

Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers
Abstract
1. Introduction
2. Materials and Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
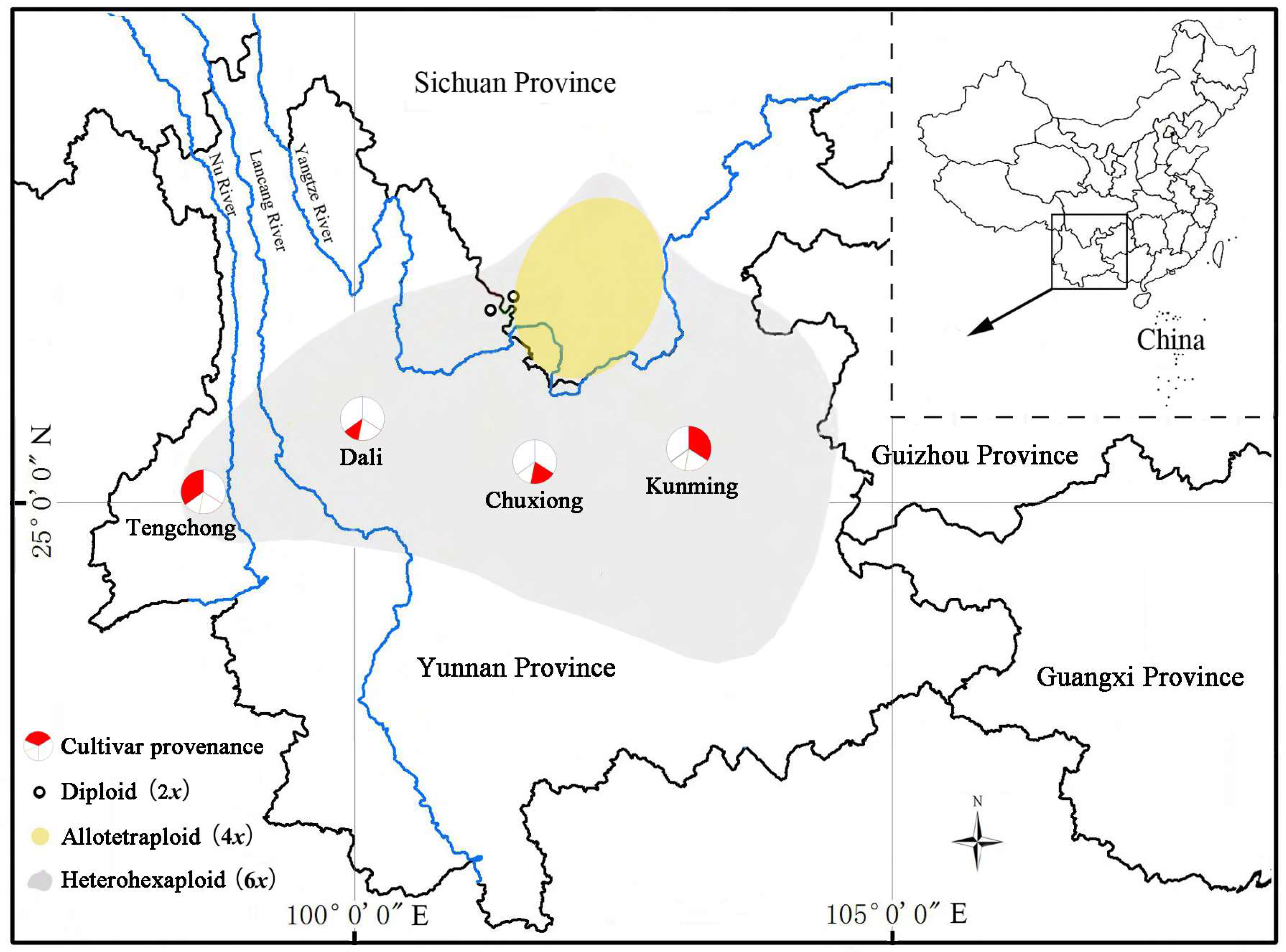
| Provenance | Cultivars |
|---|---|
| Kunming (34 cultivars) | ‘Shizitou’ (8x), ‘Juban’(Figure 2b, 8x), ‘Songzilin’ (8x), ‘Liuye Yinhong’, ‘Damanao’ (6x), ‘Maye Taohong’ (8x), ‘Yujie’, ‘Yipinhong’, ‘Xiaoguiye’ (6x), ‘Zuijiaohong’, ‘Xiaotaohong’, ‘Houye Diechi’, ‘Baoyuhong’, ‘Caiyun’, ‘Taohongpao’ (6x), ‘Dianchi Xiuqiu’, ‘Mudancha’ (8x), ‘Dianchi Mingzhu’ (8x), ‘Zipao’ (8x), ‘Zaomudan’ (8x), ‘Saitaohong’ (8x), ‘Guiye Yanghong’ (6x), ‘Dayinhong’ (8x), ‘Maye Yinhong’, ‘Dataohong’, ‘Baiyi Zaotaohong’ (8x), ‘Lianrui’, ‘Fenhudie’, ‘Dayulan’ (6x), ‘Yanhe’, ‘Jinrui Furong’, ‘Yulancha’, ‘Hongwancha’, ‘Jingancha’ (8x) |
| Chuxiong (19 cultivars) | ‘Dandinghe’ (8x), ‘Zibao’ (8x), ‘Miyilu’ (8x), ‘Chuxiongcha’, ‘Baize’ (8x), ‘Fozuolian’ (6x), ‘Zehe’ (6x), ‘Ziyu’, ‘Lifang’, ‘Weixihong’, ‘Zifen’, ‘Luchengchun’ (6x), ‘Ziyan’, ‘Liuye Meihong’, ‘Yundie’, ‘Jinrui Dahong’, ‘Jianye Diechi’, ‘Zilian’, ‘Weichu’ |
| Dali (12 cultivars) | ‘Daguiye’ (6x), ‘Saijuban’, ‘Yanhong Songzike’, ‘Meihong Guiye’ (6x), ‘Tongzimian’ (8x), ‘Baozhucha’ (6x), ‘Dalicha’, ‘Duxin Dalicha’, ‘Hentiangao’ (10x), ‘Pumencha’ (6x), ‘Songzike’ (6x), ‘Xiguiye’ |
| Tengchong (35 cultivars) | ‘Fentongcao’ (6x), ‘Mudankui’ (Figure 2a), ‘Fengshancha’, ‘Yunzhen’, ‘Wujiao Xiuqiu’ (6x), ‘Hehua Xianzi’ (8x), ‘Tuanye Diechi’ (6x), ‘Shuimeiren’ (6x), ‘Biyu’, ‘Yunfeng’ (8x), ‘Honghua Youcha’ (8x), ‘Fenzhen Mudan’, ‘Huahun’, ‘Jinrui Diechi’, ‘Xiaojiaojiao’, ‘Fenzhaoyun’, ‘Yushizi’, ‘Xianyecha’, ‘Yumeiren’, ‘Hongmei’, ‘Xiyingchun’, ‘Taohong Mudan’, ‘Yunhuacha’, ‘Fentianjiao’, ‘Saierqiao’, ‘Taihe Mudan’, ‘Manao Dahongcha’, ‘Jiaohe’, ‘Naochun Xiuqiu’, ‘Jiaoxiaohong’, ‘Fenhong Xiuqiu’, ‘Yanghong Songzike’, ‘Yulin’, ‘Nansongzi’, ‘Heidahong’ |
3. Results
3.1. Identification of Low-Copy Nuclear Genes of C. reticulata
3.2. Nucleotide and Haploid Diversity of C. reticulata Cultivars
3.3. Genetic Distance, Genetic Structure and Differentiation of C. reticulata Cultivars
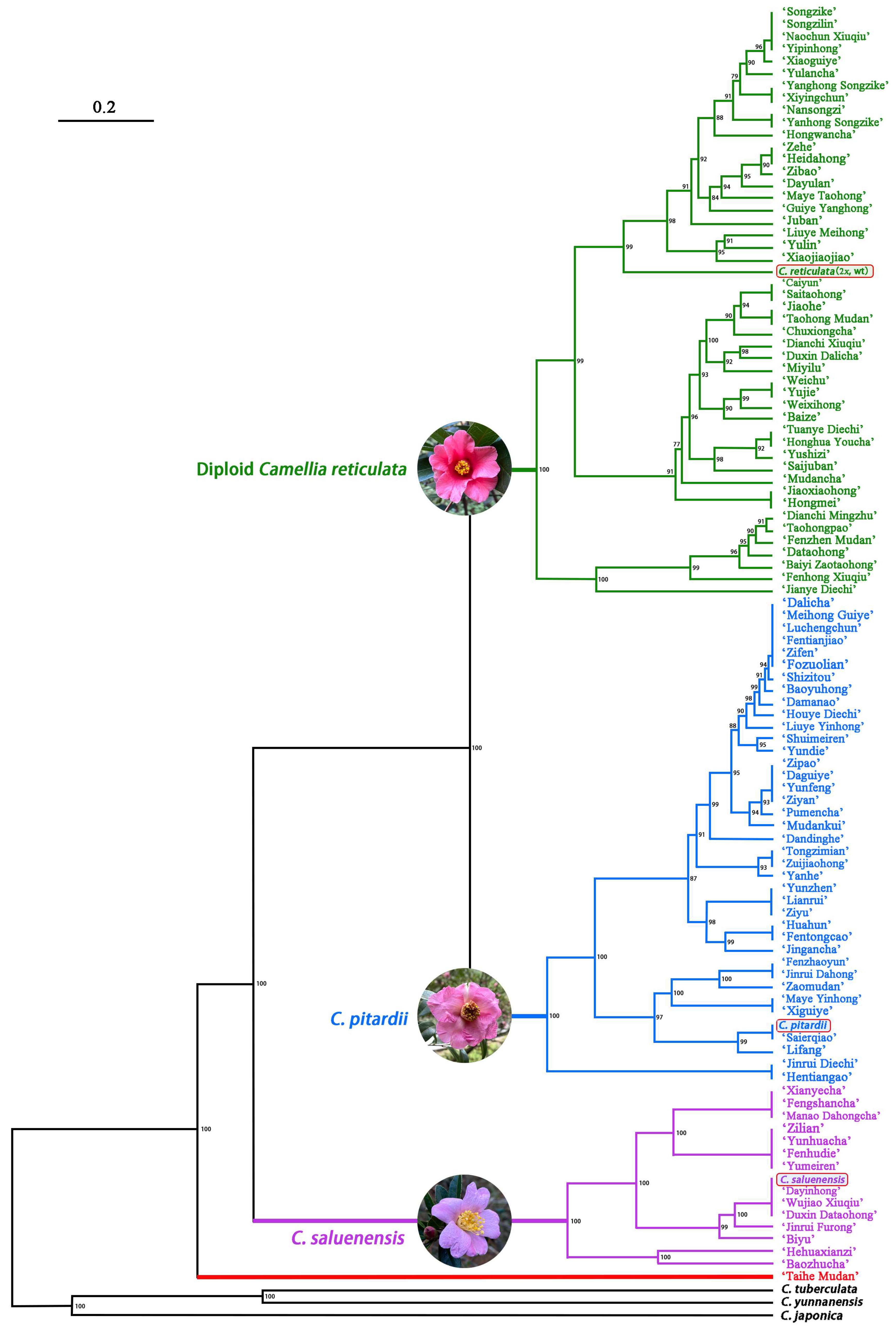
3.4. Phylogenetic Relationships of C. reticulata Cultivars
4. Discussion
4.1. Genetic Diversity of C. reticulata Cultivars
