Whole-Genome Resequencing Reveals Phylogenetic Relationships and Sex Differentiation Mechanisms Among Fujian Cycas Species

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Re-Sequencing Data Processing and Variation Detection
2.3. Genetic Evolution Analysis
3. Results
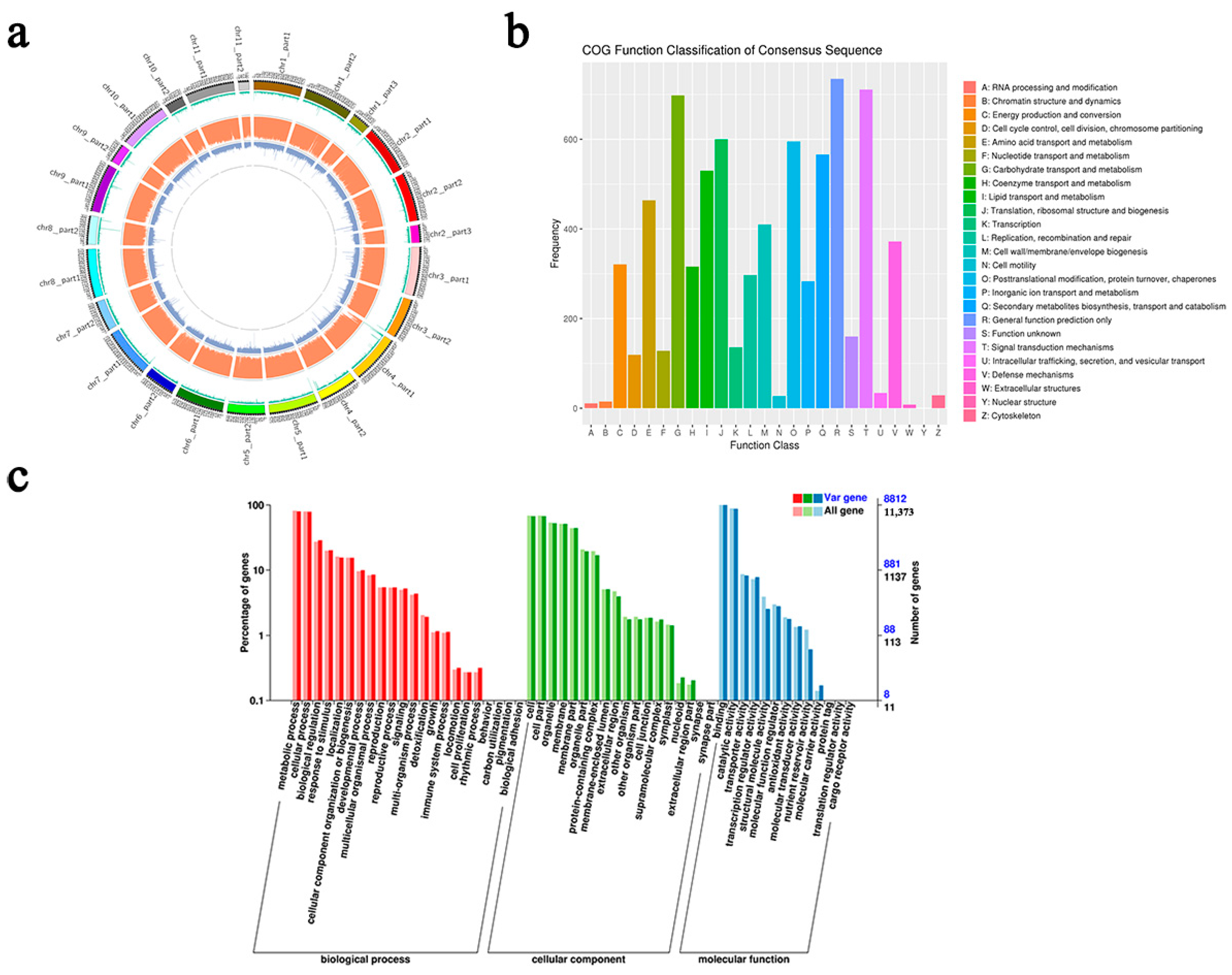
3.1. Whole-Genome Resequencing of Fujian’s Cycas
3.2. SNP and InDel Detection and Annotation
3.3. Population Structure and Genetic Analysis
3.4. Sexual Differentiation and Genomic Variation in Cycas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dehgan, B. Propagation and culture of cycads: A practical approach. In Proceedings of the II International Symposium on Ornamental Palms & Other Monocots from the Tropics 486, Tenerife, Spain, 3–6 February 1997; pp. 123–132. [Google Scholar]
- Zheng, Y.; Liu, J.; Feng, X.; Gong, X. The distribution, diversity, and conservation status of Cycas in China. Ecol. Evol. 2017, 7, 3212–3224. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lindstrom, A.J.; Gong, X. Towards the plastome evolution and phylogeny of Cycas L. (Cycadaceae): Molecular-morphology discordance and gene tree space analysis. BMC Plant Biol. 2022, 22, 116. [Google Scholar] [CrossRef] [PubMed]
- Renner, S.S. The relative and absolute frequencies of angiosperm sexual systems: Dioecy, monoecy, gynodioecy, and an updated online database. Am. J. Bot. 2014, 101, 1588–1596. [Google Scholar] [CrossRef]
- Zhang, M.; He, F. Plant sex affects the structure of plant–pollinator networks in a subtropical forest. Oecologia 2017, 185, 269–279. [Google Scholar] [CrossRef]
- Hobza, R.; Kubat, Z.; Cegan, R.; Jesionek, W.; Vyskot, B.; Kejnovsky, E. Impact of repetitive DNA on sex chromosome evolution in plants. Chromosome Res. 2015, 23, 561–570. [Google Scholar] [CrossRef]
- Pannell, J.R. Plant Sex Determination. CB/Curr. Biol. 2017, 27, R191–R197. [Google Scholar] [CrossRef] [PubMed]
- Marler, T.; Calonje, M. Stem Branching of Cycad Plants Informs Horticulture and Conservation Decisions. Horticulturae 2020, 6, 65. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Li, L.; Yang, T.; Dong, S.; Wei, T.; Wu, S.; Liu, Y.; Gong, Y.; Feng, X.; et al. The Cycas genome and the early evolution of seed plants. Nat. Plants 2022, 8, 389–401. [Google Scholar] [CrossRef]
- Harkess, A.; Zhou, J.; Xu, C.; Bowers, J.; Van der Hulst, R.; Ayyampalayam, S.; Mercati, F.; Riccardi, P.; McKain, M.R.; Kakrana, A.; et al. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 2017, 8, 1279. [Google Scholar] [CrossRef]
- Lappin, F.M.; Medert, C.M.; Hawkins, K.K.; Mardonovich, S.; Wu, M.; Moore, R.C. A polymorphic pseudoautosomal boundary in the Carica papaya sex chromosomes. Mol. Genet. Genom. MGG 2015, 290, 1511–1522. [Google Scholar] [CrossRef]
