Abstract
Camellia japonica (Naidong), a rare temperate arbor in the genus Camellia, is an ornamental plant with high economic value. To understand transcriptional changes of the drought response in C. japonica, a comparative transcriptome analysis of C. japonica (Naidong) was conducted at two drought stages (0 and 20 DAF) based on the PacBio platform. The results showed significant differences in 265 genes and 3383 lncRNAs. Of these, 150 were upregulated and 115 were downregulated. Functional analysis revealed the involvement of distinct genes in 43 pathways. The biosynthesis of amino acids and the circadian rhythm of the plant were significantly enriched, with a significant change in CjGST potentially playing an important role under drought stress. In addition, three differential protein interaction network modules composed of 45 differentially expressed genes were predicted, which involved E3 ubiquitin ligases and threonine synthetic proteins. Moreover, a transient expression experiment demonstrated that overexpression of CjGST1 in camellia leaves significantly increased leaf area compared to wild variants under drought stress, resulting in higher drought resistance. These findings provide a valuable resource for studying the genus Camellia while shedding new light on the molecular mechanisms of drought stress.
1. Introduction
In recent years, climate disasters have been on the rise in various regions, and ecosystems have shown increasing vulnerability [1]. Land use, climate change and other factors have caused droughts, which, in turn, have caused dehydration in plants, which undermines plant growth [2], resulting in a vicious circle. Plants present a variety of dynamic responses to drought stress [3], including molecular transcription factor regulation, transporters, phosphorylated modifications and physiological and biochemical aspects including malondialdehyde (MDA) and proline metabolites being generated. Analyzing the molecular mechanisms of plant resistance to drought is of significant value. Leaves, as the primary organs of transpiration and photosynthesis, have a crucial role in the use and loss of water. Through leaf wither and cuticle deposition, photosynthetic efficiency is reduced to mitigate the threat of drought. Therefore, leaves are important drought-resistant organs [4].
C. japonica (Naidong) is an evergreen tree species belonging to the Theaceae family in the genus Camellia and is considered a Tertiary relict species. Unlike most camellias, it is mainly distributed in Laoshan District in Qingdao, China (36°14′ N, 120°46′ E), adapted to an air environment of low humidity and low temperature. In addition, C. japonica (Naidong) has an important role in ornamental horticulture and is economically valuable [5]. Meanwhile, due to low precipitation and other factors, most other camellias are very difficult to introduce to arid areas. Solving the problem of lack of drought resistance in C. japonica is, therefore, a crucial task. Unfortunately, few studies have been conducted to determine the genetic resources and transcriptional regulation of protein sequences in plants of the genus Camellia under drought stress [6]. Although some research has been conducted on C. japonica (Naidong), it has mainly focused on cold resistance [7,8], and an influence on drought resistance is yet to be elucidated. Hence, it is crucial to comprehend the molecular mechanism behind C. japonica’s response to drought.
Abiotic stress activates non-enzymatic antioxidant and antioxidant enzyme defense systems in plants [9]. Reduced glutathione is the main antioxidant of non-enzymatic systems. Glutathione S-transferase (GST) catalyzes the binding of glutathione with electrophilic substrates to form conjugates that degrade harmful substances [10]. In pumpkins, genome-wide analysis shows that GST accumulates significantly under cold stress, decreasing the oxidative damage of hydrogen peroxide [11]. Molecular analysis of rice, meanwhile, demonstrates that OsSPX1 can enhance stress resistance by upregulating the expression of oxidative stress marker genes, such as glutathione S-transferase [12]. Furthermore, GST activity exhibits sensitivity and plays a critical role in nanmu’s response to drought [13]. From these findings, we can surmise that GST plays a crucial role in the plant response to abiotic stress. However, the function of CjGSTs in C. japonica has yet to be identified and confirmed.
Transcriptomic analysis is a widely used large-scale experimental approach to investigate genome function and may contribute to clarifying important pathways and genes related to the phenotype [14]. PacBio provides a new long-reading sequencing technology, which can sequence a complete single RNA molecule after it is converted to cDNA. By eliminating the need to assemble a short reading length, it overcomes some problems related to short reading methods. For example, the fuzziness in sequence reading graphs is reduced, and longer transcripts can be identified, which can obtain richer transcripts of structural difference events. Many short-reading-length RNA-Seq also reduce the high proportion of false positives through the optimization of algorithms. The Iso-Seq technology developed by PacBio can produce transcripts as long as 15 kb, which helps to find a large number of transcripts that were previously uncommented on. However, although full-length reading technology has advantages over the short reading platform in transcript structure, it also has some obvious limitations. For instance, low throughput limits the scale of experiments that can be carried out with long-read sequencing. Combining SMART’s full-length sequencing with NGS’s high-throughput advantage has proven to be an efficient and feasible solution [15]. Alternatively, 3+2 sequencing (the combination of PacBio and Illumina sequencing) provides us with a relatively low-cost solution [16,17] for large-scale high-quality transcription databases.
