
Genome-Wide Identification and Expression Analysis of the MADS-Box Gene Family in Cassava (Manihot esculenta)
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Data Resources
2.3. Identification of the MADS-Box Gene Family
2.4. Phylogenetic Tree Construction in the MADS-Box Gene Family
2.5. Analysis of Basic Physicochemical Properties of the MADS-Box Gene Family in M. esculenta
2.6. Motif and Gene Structure Analysis of the MADS-Box Gene Family in M. esculenta
2.7. Chromosome Localization and Collinearity Analysis of the MADS-Box Gene Family in M. esculenta
2.8. Analyzing Cis-Acting Elements of the MADS-Box Gene Family Promoter in M. esculenta
2.9. Expression Analysis of the MADS-Box Gene Family in M. esculenta using RNA-Seq Analysis
2.10. Time-Ordered Gene Co-Expression Network (TO-GCN) Analysis
2.11. RNA Extraction and RT-qPCR Analysis
2.12. Subcellular Localization of MeMADS12
3. Results
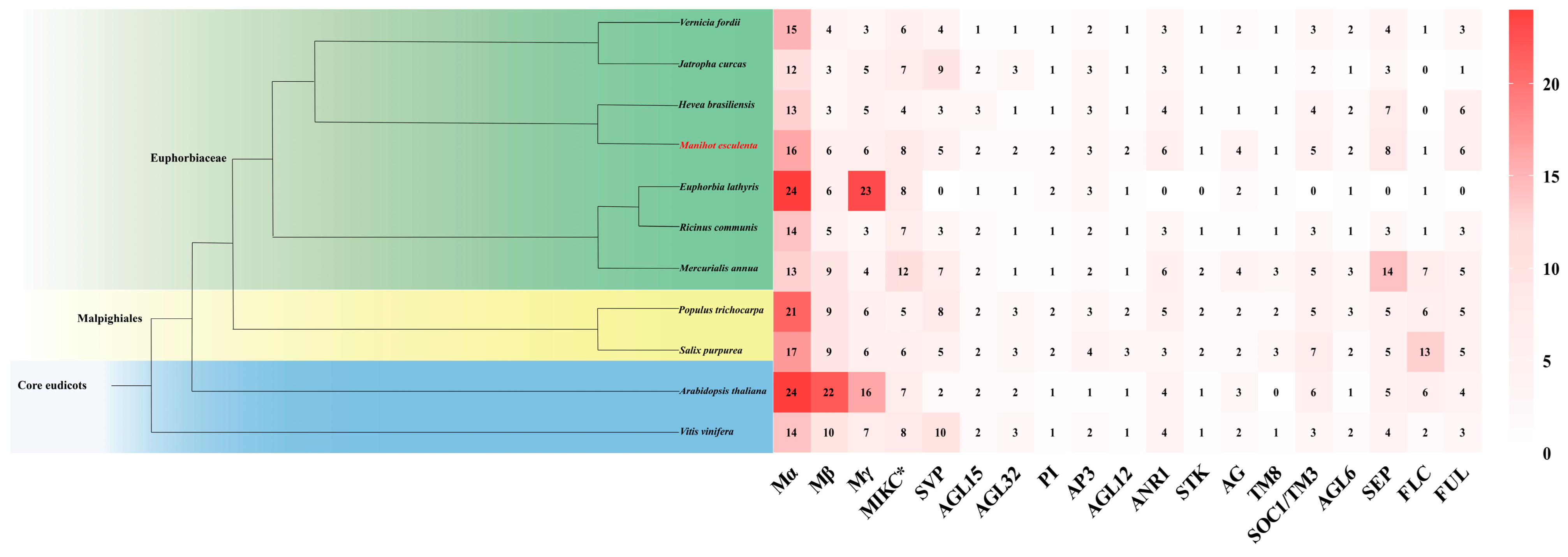
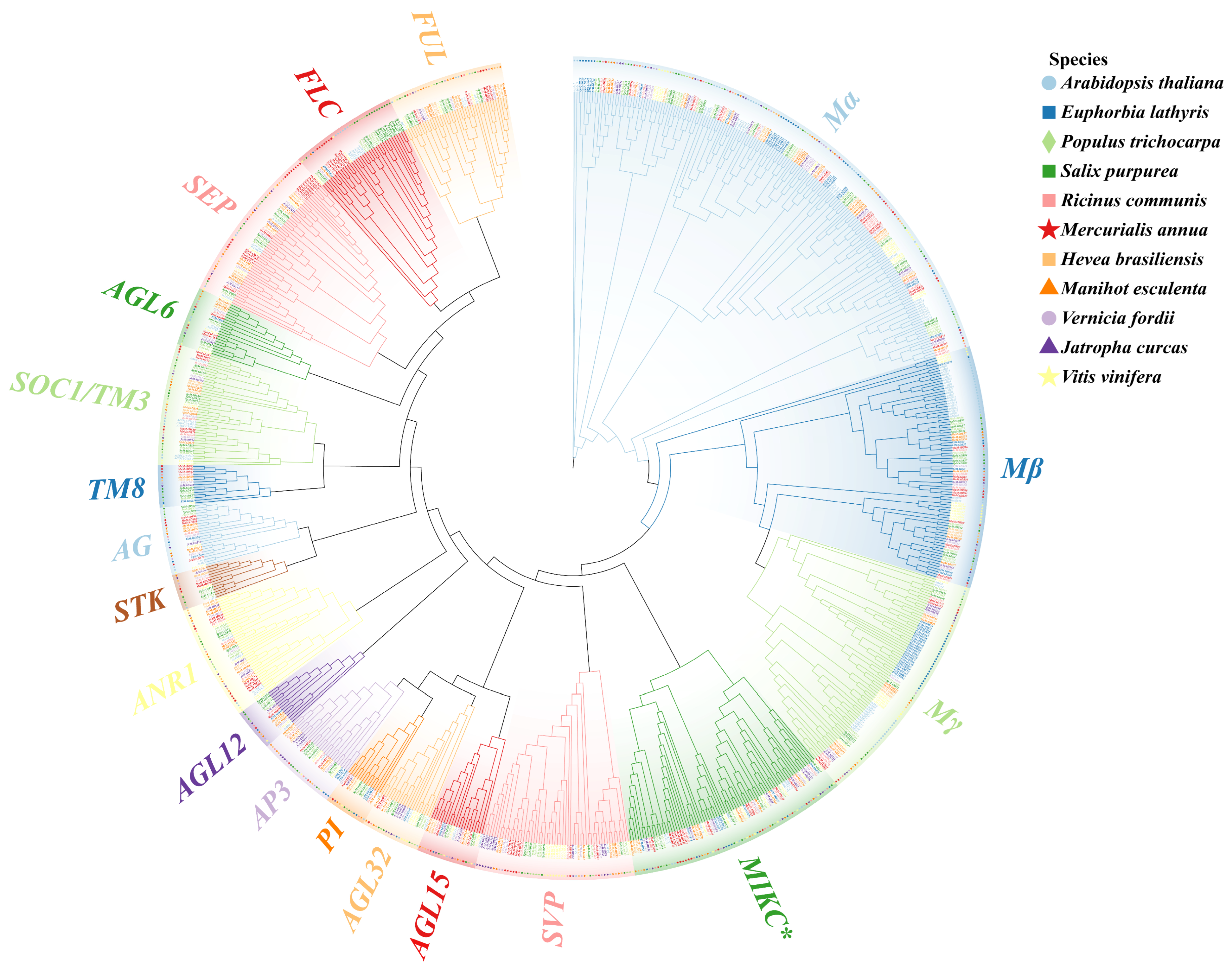
3.1. Identification and Phylogenetic Analysis of the MADS-Box Gene Family in 11 Species
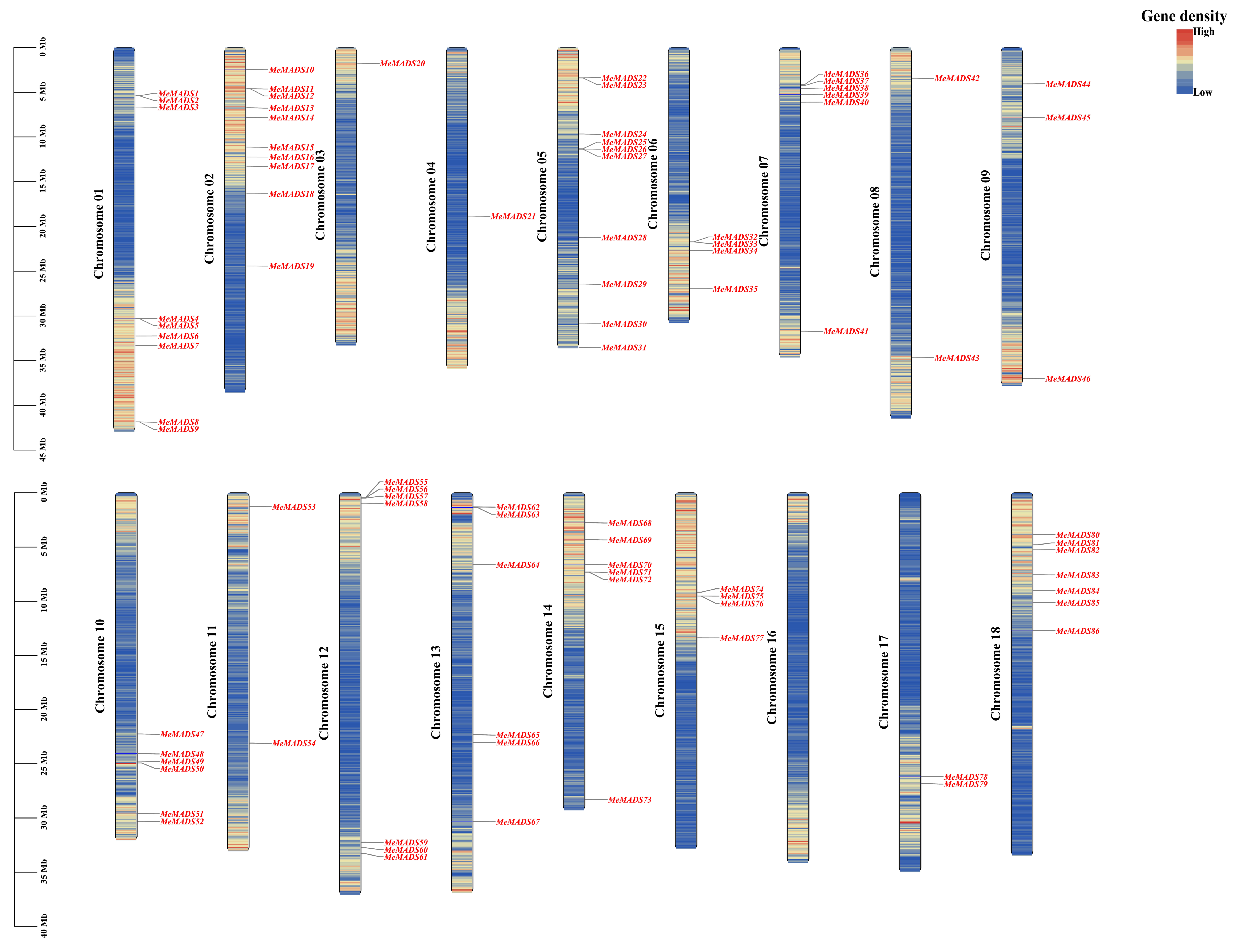
3.2. Chromosome Localization and Physicochemical Property Analysis of Protein
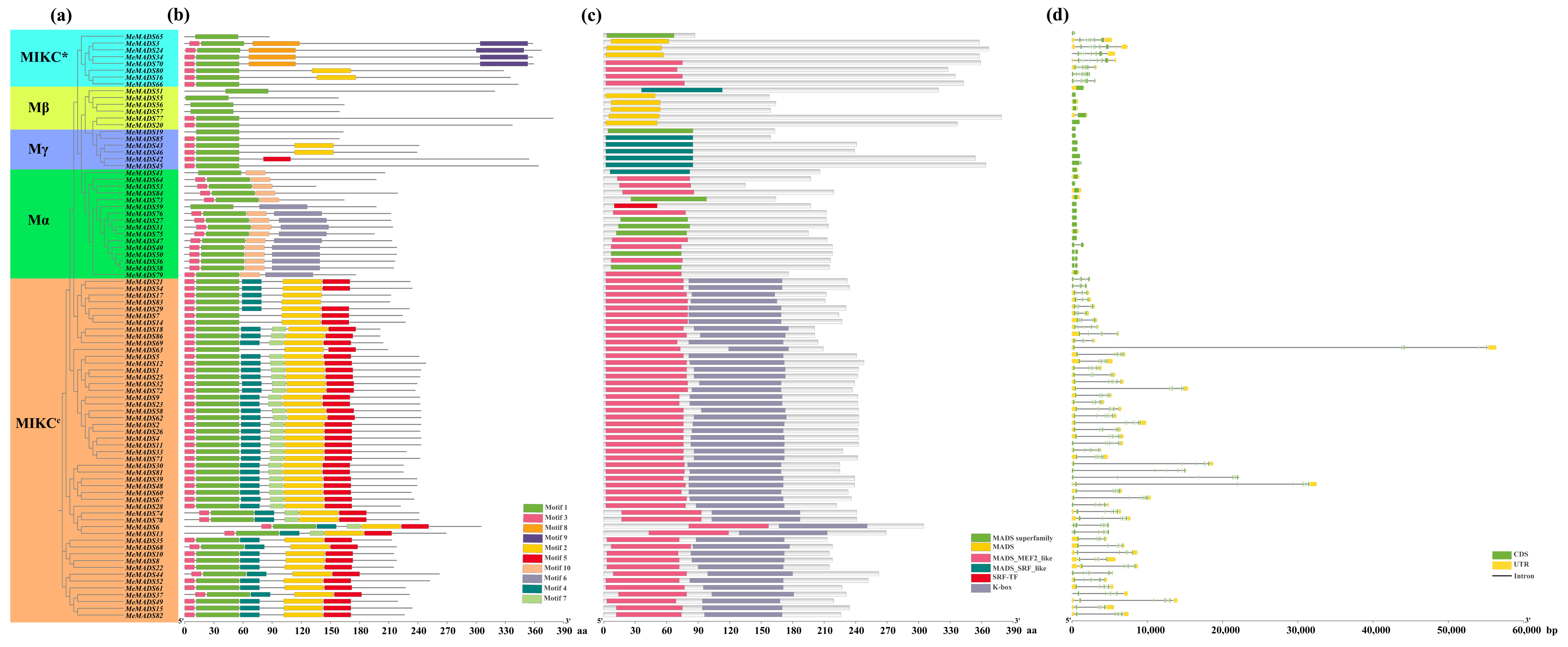
3.3. Analysis of Conserved Motif and Gene Structure of MeMADS-Box Protein
3.4. Analysis of Cis-Element of the MADS-Box Gene Promoter in M. esculenta
3.5. Gene Duplication Analysis of the MADS-Box Gene Family in M. esculenta
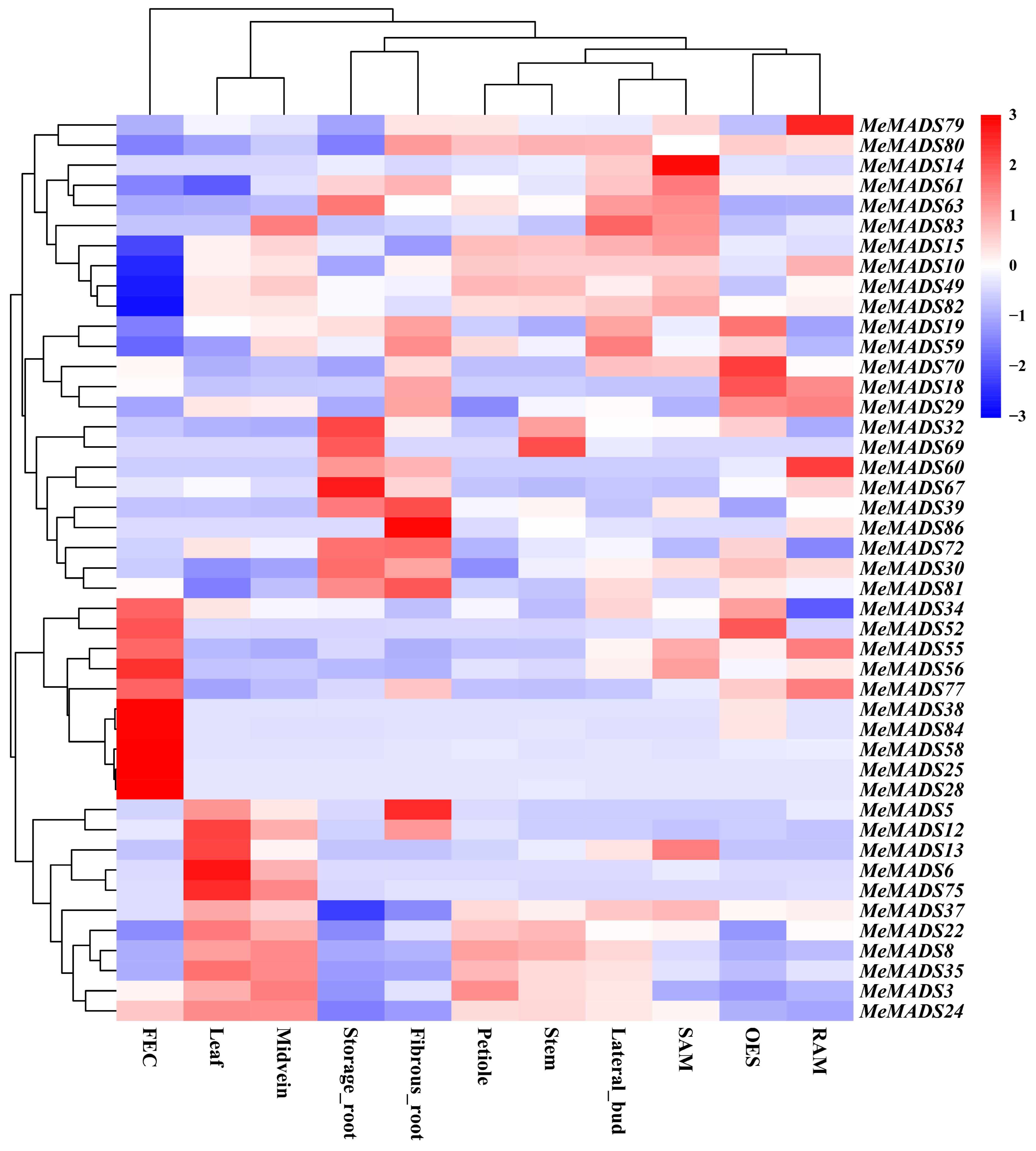
3.6. Analysis of MADS-Box Gene Expression Pattern in Tissues of M. esculenta via RNA-Seq Analysis
3.7. MADS-Box Gene Expression Pattern in M. Esculenta under Drought Stress via RNA-Seq Analysis