4.2. Genetic Relationships among C. reticulata Cultivars
4.3. Evolutionary Dynamics of C. reticulata Cultivars
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Kunming Association for Science and Technology. Wonderful Flower of Yunnan—Camellia Reticulata; Yunnan Science and Technology Press: Kunming, China, 2010. [Google Scholar]
- Wu, G.; Chen, L.; Wei, Q.; Hu, Y.; Xu, J.; Li, D.; Chen, S.; Wang, Z.; Geng, F. Statistics and phenotypic traits analysis of Camellia reticulata registered cultivars. Acta Hortic. Sin. 2023, 50, 2157–2170. [Google Scholar]
- Zhang, W.; Ming, T. A cytogeographical study of Camellia, Sect Camellia. Acta Bot. Yun. 1998, 20, 321–328. [Google Scholar]
- Vijayan, K.; Zhang, W.; Tsou, C.H. Molecular taxonomy of Camellia (Theaceae) inferred from nrITS sequences. Am. J. Bot. 2009, 96, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Min, T.; Bartholomew, B. Theaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MI, USA, 2007; Volume 12, pp. 368–372. [Google Scholar]
- Xin, T.; Huang, W.; Riek, J.D.; Zhang, S.; Ahmed, S.; Huylenbroeck, J.V.; Long, C. Genetic diversity, population structure, and traditional culture of Camellia reticulata. Ecol. Evol. 2017, 7, 8915–8926. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shao, W.; Zheng, W. Ploidy study on polyploidy cultivars of Camellia reticulata. Sci. Silvae Sin. 2018, 54, 44–48. [Google Scholar]
- Xu, X.; Zheng, W.; Harris, A.J.; Wang, W.; Shao, W.; Wen, J. Assessing the maternal origin in the polyploid complex of Camellia reticulata based on the chloroplast rpl16 intron sequences: Implications for camellia cross breeding. Mol. Breed. 2018, 38, 123. [Google Scholar] [CrossRef]
- Elizabeth, A.Z.; Wen, J. Using nuclear gene data for plant phylogenetics: Progress and prospects II. Next-gen approaches. J. Syst. Evol. 2015, 53, 371–379. [Google Scholar]
- Guo, S.; Ji, P.; Wang, J.; He, Y.; Zhang, Y.; Zhang, F.; Yun, Y.; Zhang, G. Estimation of genetic diversity between and within biparental clones and full-sib families of the Chinese pine using SSR markers. Horticulturae 2023, 9, 1205. [Google Scholar] [CrossRef]