The aim of this study was to understand the mechanism employed by C. japonica (Naidong) to counteract drought stress. This study involved observing and measuring the phenotypic and physiological changes after drought treatment. Potential regulatory pathways and key genes in the response to drought were identified from transcriptome data based on the PacBio and Illumina platforms. The drought-resistant effect of CjGST1 in camellia leaves was verified via gene-modified expression. This study provides further insight into the molecular breeding of C. japonica.
2. Materials and Methods
2.1. Plant Materials and Drought Treatment
C. japonica (Naidong) seeds were obtained from the Qingdao Botanical Garden (Qingdao Agricultural University Cooperative Research Base, 36°05′ N, 120°08′ E) in September 2018. The seeds were stored in sand in the Qingdao Agricultural University culture room (20 ± 2 °C) during winter until the fourth true leaf appeared. Upon germination, the seedlings were transferred to the artificial climate room at Qingdao Agricultural University (36°31′ N,120°39′ E) for 3 months of normal culture. The seedlings were grown in plastic pots with top and bottom diameters of 28 cm and 19 cm, respectively, and a height of 26 cm. The culture conditions consisted of a temperature of 25 ± 2 °C, relative humidity of 60%, a soil mixture of peat soil (80%) and river sand (20%), natural light, and watering every third day. Seedlings with a similar growth status were selected and divided into two groups. Subsequently, these seedlings were then exposed to drought stress for 0 (CK, no drought stress) and 20 (DS, drought stress; stop watering and ambient humidity: 30%) days, respectively. Leaves were collected from three individual plants in each group and stored at −80 °C for further analysis.
2.2. Physiological Measurement
The leaves were dried at 50 °C for 24 h, and the dry weight and fresh weight were used to calculate the relative water content. For a chlorophyll content assay, the leaves were placed in 15 mL tubes containing 10 mL 90% ethanol for 24 h in a no-light solution, after which absorbance values were detected [18]. Each parameter was measured in 6 biological replicates.
2.3. RNA Extraction and the Sequencing of the cDNA Library
The total RNA of C. japonica (Naidong) leaves was extracted according to the instructions of the AC0307 (Qingdao Haosai Company, Qingdao, China) kit. In brief, blades were first ground in liquid nitrogen followed by separation of RNA and DNA using solubility differences between the lysate and centrifugal column, and then performing separation, rinsing and, lastly, collecting RNA, which was fully dissolved in ddH2O. The purity, concentration, and integrity of the samples were assessed via Agilent2100 instruments (Agilent Technologies, Palo Alto, CA, USA) and NanoDrop2000 (ThermoFisher, Waltham, MA, USA). The cDNA library was prepared according to the requirements of smart sequencing and was sequenced using Illumina HiSeq (Illumina, San Diego, CA, USA) and Pacbio sequel II (Pacbio, Menlo Park, CA, USA).
Raw reads were processed using the Iso-seq pipeline to obtain error-corrected reads of insert (ROIs) with a minimum Full Pass of 3 and minimum Predicted Accuracy of 0.9. Subsequently, full-length, non-chimeric (FLNC) transcripts were identified by the polyA tail signal and the 5′ and 3′ cDNA primers in the ROIs. The ICE (Iterative Clustering for Error Correction) method was employed to obtain consensus isoforms, and FL consensus sequences from ICE were polished using Quiver. High-quality FL transcripts with a post-correction accuracy above 99% were classified. Iso-Seq high-quality FL transcripts were processed to remove redundancy using cd-hit (identity > 0.99) [19]. The transcriptome data obtained in this study were deposited in the NCBI SRA database (NCBI project accession number: PRJNA689105).
2.4. Gene Functional Annotation and Expression Network Construction
The unigenes were functionally annotated based on several databases: COG [20], KOG [21], GO [22], NR [23], KEGG [24], PFAM [25] and Swiss-Prot [26]. Prediction of transcription factors (TFs) and protein kinases was performed using iTAK [27]. Transcripts with a length of more than 200 nt and no protein-coding potential were screened as candidate lncRNAs using the CPC [28], CPAT [29] and PFAM databases. In order to construct the interacting protein network, the Blastx program was utilized to compare the protein sequences of different genes with interacting protein sequences in the STRING database [30]. The resulting protein network was visualized using Cytoscape 3.0 [31].
2.5. Analysis of Differentially Expressed Genes
Quantification of gene expression levels was estimated using fragments per kilobase of transcript per million fragments mapped. The edge R package was employed for normalization processing to adjust expression count. Significantly differentially expressed genes were determined based on an FDR < 0.05, and |log2 (fold change)| ≥ 1 was set as the threshold [32].