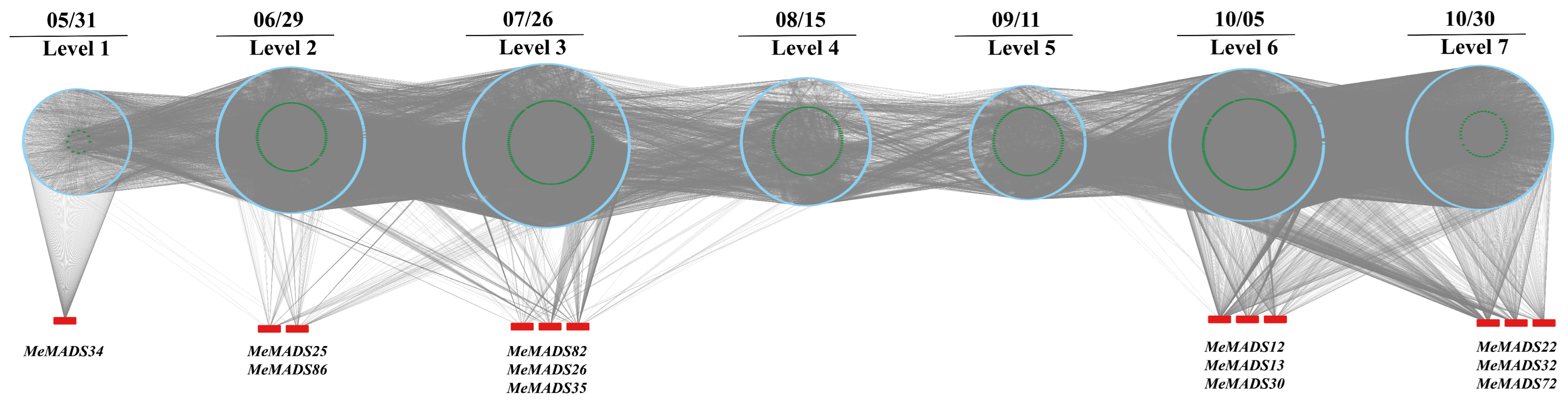
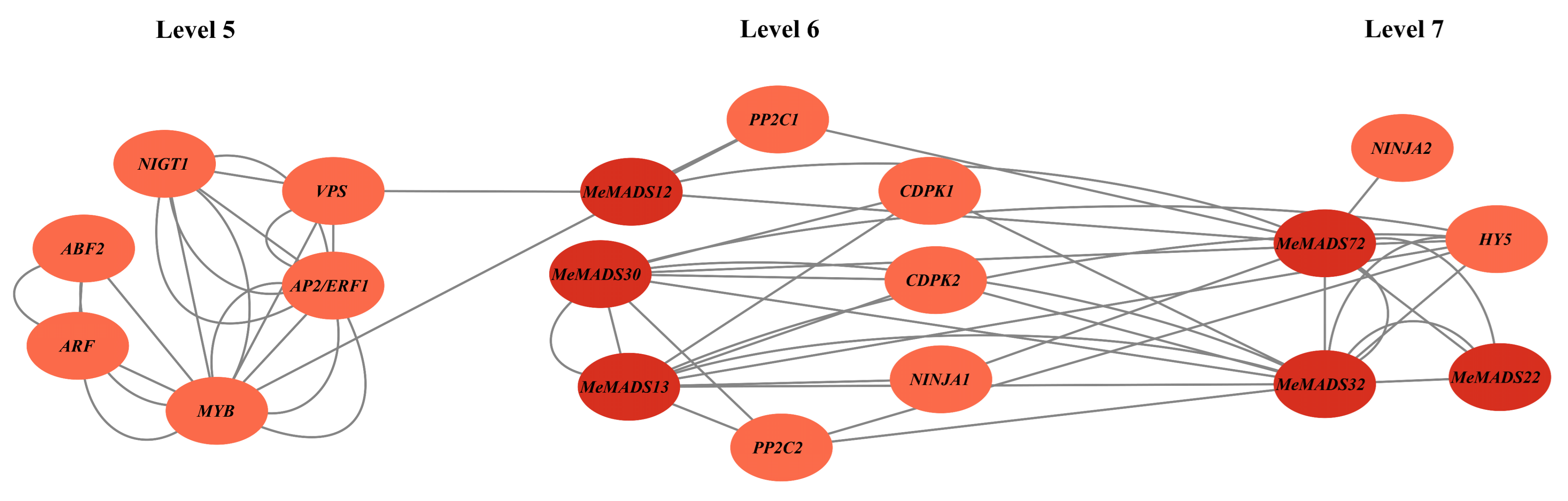
3.8. Time-Ordered MADS-Box Gene Co-Expression in M. esculenta in Response to Flowering
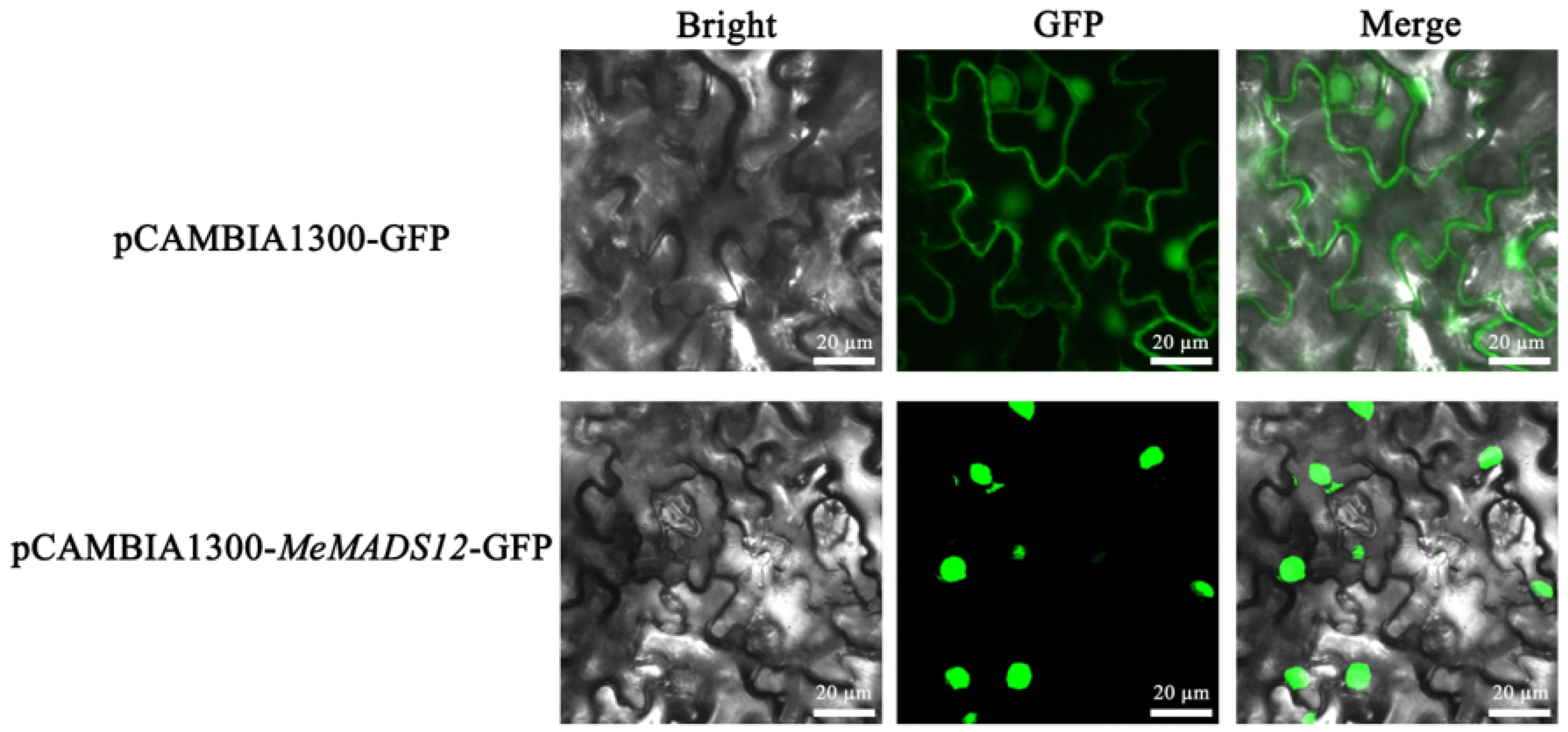
3.9. Vector Construction and Subcellular Localization of MeMADS12
3.10. Relative Expression Levels of MeMADS12 via RT-qPCR Analysis
4. Discussion
4.1. Identification and Evolutionary Analysis of Cassava MADS-Box Gene Family
4.2. MADS-Box Genes’ Regulatory Mechanisms for Flowering in Response to Drought Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Theissen, G.; Kim, J.T.; Saedler, H. Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 1996, 43, 484–516. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, C.A.; Davies, B. Gene duplication and the evolution of plant MADS-box transcription factors. J. Genet. Genom. 2012, 39, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ryu, H.S.; Chung, K.S.; Posé, D.; Kim, S.; Schmid, M.; Ahn, J.H. Regulation of temperature-responsive flowering by MADS-box transcription factor repressors. Science 2013, 342, 628–632. [Google Scholar] [CrossRef]
- Qiu, Y.; Köhler, C. Endosperm Evolution by Duplicated and Neofunctionalized Type I MADS-Box Transcription Factors. Mol. Biol. Evol. 2022, 39, msab355. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Ribas, D.P.L.; Martínez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef]
- Parenicová, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Huang, X.; Cheng, J.; Li, Z.; de Folter, S.; Huang, Z.; Jiang, X.; Pang, H.; Tao, S. Conservation and evolution in and among SRF- and MEF2-type MADS domains and their binding sites. Mol. Biol. Evol. 2011, 28, 501–511. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, F.; Zhang, X.; Li, Z.; Zhao, Y.; Lohaus, R.; Chang, X.; Dong, W.; Ho, S.; Liu, X.; et al. The water lily genome and the early evolution of flowering plants. Nature 2020, 577, 79–84. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, X.; Liu, X.; Zhang, L. Evolutionary Analysis of MIKC(c)-Type MADS-Box Genes in Gymnosperms and Angiosperms. Front. Plant Sci. 2017, 8, 895. [Google Scholar]
- Li, S.; Cui, Y.; Zhou, Y.; Luo, Z.; Liu, J.; Zhao, M. The industrial applications of cassava: Current status, opportunities and prospects. J. Sci. Food Agric. 2017, 97, 2282–2290. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, P.; Shao, J.; Li, C.; Wang, B.; Guo, X.; Yan, B.; Xia, Y.; Peng, M. Analysis of different strategies adapted by two cassava cultivars in response to drought stress: Ensuring survival or continuing growth. J. Exp. Bot. 2015, 66, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Hu, Z.; Hu, J.; Zhu, Z.; Yu, X.; Cui, B.; Chen, G. Tomato (Solanum lycopersicum) MADS-box transcription factor SlMBP8 regulates drought, salt tolerance and stress-related genes. Plant Growth Regul. 2017, 83, 55–68. [Google Scholar] [CrossRef]
- Khong, G.N.; Pati, P.K.; Richaud, F.; Parizot, B.; Bidzinski, P.; Mai, C.D.; Bès, M.; Bourrié, I.; Meynard, D.; Beeckman, T.; et al. OsMADS26 Negatively Regulates Resistance to Pathogens and Drought Tolerance in Rice. Plant Physiol. 2015, 169, 2935–2949. [Google Scholar] [CrossRef]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef]