- Fujita, N.; Ayukawa, Y.; Fuke, M.; Teraoka, T.; Watanabe, K.; Arie, T.; Komatsu, K. Rapid sex identification method of spinach (Spinacia oleracea L.) in the vegetative stage using loop-mediated isothermal amplification. Planta 2017, 245, 221–226. [Google Scholar] [CrossRef]
- Akagi, T.; Henry, I.M.; Tao, R.; Comai, L. A Y-chromosome–encoded small RNA acts as a sex determinant in persimmons. Science 2014, 346, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Akagi, T.; Kawakatsu, T.; Tao, R. Gene networks orchestrated by MeGI: A single-factor mechanism underlying sex determination in persimmon. Plant J. 2019, 98, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.; Cottrell, J.; Ennos, R.A.; Vendramin, G.G.; A’Hara, S.; King, S.; Perry, A.; Wachowiak, W.; Cavers, S. Reconstructing the plant mitochondrial genome for marker discovery: A case study using Pinus. Mol. Ecol. Resour. 2017, 17, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Zhou, Y.; Kathiresan, N.; Yu, Z.; Rivera, L.F.; Yang, Y.; Thimma, M.; Manickam, K.; Chebotarov, D.; Mauleon, R.; Chougule, K.; et al. A high-performance computational workflow to accelerate GATK SNP detection across a 25-genome dataset. BMC Biol. 2024, 22, 13. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Scheible, M.K.R.; Boggs, L.M.; Dunn, R.R.; Ricke, D.O. Using FastID to analyze complex SNP mixtures from indoor dust. J. Forensic Sci. 2023, 68, 768–779. [Google Scholar] [CrossRef]
- Elhaik, E. Principal Component Analyses (PCA)-based findings in population genetic studies are highly biased and must be reevaluated. Sci. Rep. 2022, 12, 14683. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Y.; Gong, X. Genetic diversity, genetic structure and demographic history of Cycas simplicipinna (Cycadaceae) assessed by DNA sequences and SSR markers. BMC Plant Biol. 2014, 14, 187. [Google Scholar] [CrossRef]
- Wang, W.; Ma, L.; Becher, H.; Garcia, S.; Kovarikova, A.; Leitch, I.J.; Leitch, A.R. Astonishing 35S rDNA diversity in the gymnosperm species Cycas revoluta Thunb. Chromosoma 2016, 125, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Leuschner, C.; Weithmann, G.; Weigel, R.; Banzragch, B.; Steiner, W.; Gailing, O. A genome-wide genetic association study reveals SNPs significantly associated with environmental variables and specific leaf area in European beech. Physiol. Plant. 2024, 176, e14334. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Shen, X.; Li, Y. Variation Pattern and Genome-Wide Association Study of Leaf Phenotypic Traits among Ancient Ginkgo biloba L. Populations. Forests 2022, 13, 1764. [Google Scholar] [CrossRef]
- Li, S.; Lv, C.; Lan, L.; Jiang, K.; Zhang, Y.; Li, N.; Deng, C.-L.; Gao, W.-J. DNA methylation is involved in sexual differentiation and sex chromosome evolution in the dioecious plant garden asparagus. Hortic. Res. 2021, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.F.; Mohamoud, Y.A.; Younuskunju, S.; Suhre, K.; Malek, J.A. Evidence of Recombination Suppression Blocks on the Y Chromosome of Date Palm (Phoenix dactylifera). Front. Plant Sci. 2021, 12, 634901. [Google Scholar] [CrossRef]