2.6. RT-qPCR Analysis
Fourteen randomly selected genes with significant differences were subjected to quantitative real-time PCR (qRT-PCR) analysis using the same operational approaches as described previously [33]. Primer design used conventional primer design standards. The 18sRNA gene was used as an internal control. The experiment was repeated three times. The relative expression levels of genes were calculated using the 2−ΔΔCt method [33]. The experiment was repeated three times. Primers for the qRT-PCR were designed using Premier 5.0 software, and the sequences are provided in Supplementary Table S1.
2.7. Statistical Analysis
All measurements in this study were conducted with a minimum of three biological and three technique replicates. Statistical analysis was performed using SPSS (v22.0). A p-value of less than 0.05 (*) indicated a significant difference between the mean values. The data are presented as the mean ± standard deviation (SD) from three independent experiments.
2.8. Transient Expression of CjGST1 in Camellia Leaves
The coding sequence of CjGST1 was amplified from the cDNA of camellia leaves using gene-specific primers. It was then inserted into a super1300 vector to generate 35S::CjGST1 constructs. Transformation of camellia leaves using the Agrobacterium-mediated osmotic infection method [34] included immersing the leaves in bacterial liquid, applying vacuum for 30 min, slowly releasing pressure and rinsing the infected leaves with distilled water. Next, these leaves were sown on MS solid medium and incubated for 48 h at 25 °C. At least nine pots of each of the control groups and CjGST1-OE leaves were dehydrated for 24 h (in a dry environment), then rehydrated for 24 h (put back into water). The transformed leaves were used to observe phenotypes and determine gene expression.
3. Results
3.1. Physiological Responses of C. japonica (Naidong) Seedlings under Drought Stress
To observe the response of C. japonica (Naidong) to drought stress, we monitored phenotypic and physiological changes in C. japonica (Naidong) leaves during drought treatment. After 20 days of drought treatment, the leaves of C. japonica (Naidong) exhibited signs of withering, chlorosis and sagging. These phenotypic and structural changes of leaves may lead to alterations in physiological and biochemical characteristics. The relative water content of leaves in the drought group was lower compared to the control group. Additionally, the chlorophyll content decreased significantly from 1.224782 mg/g in the control group to 0.687788 mg/g in the drought treatment group, indicating a reduction in chlorophyll content due to drought stress (Figure 1). These results showed that drought stress affects the fundamental physiological activities of C. japonica (Naidong).
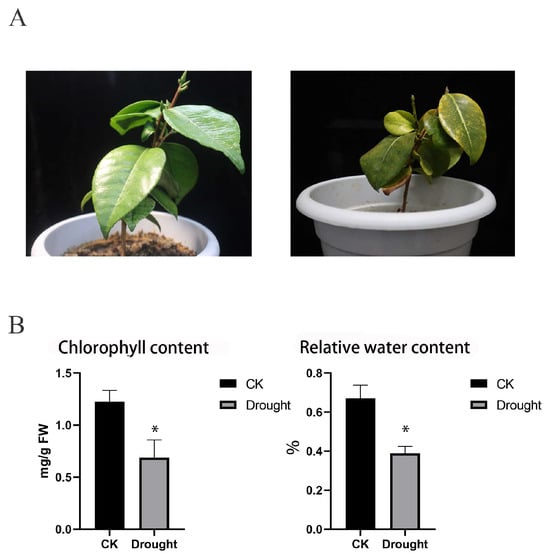
Figure 1.
The physiological response to drought in C. japonica (Naidong). (A) Phenotypical changes in camellia leaves under drought stress, the left: CK, the right: drought treatment. (B) The content of chlorophyl and relative water. Data are mean ± SD of three independent experiments, and * indicates a significant difference at p < 0.05 between CK and drought treatment groups using Student’s t-test.
3.2. Analysis of Illumina and PacBio Transcriptome Data
From the Illumina platform, we obtained 37.82 Gb of clean data with a GC content > 44% from six samples. In addition, the PacBio platform generated 20.7 Gb of clean data from the control group and the drought stress group. The sequencing quality was sufficient for further analysis. After data filtering, we obtained 52,594 transcribed sequences from high-quality, consistent sequences. Among these, 48,690 (92.5%) of 52,594 unigenes were annotated in various databases: COG (20,348), GO (38,413), KEGG (20,861), KOG (31,767), Pfam (38,829), Swiss-Prot (35,479), eggnog (47,679) and NR (48,554). The BLAST results of the KEGG database showed that 20,861 unigenes were assigned to 129 pathways. In addition, BUSCO analysis showed high homology with Vitis vinifera, Coffea canephora, Sesamum indicum, Theobroma cacao and Nelumbo nucifera as the top five species (Figure S1).