- Criscuolo, A. A fast alignment-free bioinformatics procedure to infer accurate distance-based phylogenetic trees from genome assemblies. Res. Ideas Outcomes 2019, 5, e36178. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performanc and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms. Nat. Protoc. 2008, 3, 153–162. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Wilson, M.C.; Mutka, A.M.; Hummel, A.W.; Berry, J.; Chauhan, R.D.; Vijayaraghavan, A.; Taylor, N.J.; Voytas, D.F.; Chitwood, D.H.; Bart, R.S. Gene expression atlas for the food security crop cassava. New Phytol. 2017, 213, 1632–1641. [Google Scholar] [CrossRef]
- Ding, Z.; Tie, W.; Fu, L.; Yan, Y.; Liu, G.; Yan, W.; Li, Y.; Wu, C.; Zhang, J.; Hu, W. Strand-specific RNA-seq based identification and functional prediction of drought-responsive lncRNAs in cassava. BMC Genom. 2019, 20, 214. [Google Scholar] [CrossRef]
- Tokunaga, H.; Quynh, D.; Anh, N.H.; Nhan, P.T.; Matsui, A.; Takahashi, S.; Tanaka, M.; Anh, N.M.; Van Dong, N.; Ham, L.H.; et al. Field transcriptome analysis reveals a molecular mechanism for cassava-flowering in a mountainous environment in Southeast Asia. Plant Mol. Biol. 2022, 109, 233–248. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.S.; Chang, Y.M.; Lim, Y.M.; Cheong, S.K.; Chung, I.F.; Wong, C.Y. Generating transcriptional regulatory networks from time-ordered stem cell differentiation RNA sequencing data. STAR Protoc. 2022, 3, 101541. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Mccarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Filleur, S.; Rahman, A.; Gotensparre, S.; Forde, B.G. Nutritional regulation of ANR1 and other root-expressed MADS-box genes in Arabidopsis thaliana. Planta 2005, 222, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.H.; Yu, J.Q.; Duan, X.; Wang, J.H.; Zhang, Q.Y.; Gu, K.D.; Hu, D.G.; Zheng, C.S. The MADS transcription factor CmANR1 positively modulates root system development by directly regulating CmPIN2 in chrysanthemum. Hortic. Res. 2018, 5, 52. [Google Scholar] [CrossRef]