- Wang, P.; Meng, F.; Yang, Y.; Ding, T.; Liu, H.; Wang, F.; Wang, X. De novo assembling a high-quality genome sequence of Amur grape (Vitis amurensis Rupr.) gives insight into Vitis divergence and sex determination. Hortic. Res. 2024, 11, uhae117. [Google Scholar] [CrossRef]
- Akagi, T.; Varkonyi-Gasic, E.; Shirasawa, K.; Catanach, A.; Henry, I.M.; Mertten, D.; Datson, P.; Masuda, K.; Fujita, N.; Kuwada, E.; et al. Recurrent neo-sex chromosome evolution in kiwifruit. Nat. Plants 2023, 9, 393–402. [Google Scholar] [CrossRef]
- Qiu, T.; Qi, M.; Ding, X.; Zheng, Y.; Zhou, T.; Chen, Y.; Han, N.; Zhu, M.; Bian, H.; Wang, J. The SAUR41 subfamily of SMALL AUXIN UP RNA genes is abscisic acid inducible to modulate cell expansion and salt tolerance in Arabidopsis thaliana seedlings. Ann. Bot. 2020, 125, 805–819. [Google Scholar] [CrossRef]
- Stamm, P.; Kumar, P.P. Auxin and gibberellin responsive Arabidopsis SMALL AUXIN UP RNA36 regulates hypocotyl elongation in the light. Plant Cell Rep. 2013, 32, 759–769. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Z.; Wu, H.; Xu, Z.; Zhang, H.; Qian, W.; Gao, W.; She, H. Genome-Wide Identification and Characterization of MYB Gene Family and Analysis of Its Sex-Biased Expression Pattern in Spinacia oleracea L. Int. J. Mol. Sci. 2024, 25, 795. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, L.; Zhou, P.; Liao, Z.; Lin, H.; Yu, Q.; Ming, R. Sex biased expression of hormone related genes at early stage of sex differentiation in papaya flowers. Hortic. Res. 2021, 8, 147. [Google Scholar] [CrossRef]
- Wu, L.; Xu, H.; Jian, S.; Gong, X.; Feng, X. Geographic factors and climatic fluctuation drive the genetic structure and demographic history of Cycas taiwaniana (Cycadaceae), an endemic endangered species to Hainan Island in China. Ecol. Evol. 2022, 12, e9508. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Chen, B.; Kang, M.; Liu, Y.; Wang, J. Genome-Wide Evidence for Complex Hybridization and Demographic History in a Group of Cycas from China. Front. Genet. 2021, 12, 717200. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Liu, N. New Discoveries of Cycads and Advancement of Conservation of Cycads in China. Bot. Rev. 2004, 70, 93–100. [Google Scholar] [CrossRef]
- Liu, J.; Lindstrom, A.J.; Gong, Y.; Dong, S.; Liu, Y.; Zhang, S.; Gong, X. Eco-evolutionary evidence for the global diversity pattern of Cycas (Cycadaceae). J. Integr. 2024, 66, 1170–1191. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | CR-FZ-18 | CR-FZ-19 | CR-FZ-20 | CS-SX-28 | CS-YT-09 | CT-ZARG01 |
|---|---|---|---|---|---|---|
| Species | C. revoluta | C. revoluta | C. revoluta | C. szechuanensis | C. Szechuaneses | C. taiwaniana |