3.3. Identification of Differentially Expressed Genes (DEGs) in C. japonica (Naidong) Leaves under Drought
Principal component analysis (PCA) of all the unigenes indicated that the six samples formed two distinct groups, with the drought stress group being notably different from the control group (Figure 2A). In order to obtain the core response genetic components, based on fold change ≥ 2 and FDR < 0.01 criteria, 265 genes were identified as significantly differentially expressed during drought stress, with 150 genes upregulated and 115 downregulated. Furthermore, among these upregulated genes, CjGST1, which we are particularly interested in, was significantly altered. Heatmap analysis displayed the expression changes of these differentially expressed genes (Figure 2B).
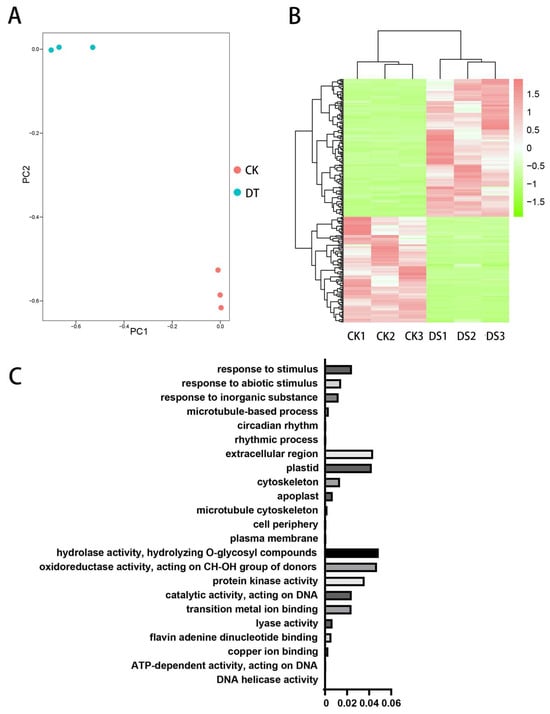
Figure 2.
Transcriptome analysis of the DEGs. (A) The PCA of global gene expression, CK: control group, DT: drought group. (B) Heatmap analysis of DEGs. The expression patterns of DEGs in the CK groups and DS groups using the RNA data. Red and green indicate upregulated and downregulated, respectively. (C) Plot shows the significantly enriched GO terms for the higher expression of DEGs in C. japonica (Naidong). The abscissa represents the p-value.
To explore the molecular mechanism underlying drought resistance, the functional categories of the DEGs were classified through GO analysis. The results of this classification showed that differentially expressed genes in cellular component GO terms were primarily enriched in cell (GO:0005623), membrane (GO:0016020), intracellular (GO:0005622), macromolecular complex (GO:0032991) and organelle (GO:0043226). In terms of molecular functions, they were primarily enriched in catalytic activity (GO:0003824), binding (GO:0005488), nucleic acid binding transcription factor activity (GO:0001071), signal transducer activity (GO:0004871), structural molecule activity (GO:0005198), electron carrier activity (GO:0009055) and antioxidant activity (GO:0016209), along with another 11 terms. In terms of biological processes, they were primarily enriched in metabolic process (GO:0008152), cellular process (GO:0009987), reproductive process (GO:0022414), signaling (GO:0023052), multicellular organismal process (GO:0032501), developmental process (GO:0032502), single-organism process (GO:0044699), response to stimulus (GO:0050896), localization (GO:0051179) and biological regulation (GO:0065007), along with another 15 terms (Figure 2C).
The DEGs involved in various biological metabolic pathways were analyzed using the KEGG database. This analysis revealed that 104 DEGs were involved in 43 biological metabolic pathways (Figure S2), including alanine, aspartate and glutamate metabolism, arginine and proline metabolism, arginine biosynthesis, cysteine and methionine metabolism and glycine metabolism. The synthesis of metabolites, such as serine and threonine metabolism, histidine metabolism and valine, leucine and isoleucine degradation, was also involved in carbon metabolism and in leucine and plant hormone signal transduction pathways. One of the genes encoding ethylene receptors was significantly downregulated, and along with two Arr-a genes.
3.4. Transcription Factors and lncRNA
Transcription factors play a crucial role in plant growth, development and stress responses. Using iTAK (1.4) software, we predicted the transcription factors present in C. japonica (Naidong). The column diagram in Figure 3A displays the numbers of the top 13 largest annotated transcription factor families, with bZIP, C3H, MYB, bHLH and NAC found to be the five largest. Heatmap analysis of the differentially expressed transcription factors (DETs) is shown in Figure 3B. Most transcription factors were upregulated as a result of drought treatment. Four database schemes were used to predict the lncRNAs of the de-redundant transcripts (Figure 3C), and the expression of 11 possible lncRNAs showed significant differences (Figure 3D).