- Zidenga, T.; Leyva-Guerrero, E.; Moon, H.; Siritunga, D.; Sayre, R. Extending cassava root shelf life via reduction of reactive oxygen species production. Plant Physiol. 2012, 159, 1396–1407. [Google Scholar] [CrossRef]
- Xu, J.; Duan, X.; Yang, J.; Beeching, J.R.; Zhang, P. Enhanced reactive oxygen species scavenging by overproduction of superoxide dismutase and catalase delays postharvest physiological deterioration of cassava storage roots. Plant Physiol. 2013, 161, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nie, S.; Xu, C.Q.; Liu, H.; Jia, K.H.; Zhou, S.S.; Zhao, W.; Zhou, X.Q.; El-Kassaby, Y.A.; Wang, X.R.; et al. UV-B-induced molecular mechanisms of stress physiology responses in the major northern Chinese conifer Pinus tabuliformis Carr. Tree Physiol. 2021, 41, 1247–1263. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, R. Abscisic Acid synthesis and response. Arab. Book 2013, 11, e166. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.N.; Zeng, B.; Liu, H.S.; Qi, H.; Xie, L.J.; Yu, L.J.; Chen, Q.F.; Li, J.F.; Chen, Y.Q.; Jiang, L.; et al. SINAT E3 Ubiquitin Ligases Mediate FREE1 and VPS23A Degradation to Modulate Abscisic Acid Signaling. Plant Cell 2020, 32, 3290–3310. [Google Scholar] [CrossRef]
- Belda-Palazon, B.; Rodriguez, L.; Fernandez, M.A.; Castillo, M.C.; Anderson, E.M.; Gao, C.; Gonzalez-Guzman, M.; Peirats-Llobet, M.; Zhao, Q.; De Winne, N.; et al. FYVE1/FREE1 Interacts with the PYL4 ABA Receptor and Mediates Its Delivery to the Vacuolar Degradation Pathway. Plant Cell 2016, 28, 2291–2311. [Google Scholar] [CrossRef]
