species (Juniperus phoenicea ssp. turbinata). Tree Genet. Genomes 2018, 14, 87. [Google Scholar] [CrossRef]
- Nam, D.; Cha, M.J.; Kim, Y.D.; Awasthi, M.; Do, Y.; Kong, S.G.; Chung, K.W. Microsatellite Dataset for Cultivar Discrimination in Spring Orchid (Cymbidium goeringii). Genes 2023, 14, 1610. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Shen, A.; Tan, Y.; Liu, L.; Li, S.; Tan, Z. Development of KASP markers, SNP fingerprinting and population genetic analysis of Cymbidium ensifolium (L.) Sw. germplasm resources in China. Front. Plant Sci. 2025, 15, 1460603. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.M.; Pereira, R.J.; Coelho, P.S.; Leitao, J.M. Assessment of Wild Rocket (Diplotaxis tenuifolia (L.) DC.) Germplasm Accessions by NGS Identified SSR and SNP Markers. Plants 2022, 11, 3482. [Google Scholar] [CrossRef] [PubMed]
- Moreau, E.L.P.; Medberry, A.N.; Honig, J.A.; Molnar, T.J. Genetic diversity analysis of big-bracted dogwood (Cornus florida and C. kousa) cultivars, interspecific hybrids, and wild-collected accessions using RADseq. PLoS ONE 2024, 19, e0307326. [Google Scholar] [CrossRef]
- Shen, B.; Shen, A.; Liu, L.; Tan, Y.; Li, S.; Tan, Z. Assembly and comparative analysis of the complete multichromosomal mitochondrial genome of Cymbidium ensifolium, an orchid of high economic and ornamental value. BMC Plant Biol. 2024, 24, 255. [Google Scholar] [CrossRef]
- Ai, Y.; Li, Z.; Sun, W.H.; Chen, J.; Zhang, D.Y.; Ma, L.; Zhang, Q.H.; Chen, M.K.; Zheng, Q.D.; Liu, J.F.; et al. The Cymbidium genome reveals the evolution of unique morphological traits. Hortic. Res. 2021, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, G.Z.; Huang, J.; Liu, D.K.; Xue, F.; Chen, X.L.; Chen, S.Q.; Liu, C.G.; Liu, H.; Ma, H.; et al. The Cymbidium goeringii genome provides insight into organ development and adaptive evolution in orchids. Ornament. Plant Res. 2021, 1, 1–13. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, C.; An, Y.; Zhang, X.; Zhou, Y. Integrated transcriptome and metabolome analysis reveals the anthocyanin biosynthesis mechanisms in blueberry (Vaccinium corymbosum L.) leaves under different light qualities. Front. Plant Sci. 2022, 13, 1073332. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Jiang, X. Construction of Bin Map and QTL Mapping of Flower Traits in F1 Population of Cymbidium goeringii ‘Huanghemei’ × C. faberi ‘Laojipin’; Fujian Agriculture and Forestry University: Fuzhou, China, 2024. [Google Scholar]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Ouellette, L.A.; Reid, R.W.; Blanchard, S.G.; Brouwer, C.R. LinkageMapView—Rendering high-resolution linkage and QTL maps. Bioinformatics 2018, 34, 306–307. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Moe, K.T.; Zhao, W.; Song, H.-S.; Kim, Y.-H.; Chung, J.-W.; Cho, Y.-I.; Park, P.H.; Park, H.-S.; Chae, S.-C.; Park, Y.-J. Development of SSR markers to study diversity in the genus Cymbidium. Biochem. Syst. Ecol. 2010, 38, 585–594. [Google Scholar] [CrossRef]
- Li, X.; Jin, F.; Jin, L.; Jackson, A.; Huang, C.; Li, K.; Shu, X. Development of Cymbidium ensifolium genic-SSR markers and their utility in genetic diversity and population structure analysis in Cymbidiums. BMC Genet. 2014, 15, 124. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Shih, H.-C.; Wang, H.-V.; Lin, Y.-S.; Chang, C.-H.; Chiang, Y.-C.; Chou, C.-H. RNA-Seq SSRs of Moth Orchid and Screening for Molecular Markers across Genus Phalaenopsis (Orchidaceae). PLoS ONE 2015, 10, e0141761. [Google Scholar] [CrossRef]