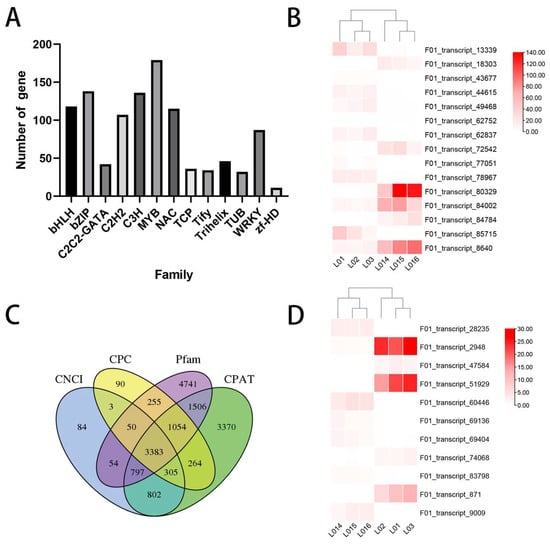
Figure 3.
General analysis of transcriptional factors and lncRNAs. (A) The number of TFs in different gene families. (B) Heatmap analysis of DETs. The expression patterns of DETs in the CK and drought treatment groups using the RNA data. Red and white indicate upregulated and downregulated, respectively. (C) Venn diagram of the number of lncRNAs predicted using the CPC Calculator, the Coding-Non-Coding Index (CNCI), the Coding Potential Assessment Tool (CPAT), and pfam protein structure domain analysis. (D) The expression patterns of DELs in the CK and DS groups using the RNA data. Red and white indicate upregulated and downregulated, respectively.
3.5. Gene Network Response to Drought
The STRING database was combined with our expression profile to construct three differential protein interaction network modules composed of 45 DEGs. Module 1 (Figure 4), Module 2 and Module 3 were composed of 23 proteins, including E3 ubiquitin ligase Ari2 (F01_transcript_7694), light-mediated developmental protein (F01_transcript_68168), serine/threonine synthetic protein (F01_transcript_69345) and phosphatase 2C protein (F01_transcript_12447). Of these, the most notable are the splicing factor protein (F01_transcript_10548) and the precursor mRNA receptor protein (F01_transcript_46000). Phosphatase and ubiquitin ligase seem closely related to mRNA splicing, but the specific mechanism for this remains unclear. Therefore, further verification is needed.
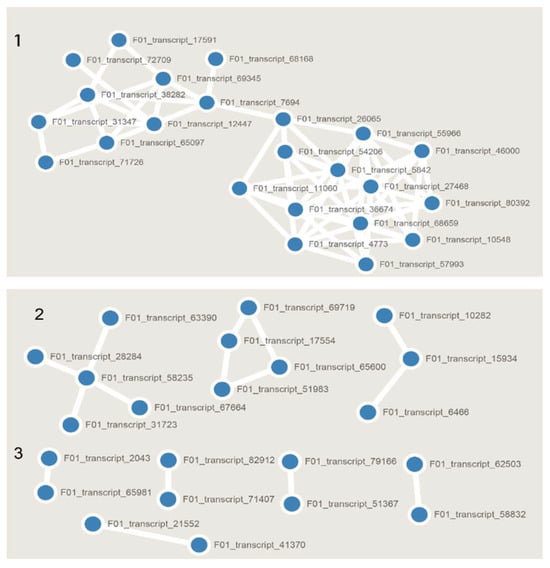
Figure 4.
The predicted base on the STRING database for the gene expression network, divided into three modules according to gene connection relationships. 1–3: Predicted gene networks.
3.6. Validation of Transcriptome Data Using RT-qPCR Analyses
To validate the reliability of the RNA-seq data, we randomly selected 14 candidate genes for quantitative real-time PCR (qRT-PCR) analyses (Figure 5). The expression patterns of these genes were largely consistent with the FPKM values obtained from the RNA-seq data. The results provide further evidence of the reliability of the transcriptome results.
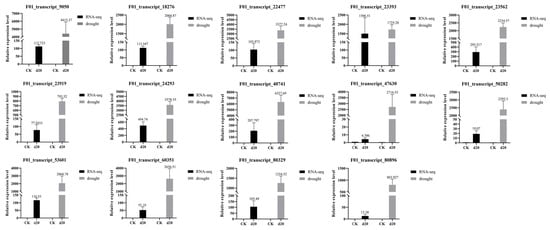
Figure 5.
Verification of transcript factor expression in transcriptomes using qRT-PCR, where CK indicates CK group and d20 indicates drought treatment group.