- Yu, F.; Lou, L.; Tian, M.; Li, Q.; Ding, Y.; Cao, X.; Wu, Y.; Belda-Palazon, B.; Rodriguez, P.L.; Yang, S.; et al. ESCRT-I Component VPS23A Affects ABA Signaling by Recognizing ABA Receptors for Endosomal Degradation. Mol. Plant 2016, 9, 1570–1582. [Google Scholar] [CrossRef]
- Mankotia, S.; Jakhar, P.; Satbhai, S.B. HY5: A key regulator for light-mediated nutrient uptake and utilization by plants. New Phytol. 2024, 241, 1929–1935. [Google Scholar] [CrossRef]
- Pauwels, L.; Barbero, G.F.; Geerinck, J.; Tilleman, S.; Grunewald, W.; Pérez, A.C.; Chico, J.M.; Bossche, R.V.; Sewell, J.; Gil, E.; et al. NINJA connects the co-repressor TOPLESS to jasmonate signalling. Nature 2010, 464, 788–791. [Google Scholar] [CrossRef]
- Melzer, S.; Lens, F.; Gennen, J.; Vanneste, S.; Rohde, A.; Beeckman, T. Flowering-time genes modulate meristem determinacy and growth form in Arabidopsis thaliana. Nat. Genet. 2008, 40, 1489–1492. [Google Scholar] [CrossRef]
- Borner, R.; Kampmann, G.; Chandler, J.; Gleissner, R.; Wisman, E.; Apel, K.; Melzer, S. A MADS domain gene involved in the transition to flowering in Arabidopsis. Plant J. 2000, 24, 591–599. [Google Scholar] [CrossRef]
- Jiang, X.; Lubini, G.; Hernandes-Lopes, J.; Rijnsburger, K.; Veltkamp, V.; de Maagd, R.A.; Angenent, G.C.; Bemer, M. FRUITFULL-like genes regulate flowering time and inflorescence architecture in tomato. Plant Cell 2022, 34, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Jia, D.; Zhang, X.; Li, S.; Su, J.; Jiang, J.; Chen, S.; Chen, F.; Ding, L. FUL homologous gene CmFL1 is involved in regulating flowering time and floret numbers in Chrysanthemum morifolium. Plant Sci. 2023, 336, 111863. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Hsu, C.T.; Liao, D.C.; Chang, W.J.; Chou, M.L.; Huang, Y.T.; Chen, J.J.; Ko, S.S.; Chan, M.T.; Shih, M.C. Transcriptome-wide analysis of the MADS-box gene family in the orchid Erycina pusilla. Plant Biotechnol. J. 2016, 14, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef]
- Kong, H.; Leebens-Mack, J.; Ni, W.; Depamphilis, C.W.; Ma, H. Highly heterogeneous rates of evolution in the SKP1 gene family in plants and animals: Functional and evolutionary implications. Mol. Biol. Evol. 2004, 21, 117–128. [Google Scholar] [CrossRef]
- De Bodt, S.; Raes, J.; Van de Peer, Y.; Theissen, G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003, 8, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Chen, C.; Song, J.; Zheng, P.; Wang, J.; Wei, J.; Cai, W.; Chen, S.; Cai, Y.; Yuan, Y.; et al. The Capsicum MYB31 regulates capsaicinoid biosynthesis in the pepper pericarp. Plant Physiol. Biochem. 2022, 176, 21–30. [Google Scholar] [CrossRef]
- Zhu, Z.; Sun, B.; Cai, W.; Zhou, X.; Mao, Y.; Chen, C.; Wei, J.; Cao, B.; Chen, C.; Chen, G.; et al. Natural variations in the MYB transcription factor MYB31 determine the evolution of extremely pungent peppers. New Phytol. 2019, 223, 922–938. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, S.; Xu, L.; Chu, S.; Yan, X.; Lin, L.; Wen, J.; Zheng, B.; Chen, S.; Li, Q. Transcription factor PagMYB31 positively regulates cambium activity and negatively regulates xylem development in poplar. Plant Cell 2024, 36, 1806–1828. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef]