| Clean_Reads | 195,298,221 | 189,740,456 | 241,606,263 | 199,116,827 | 201,245,198 | 203,092,913 |
| Clean_Base | 58,284,172,560 | 56,681,698,614 | 71,996,807,318 | 59,685,488,476 | 60,324,130,511 | 60,877,892,614 |
| Q30 (%) | 93.31 | 92.50 | 94.07 | 97.96 | 98.10 | 97.95 |
| GC (%) | 36.95 | 37.36 | 37.42 | 34.78 | 35.15 | 34.66 |
| Mapped (%) | 96.43 | 95.83 | 95.94 | 99.36 | 98.98 | 99.39 |
| Properly_mapped (%) | 78.91 | 77.28 | 79.27 | 78.71 | 77.15 | 78.62 |
| Average depth | 5 | 5 | 6 | 5 | 5 | 5 |
| Coverage_1X (%) | 81.55 | 81.14 | 82.63 | 79.52 | 79.91 | 79.97 |
| Sample ID | Species | Q1 | Q2 | Q3 | Q4 | Q5 | Q6 | Q7 | Q8 | Group |
|---|---|---|---|---|---|---|---|---|---|---|
| CR-FZ-18 | C. revoluta | 0.99993 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | Q1 |
| CR-FZ-19 | C. revoluta | 0.00001 | 0.00001 | 0.00001 | 0.99993 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | Q4 |
| CR-FZ-20 | C. revoluta | 0.99993 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | Q1 |
| CS-SX-28 | C. szechuanensis | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.875159 | 0.00001 | 0.00001 | 0.124781 | Q5 |
| CS-YT-09 | C. szechuanensis | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.99993 | Q8 |
| CT-ZARG01 | C. taiwaniana | 0.00001 | 0.584212 | 0.00001 | 0.00001 | 0.00001 | 0.379774 | 0.035964 | 0.00001 | Q2 |
| CP-PRJNA734434 | C. panzhihuaensis | 0.00001 | 0.00001 | 0.99993 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | 0.00001 | Q3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; A. El-Kassaby, Y.; Liu, S.; Zhang, J.; Zhang, L.; Li, J.; Li, W.; Zhang, K.; Zou, M.; Lai, Z.; et al. Whole-Genome Resequencing Reveals Phylogenetic Relationships and Sex Differentiation Mechanisms Among Fujian Cycas Species. Horticulturae 2025, 11, 488. https://doi.org/10.3390/horticulturae11050488
Xu X, A. El-Kassaby Y, Liu S, Zhang J, Zhang L, Li J, Li W, Zhang K, Zou M, Lai Z, et al. Whole-Genome Resequencing Reveals Phylogenetic Relationships and Sex Differentiation Mechanisms Among Fujian Cycas Species. Horticulturae. 2025; 11(5):488. https://doi.org/10.3390/horticulturae11050488
Chicago/Turabian StyleXu, Xinyu, Yousry A. El-Kassaby, Sijia Liu, Juan Zhang, Lanqi Zhang, Junnan Li, Wenkai Li, Kechang Zhang, Minghai Zou, Zhiru Lai, and et al. 2025. "Whole-Genome Resequencing Reveals Phylogenetic Relationships and Sex Differentiation Mechanisms Among Fujian Cycas Species" Horticulturae 11, no. 5: 488. https://doi.org/10.3390/horticulturae11050488
APA StyleXu, X., A. El-Kassaby, Y., Liu, S., Zhang, J., Zhang, L., Li, J., Li, W., Zhang, K., Zou, M., Lai, Z., Lin, L., Zhang, Y., Wu, S., & Chen, B. (2025). Whole-Genome Resequencing Reveals Phylogenetic Relationships and Sex Differentiation Mechanisms Among Fujian Cycas Species. Horticulturae, 11(5), 488. https://doi.org/10.3390/horticulturae11050488