3.7. Transient Expression of CjGST1 Enhanced the Drought Resistance of Camellia Leaves
During the drought stress period, CjGST1 in C. japonica was significantly upregulated. To further examine gene function, CjGST1 driven by the cauliflower mosaic virus (CaMV) 35S promoter was introduced into C. japonica leaves. We observed phenotypic and physiological changes under drought treatment (Figure 6). The area of the leaves with overexpression of CjGST1 was larger than in the control group, and the dry weight increased significantly after 24 h of dehydration compared to the control group. These results show that CjGST1 can prevent leaf dehydration to some extent and alleviate drought stress in C. japonica leaves.
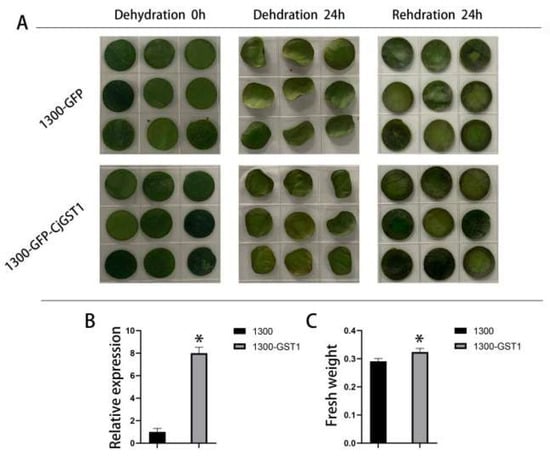
Figure 6.
Dehydration stress in CjGST1-overexpressed camellia leaves, where each treatment has 9 biological repeats. (A) Phenotypes of 1300-GFP and 1300-CjGST1 leaves after dehydration and rehydration. (B) Relative expression levels of CjGST1 in 1300-GFP and 1300-CjGST1 under dehydration stress. (C) Fresh weights of 1300-GFP and 1300-CjGST1 leaves after dehydration and rehydration. * indicates a significant difference at p < 0.05.
4. Discussion
Camellias are mainly distributed in the subtropics and other areas characterized by high air humidity and heavy rainfall. They have good ornamental value and resistance to adversity. However, temperature, drought and other stresses restrict their normal growth and distribution. Fortunately, C. japonica (Naidong) is unusual for a plant of this genus in that it can grow naturally in temperate regions [35]. Therefore, it is necessary to thoroughly analyze the adaptive drought resistance of C. japonica (Naidong), and thereby enable drought-resistant transgenic varieties to be bred.
Under drought stress, plants have developed various physiological and biochemical mechanisms to cope with adversity, such as peroxide removal, proline, soluble protein, and trehalose synthesis to adjust intracellular osmotic pressure [36]. Additionally, transcriptional regulation controls gene expression, while cutin and wax formation helps adjust leaf surface properties to minimize moisture loss and optimize water utilization during drought. In our study, several transcription factors related to stress responses were significantly upregulated in the drought treatment group, including families such as AP2, NAC, MYB, BHLH, and WRKY. [37] Previous studies have shown that the RhNAAC31 transcription factor improves drought resistance, cold resistance and salt tolerance in transgenic Arabidopsis thaliana [38]. These differentially regulated transcription factors may play crucial roles in the abiotic stress response of C. japonica (Naidong), but their specific regulatory mechanisms require further investigation. The RLK-Pelle-DLSV family is commonly associated with early drought-responsive protein kinases. Notably, glutathione transferase (GST) was upregulated under drought stress in our results. Earlier studies have demonstrated the importance of GST in walnut’s response to drought stress [39]. Furthermore, the overexpression of CjGST1 in C. japonica (Naidong) leaves significantly alleviated the pressure of drought stress. Additionally, lncRNAs have been identified as potential regulatory factors in plant stress responses [40,41]. We screened and predicted 11 significantly differentially expressed lncRNAs during drought stress, which likely play roles in the drought stress response of C. japonica (Naidong). However, further studies are needed to elucidate the specific functions of these differentially expressed lncRNAs as they represent valuable genetic resources.
In recent years, researchers have discovered that transcription before processing, processing after transcription and translation modifications play important roles in the stress response. This is further supported by our results. E3 ubiquitin ligase interactions of protein, such as protein and protein phosphatase 2 c, light-mediated development serine/threonine synthase and alternative splicing are important transcriptional mechanisms, which have drastically increased our knowledge of the complexity of eukaryotes [42]. Different coding is formed by combining different splicing receptor proteins that work differently in relation to diverse living environments and the dynamic regulation of plant growth. Alternative splicing is known to be regulated by a variety of mechanisms [43], and we found that splicing factor SF3A60 may interact with mRNA precursor processing proteins, leucine-receptor-rich serine proteins and cytoplasmic kinetin. Our research results help to fill a significant research gap in the drought tolerance of C. japonica (Naidong), providing the basis for the genetic improvement of C. japonica.