- Chalbi, A.; Chikh-Rouhou, H.; Mezghani, N.; Slim, A.; Fayos, O.; Bel-Kadhi, M.S.; Garcés-Claver, A. Genetic diversity analysis of onion (Allium cepa L.) from the arid region of Tunisia using phenotypic traits and SSR markers. Horticulturae 2023, 9, 1098. [Google Scholar] [CrossRef]
- Zhou, A.; Yue, L.; Li, M.; Liu, D.; Ding, Y. Intra-genomic polymorphism in the nrDNA ITS sequence of Camellia reticulata. Plant Sci. J. 2013, 31, 1–10. [Google Scholar] [CrossRef]
- Yang, J.; Li, H.; Yang, S.; Li, D. The application of four DNA sequences to studying molecular phylogeny of Camellia (Theaceae). Acta Bot. Yun. 2006, 28, 108–114. [Google Scholar]
- Vijayan, K.; Tsou, C.H. Technical report on the molecular phylogeny of Camellia with nrITS: The need for high quality DNA and PCR amplification with Pfu-DNA polymerase. Bot. Stud. 2008, 49, 177–188. [Google Scholar]
- Meseguer, A.S.; Sanmartín, I.; Marcussen, T.; Pfeil, B.E. Utility of low-copy nuclear markers in phylogenetic reconstruction of Hypericum L. (Hypericaceae). Plant Syst. Evol. 2014, 300, 1503–1514. [Google Scholar] [CrossRef]
- Chery, J.G.; Sass, C.; Specht, C.D. Development of single-copy nuclear intron markers for species-level phylogenetics: Case study with Paullinieae (Sapindaceae). Appl. Plant Sci. 2017, 5, 1700051. [Google Scholar] [CrossRef] [PubMed]
- Du, S.H.; Wang, Z.S.; Zhang, J.G. A novel set of single-copy nuclear DNA markers for the genetic study of Salicaceae. Genet. Mol. Res. 2014, 13, 4911–4917. [Google Scholar] [CrossRef]
- Wen, Q.; Zhu, H.; Ye, J.; Xu, L.; Xu, L.; Jiang, X. Authentication of Camellia chekiangoleosa and its close species using RPB2 gene sequences. Mol. Plant Breed. 2015, 13, 2559–2565. [Google Scholar]
- Liu, Y.; Yang, S.; Ji, P.; Gao, L. Phylogeography of Camellia taliensis (Theaceae) inferred from chloroplast and nuclear DNA: Insights into evolutionary history and conservation. BMC Evol. Biol. 2012, 12, 92. [Google Scholar] [CrossRef]
- Wei, S.; Lu, Y.; Ye, Q.; Tang, S. Population genetic structure and phylogeography of Camellia flavida (Theaceae) based on chloroplast and nuclear DNA sequences. Front. Plant Sci. 2017, 8, 00718. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, L.; Folk, R.A.; Zhao, J.; Zamora, N.A.; Yang, S.; Soltis, D.E.; Soltis, P.S.; Gao, L.; Peng, H.; et al. Phylotranscriptomics of Theaceae: Generic-level relationships, reticulation and whole-genome duplication. Ann. Bot. 2022, 129, 457–471. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 39–40. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Narnia 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. FastaSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, L. Clumpak: A program for identifying clustering modes and packaging population structure inference across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A. Admixture models and the breeding systems of H. S. Jennings: A GENETICS connection. Genetics 2016, 202, 9–13. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Wang, B.; Ruan, Z. Genetic diversity and differentiation in Camellia reticulata (Theaceae) polyploid complex revealed by ISSR and ploidy. Genet. Mol. Res. 2012, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, W.; Chen, L.; Wen, J. Genetic diversity and population structure of Gerbera delavayi (Asteraceae) in southwest china: Implications for conservation. Ann. Bot. Fenn. 2017, 54, 409–422. [Google Scholar] [CrossRef]
- Huang, H.; Tong, Y.; Zhang, Q.; Gao, L. Genome size variation among and within Camellia species by using flow cytometric analysis. PLoS ONE 2013, 8, e64981. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Gu, Z.; Wang, Z.; Xiao, D.; Wang, L.; Kondo, K. Dawn on the origin of Camellia reticulata—The new discovery of its wild diploid in Jinshajiang Valley. Acta Bot. Yun. 1994, 16, 255–262. [Google Scholar]
- Gu, Z. The discovery of tetraploid Camellia reticulata and its implication in studies on the origin this species. Acta Phytotaxon. Sin. 1997, 35, 107–116. [Google Scholar]
- Liu, L.; Gu, Z. Genomic in situ hybridization identifies genome donors of Camellia reticulata (Theaceae). Plant Sci. 2011, 180, 554–559. [Google Scholar] [CrossRef]



| Primer Code | Primer Sequence (5′-3′) | Amplicon Length (bp) | Associated mRNAs | GenBank No |
|---|---|---|---|---|
| c1179-F c1179-R | ATCGCCAACAGAAACAACACGC ATTCACATCTAATGAGCGAAGGTTG | 700 | Disease resistance protein RPM1 | MH257926 |
| c10316-F c10316-R | CTCCCAACCCATCGTCCTTT CCTTGCCGCTCTTGCAATC | 1038 | Endo-1,3(4)-beta-glucanase | MH257929 |
| c10214-F c10214-R | TGGAAGCTCGGCAATACCAG CCTTTGCGTTCATGGGCATT | 538 | Myosin-binding protein | MH257931 |