- Yi, S.S.; Huang, M.Z.; Yang, G.S.; Niu, J.H.; Lu, S.J.; Yin, J.M.; Zhang, Z.Q. Development and characterization of expressed sequence-tagged simple sequence repeat markers in Anoectochilus roxburghii. J. Am. Soc. Hortic. Sci. 2022, 147, 349–357. [Google Scholar] [CrossRef]
- Li, X.B.; Luo, J.; Yan, T.L.; Xiang, L.; Jin, F.; Qin, D.H.; Sun, C.B.; Xie, M. Deep sequencing-based analysis of the Cymbidium ensifolium floral transcriptome. PLoS ONE 2013, 8, e85480. [Google Scholar] [CrossRef] [PubMed]
- Azodi, C.B.; Pardo, J.; VanBuren, R.; de los Campos, G.; Shiu, S.H. Transcriptome-based prediction of complex traits in maize. Plant Cell 2020, 32, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Park, H.R.; Lee, A.J.; Nam, D.; Lee, D.G.; Do, Y.; Chung, K.W. Genetic authentication of cultivars with flower-variant types using SSR markers in spring orchid, Cymbidium goeringii. Hortic. Environ. Biotechnol. 2020, 61, 577–590. [Google Scholar] [CrossRef]
- Schulz, D.; Linde, M.; Debener, T. Detection of reproducible major effect QTL for petal traits in garden roses. Plants 2021, 10, 897. [Google Scholar] [CrossRef]
- Chutimanitsakun, Y.; Nipper, R.W.; Cuesta-Marcos, A.; Cistué, L.; Corey, A.; Filichkina, T.; Johnson, E.A.; Hayes, P.M. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genom. 2011, 12, 4. [Google Scholar] [CrossRef]
- Fuentes-Utrilla, P.; Goswami, C.; Cottrell, J.E.; Pong-Wong, R.; Law, A.; A’Hara, S.W.; Lee, S.J.; Woolliams, J.A. QTL analysis and genomic selection using RADseq derived markers in Sitka spruce: The potential utility of within family data. Tree Genet. Genomes 2017, 13, 33. [Google Scholar] [CrossRef]
- Teo, X.; Toh, J.R.H.; Yeoh, H.H.; Su, M.M.J.; Yu, Y.; Chua, H.W.; Lim, C.K.; Kwan, K.M.; Yu, Y.; Wang, M.C. Dissecting the function of MADS-box transcription factors in orchid reproductive development. Front. Plant Sci. 2019, 10, 1474. [Google Scholar] [CrossRef]
- Wang, S.L.; Viswanath, K.K.; Tong, C.G.; An, H.R.; Chen, F.C. Floral induction and flower development of orchids. Front. Plant Sci. 2019, 10, 1258. [Google Scholar] [CrossRef]
- Rasoamanalina, R.O.L.; Mirzaei, K.; El Jaziri, M.; Rasamiravaka, T.; Ramarosandratana, A.V.; Bertin, P. Bertin, Diversity and structure assessment of the genetic resources in a germplasm collection from a vanilla breeding programme in Madagascar. Genet. Resour. Crop Evol. 2023, 70, 548–557. [Google Scholar]
- Baloch, F.S.; Altaf, M.T.; Liaqat, W.; Bedir, M.; Nadeem, M.A.; Cömertpay, G.; Çoban, N.; Habyarimana, E.; Barutçular, C.; Cerit, I.; et al. Recent advancements in the breeding of sorghum crop: Current status and future strategies for marker-assisted breeding. Front. Genet. 2023, 14, 1150616. [Google Scholar] [CrossRef]
- Yang, F.X.; Gao, J.; Wei, Y.L.; Ren, R.; Zhang, G.Q.; Lu, C.Q.; Jin, J.P.; Ai, Y.; Wang, Y.Q.; Chen, L.J.; et al. The genome of Cymbidium sinense revealed the evolution of orchid traits. Plant Biotechnol. J. 2021, 19, 2501–2516. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zong, J.; Luo, Z.J.; Chen, M.J.; Zhao, X.X.; Zhang, D.B.; Qi, Y.P.; Yuan, Z. Development of a RAD-seq based DNA polymorphism identification software, AgroMarker Finder, and its application in rice marker-assisted breeding. PLoS ONE 2016, 11, e0147187. [Google Scholar] [CrossRef] [PubMed]