5. Conclusions
In response to drought stress, transcriptional regulation mechanisms play a vital role in plant adaptation. In this study, a comprehensive analysis of the drought-resistance transcriptional response and physiological changes was conducted in C. japonica (Naidong). This analysis included the identification of significant DEGs between the control and drought treatment, the regulation of photosynthesis, and multiple pathways related to arginine synthesis. We found that CjGST plays a crucial role in the drought stress response in C. japonica (Naidong). The function of CjGST1 in relieving drought in camellia leaves was further confirmed through overexpression experiments. These findings enhance our understanding of plant drought-resistance mechanisms and provide valuable resources in terms of genetic information and theoretical bases for improving the drought tolerance of C. japonica through molecular breeding, which may be utilized in future research.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/horticulturae10020114/s1, Supplementary Figure S1: The BUSCO analysis of transcriptome data; Supplementary Figure S2: The KEGG annotation of unigenes; Supplementary Table S1: The primers for qRT-PCR.
Author Contributions
Y.S. and M.F. conceived and designed the experiments; R.Z. and L.W. performed the experiments; H.T. conducted the data analysis and wrote the manuscript; X.G. and X.J. contributed plant material. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the “Natural Science Foundation of Shandong Province (No. ZR2023MC156)”, the “Talent Introduction Program for Youth Innovation Team of Shandong Higher Learning (No. 018-1622001)”, and the “National Natural Science Foundation of China (No. 32271588)”.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Knapp, A.; Carroll, C.; Denton, E.; La Pierre, K.; Collins, S.; Smith, M. Differential sensitivity to regional-scale drought in six central US grasslands. Oecologia 2015, 177, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Kim, Y.; Chung, Y.S.; Lee, E.; Tripathi, P.; Heo, S.; Kim, K.H. Root response to drought stress in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2020, 21, 1513. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, L.F.; Geng, Z.W.; Zhao, X.J.; Liu, Q.H.; Jiang, X.Q. Physiological characteristic changes and full-length transcriptome of rose (Rosa chinensis) roots and leaves in response to drought stress. Plant Cell Physiol. 2020, 61, 2153–2166. [Google Scholar] [CrossRef] [PubMed]
- Akanda, M.R.; Park, B.Y. Involvement of MAPK/NF-κB signal transduction pathways: Camellia japonica mitigates inflammation and gastric ulcer. Biomed. Pharmacother. 2017, 95, 1139–1146. [Google Scholar] [CrossRef]
- Fan, M.L.; Yang, K.; Zhou, R.; Liu, Q.H.; Guo, X.; Sun, Y.K. Temporal transcriptome profiling reveals candidate genes involved in cold acclimation of Camellia japonica (Naidong). Plant Physiol. Biochem. 2021, 167, 795–805. [Google Scholar] [CrossRef]
- Li, Q.Y.; Lei, S.; Du, K.B.; Li, L.Z.; Pang, X.F.; Wang, Z.C.; Wei, M.; Fu, S.; Hu, L.M.; Xu, L. RNA-seq based transcriptomic analysis uncovers α-linolenic acid and jasmonic acid biosynthesis pathways respond to cold acclimation in Camellia japonica. Sci. Rep. 2016, 6, 36463. [Google Scholar] [CrossRef]
- Wu, Y.W.; Müller, M.; Bai, T.; Yao, S.Y.; Gailing, O.; Liu, Z. Transcriptome profiling in Camellia japonica var. decumbens for the discovery of genes involved in chilling tolerance under cold stress. Ann. For. Res. 2019, 62, 51–68. [Google Scholar] [CrossRef]
- Kim, S.; Jung, E.; Shin, S.; Kim, M.; Kim, Y.S.; Lee, J.; Park, D. Anti-inflammatory activity of Camellia japonica oil. BMB Rep. 2012, 45, 177–182. [Google Scholar] [CrossRef]
- Edwards, R.; Dixon, D.P.; Walbot, V. Plant glutathione S-transferases: Enzymes with multiple functions in sickness and in health. Trends Plant Sci. 2000, 5, 193–198. [Google Scholar] [CrossRef]