| c11230-F c11230-R | TCTTGTGAGGTTAAGAGGGTTT CTTGGACATTATCATTGGAGCA | 800 | S-receptor-like serine/threonine protein kinase | MH257925 |
| c11847-F c11847-R | ACATCGAAGAGCATGGCACA GCCCCAAACTAGCACTCTCT | 786 | Myb-like transcription factor | MH257932 |
| Provenance | Number of Cultivars | Average Number of Different Nucleotides (k) | Diversity of Nucleotide (Pi) | Number of Haplotypes (h) | Diversity of Haplotype (Hd) | Variance of Haplotype Diversity (Vh) | Standard Deviation of Haplotype Diversity (Sh) |
|---|---|---|---|---|---|---|---|
| Kunming | 34 | 23.5544 | 0.0068 | 35 | 1.0000 | 0.0001 | 0.0070 |
| Chuxiong | 19 | 22.8070 | 0.0066 | 14 | 0.9825 | 0.0007 | 0.0260 |
| Dali | 12 | 28.0833 | 0.0081 | 10 | 1.0000 | 0.0027 | 0.0520 |
| Tengchong | 35 | 29.2404 | 0.0084 | 36 | 0.9958 | 0.0001 | 0.0070 |
| Total | 100 | 26.6465 | 0.0077 | 91 | 0.9974 | 0.0000 | 0.0020 |
| Source of Variation | df | SSD | MSD | Variance Component | Total Variance (%) | p-Value |
|---|---|---|---|---|---|---|
| Among populations | 3 | 71.8090 | 23.9630 | 0.4719 | 3.50 | <0.0010 |
| Within populations | 99 | 1247.1911 | 12.5980 | 12.9916 | 96.50 | <0.0010 |
| Total | 102 | 1319.0001 | 36.5610 | 13.4635 | 100 |
| Provenance | Kunming | Chuxiong | Dali | Tengchong |
|---|---|---|---|---|
| Kunming | – | 0.0911 | −0.0183 | 0.0331 |
| Chuxiong | 0.0070 | – | −0.0040 | 0.0471 |
| Dali | 0.0070 | 0.0070 | – | −0.0019 |
| Tengchong | 0.0080 | 0.0080 | 0.0080 | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Gao, Y.; Zheng, W. Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers. Horticulturae 2024, 10, 303. https://doi.org/10.3390/horticulturae10030303
Xu X, Gao Y, Zheng W. Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers. Horticulturae. 2024; 10(3):303. https://doi.org/10.3390/horticulturae10030303
Chicago/Turabian StyleXu, Xiaodan, Ya Gao, and Wei Zheng. 2024. "Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers" Horticulturae 10, no. 3: 303. https://doi.org/10.3390/horticulturae10030303
APA StyleXu, X., Gao, Y., & Zheng, W. (2024). Exploring Genetic Diversity and Phylogenetic Relationships in Camellia reticulata Cultivars Using Novel Low-Copy Nuclear Gene Markers. Horticulturae, 10(3), 303. https://doi.org/10.3390/horticulturae10030303