- Li, C.R.; Dong, N.; Zhao, Y.; Wu, S.S.; Liu, Z.J.; Zhai, J. A review for the breeding of orchids: Current achievements and prospects. Hortic. Plant J. 2021, 7, 380–392. [Google Scholar] [CrossRef]
- Bhattarai, K.; Van Huylenbroeck, J. Breeding, genetics, and genomics of ornamental plants. Horticulturae 2022, 8, 148. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Chen, S.-Y.; Chiu, S.-Y.; Lai, C.-Y.; Lai, P.-H.; Shehzad, T.; Wu, W.-L.; Chen, W.-H.; Paterson, A.H.; Chen, H.-H. High-density genetic map and genome-wide association studies of aesthetic traits in Phalaenopsis orchids. Sci. Rep. 2022, 12, 3346. [Google Scholar] [CrossRef]
- Li, D.-M.; Zhu, G.-F. High-density genetic linkage map construction and QTLs Identification Associated with four leaf-related traits in lady’s slipper orchids (Paphiopedilum concolor × Paphiopedilum hirsutissimum). Horticulturae 2022, 8, 842. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSR Loci | Estimation of Hybrids | |
|---|---|---|---|
| P1/Chr01:78857480-78860461 | P8/Chr01:93382182-93384869 | ||
| C. goeringii (Female) | 141,144 | 132 | P1 |
| C. faberi (Male) | 144 | 138,148 | P2 |
| CH042 | 129,141 | 138,154 | F |
| CH113 | 141,144 | 132,148 | T |
| CH052 | 141,144 | 132,148 | T |
| CH044 | 141,144 | 132,138 | T |
| CH133 | 129,141 | 138,154 | F |
| CH136 | 129,144 | 132 | F |
| CH147 | 141,144 | 132 | F |
| CH178 | 129,144 | 132,154 | F |
| CH099 | 144 | 132 | F |
| CH192 | 129,144 | 132,154 | F |
| CH375 | 129,144 | 132,154 | F |
| CH366 | 144 | 132 | F |
| CH346 | 144 | 132 | F |
| CH454 | 144 | 132 | F |
| CH199 | 141,144 | 132,148 | T |
| CH093 | 141,144 | 132,138 | T |
| CH064 | 141,144 | 132,138 | T |
| CH051 | 129,144 | 132,154 | F |
| CH118 | 141,144 | 132,138 | T |
| CH009 | 141,144 | 132,138 | T |
| CH081 | 144 | 132 | F |
| CH087 | 141,144 | 132,138 | T |
| CH142 | 141,144 | 132,138 | T |
| CH057 | 141,144 | 132,138 | T |
| CH067 | 129,141 | 132,138 | F |
| CH285 | 129,141 | 138,154 | F |
| CH310 | 141,144 | 132,138 | T |
| CH304 | 129,141 | 138,154 | F |
| CH196 | 129,141 | 138,154 | F |
| CH001 | 129,141 | 138,154 | F |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.; Liu, Y.; Jin, C.; Jiang, X.; Chen, X.; Wang, F.; Duan, Y.; Zhuo, X.; Peng, D. Genome-Wide Identification of SNP and SSR Markers from Cymbidium goeringii and C. faberi for Their Potential Application in Breeding. Horticulturae 2025, 11, 622. https://doi.org/10.3390/horticulturae11060622
Cheng M, Liu Y, Jin C, Jiang X, Chen X, Wang F, Duan Y, Zhuo X, Peng D. Genome-Wide Identification of SNP and SSR Markers from Cymbidium goeringii and C. faberi for Their Potential Application in Breeding. Horticulturae. 2025; 11(6):622. https://doi.org/10.3390/horticulturae11060622
Chicago/Turabian StyleCheng, Mengya, Yingqi Liu, Chentai Jin, Xiao Jiang, Xiuming Chen, Fei Wang, Yanru Duan, Xiaokang Zhuo, and Donghui Peng. 2025. "Genome-Wide Identification of SNP and SSR Markers from Cymbidium goeringii and C. faberi for Their Potential Application in Breeding" Horticulturae 11, no. 6: 622. https://doi.org/10.3390/horticulturae11060622
APA StyleCheng, M., Liu, Y., Jin, C., Jiang, X., Chen, X., Wang, F., Duan, Y., Zhuo, X., & Peng, D. (2025). Genome-Wide Identification of SNP and SSR Markers from Cymbidium goeringii and C. faberi for Their Potential Application in Breeding. Horticulturae, 11(6), 622. https://doi.org/10.3390/horticulturae11060622