- Kayum, M.A.; Nath, U.K.; Park, J.I.; Biswas, M.K.; Choi, E.K.; Song, J.Y.; Kim, H.T.; Nou, I.S. Genome-wide identification, characterization, and expression profiling of glutathione S-Transferase (GST) Family in pumpkin reveals likely role in cold-stress tolerance. Genes 2018, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Song, Q.; Wei, Q.; Wang, C.C.; Zhang, L.W.; Xu, W.Y.; Su, Z. Down-regulation of OsSPX1 caused semi-male sterility, resulting in reduction of grain yield in rice. Plant Biotechnol. J. 2016, 14, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Li, B.; Yu, J.; Shi, R.X.; Zeng, Q.; Jiang, Y.L.; Zhao, D. Transcriptomic and proteomic analyses uncover the drought adaption landscape of Phoebe zhennan. BMC Plant Biol. 2022, 22, 95. [Google Scholar] [CrossRef] [PubMed]
- Raherison, E.S.M.; Giguère, I.; Caron, S.; Lamara, M.; Mackay, J.J. Modular organization of the white spruce (Picea glauca) transcriptome reveals functional organization and evolutionary signatures. New Phytol. 2015, 207, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Gierahn, T.M.; Wadsworth, M.H., II; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-well: Portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef]
- Adams, I.P.; Fox, A.; Boonham, N.; Massart, S.; De Jonghe, K. The impact of high throughput sequencing on plant health diagnostics. Eur. J. Plant Pathol. 2018, 152, 909–919. [Google Scholar] [CrossRef]
- Xu, Z.C.; Peters, R.J.; Weirather, J.; Luo, H.M.; Liao, B.S.; Zhang, X.; Zhu, Y.J.; Ji, A.J.; Zhang, B.; Hu, S.N.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Arnon, D.I. Copper enzymesin isolated chloroplasts phenoloxi dases in Beta Vulgaris. Plant Physiol. 1949, 24, 1–15. [Google Scholar] [CrossRef]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.; He, F.C. Integrated NR database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.F.; Banf, M. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.P.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wang, L.G.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-deltadeltact method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.L.; Zhang, Y.; Li, X.L.; Wu, S.; Yang, M.Y.; Yin, H.F.; Liu, W.X.; Fan, Z.Q.; Li, J.Y. Multi-approach analysis reveals pathways of cold tolerance divergence in Camellia japonica. Front. Plant Sci. 2022, 13, 811791. [Google Scholar] [CrossRef]
- Guo, X.; Hu, Y.; Ma, J.Y.; Wang, H.; Wang, K.L.; Wang, T.; Jiang, S.Y.; Jiao, J.B.; Sun, Y.K.; Jiang, X.L.; et al. Nitrogen deposition effects on invasive and native plant competition: Implications for future invasions. Ecotoxicol. Environ. Saf. 2023, 259, 115029. [Google Scholar] [CrossRef]
- Jiang, G.M.; Jiang, X.Q.; Lü, P.T.; Liu, J.T.; Gao, J.P.; Zhang, C.Q. The rose (Rosa hybrida) NAC transcription factor 3 gene, RhNAC3, involved in ABA signaling pathway both in rose and Arabidopsis. PLoS ONE 2014, 9, e109415. [Google Scholar] [CrossRef]
- Ding, A.Q.; Li, S.; Li, W.; Hao, Q.; Wan, X.L.; Wang, K.L.; Liu, Q.C.; Liu, Q.H.; Jiang, X.Q. RhNAC31, a novel rose NAC transcription factor, enhances tolerance to multiple abiotic stresses in Arabidopsis. Acta Physiol. Plant. 2019, 41, 75. [Google Scholar] [CrossRef]
- Yang, G.Y.; Li, D.P.; Peng, S.B.; Gao, X.Q.; Chen, S.W.; Wang, T.Y.; Su, L.Y.; Xu, Z.G.; Zhai, M.Z. Walnut JrGSTU23 and JrVHAc4 involve in drought tolerance via JrWRKY2-mediated upstream regulatory pathway. Sci. Hortic. 2022, 295, 110871. [Google Scholar]
- Quan, M.Y.; Chen, J.H.; Zhang, D.Q. Exploring the secrets of long noncoding RNAs. Int. J. Mol. Sci. 2015, 16, 5467–5496. [Google Scholar] [CrossRef]
- Zhang, W.; Han, Z.X.; Guo, Q.L.; Liu, Y.; Zheng, Y.X.; Wu, F.L.; Jin, W.B. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 2014, 9, e98958. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.B.; Wang, T.T.; Wei, W.; Lou, S.T.; Lan, F.X.; Zhu, S.; Li, Q.Z.; Ji, G.L.; Lin, C.T.; Wu, X.H.; et al. The full-length transcriptome of Spartina alterniflora reveals the complexity of high salt tolerance in monocotyledonous halophyte. Plant Cell Physiol. 2020, 61, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, P.; Jin, G.H.; Gui, D.P.; Li, L.; Zhang, C.J. Temporal regulation of alternative splicing events in rice memory under drought stress. Plant Divers. 2020, 44, 116–125. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).