- Lata, C.; Prasad, M. Role of DREBs in regulation of abiotic stress responses in plants. J. Exp. Bot. 2011, 62, 4731–4748. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Yamaguchi-Shinozaki, K.; Urao, T.; Iwasaki, T.; Hosokawa, D.; Shinozaki, K. Role of arabidopsis MYC and MYB homologs in drought- and abscisic acid-regulated gene expression. Plant Cell 1997, 9, 1859–1868. [Google Scholar] [PubMed]
- Busk, P.K.; Pagès, M. Regulation of abscisic acid-induced transcription. Plant Mol. Biol. 1998, 37, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.M.; Boisson-Dernier, A.; Dizon, M.B.; Maktabi, M.H.; Schroeder, J.I. The protein phosphatase AtPP2CA negatively regulates abscisic acid signal transduction in Arabidopsis, and effects of abh1 on AtPP2CA mRNA. Plant Physiol. 2006, 140, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Tähtiharju, S.; Palva, T. Antisense inhibition of protein phosphatase 2C accelerates cold acclimation in Arabidopsis thaliana. Plant J. 2001, 26, 461–470. [Google Scholar] [CrossRef]
- Yoshida, T.; Nishimura, N.; Kitahata, N.; Kuromori, T.; Ito, T.; Asami, T.; Shinozaki, K.; Hirayama, T. ABA-hypersensitive germination3 encodes a protein phosphatase 2C (AtPP2CA) that strongly regulates abscisic acid signaling during germination among Arabidopsis protein phosphatase 2Cs. Plant Physiol. 2006, 140, 115–126. [Google Scholar] [CrossRef]
- Urao, T.; Katagiri, T.; Mizoguchi, T.; Yamaguchi-Shinozaki, K.; Hayashida, N.; Shinozaki, K. Two genes that encode Ca (2+)-dependent protein kinases are induced by drought and high-salt stresses in Arabidopsis thaliana. Mol. Gen. Genet. 1994, 244, 331–340. [Google Scholar] [CrossRef]
- Zou, J.J.; Wei, F.J.; Wang, C.; Wu, J.J.; Ratnasekera, D.; Liu, W.X.; Wu, W.H. Arabidopsis calcium-dependent protein kinase CPK10 functions in abscisic acid-and Ca2+-mediated stomatal regulation in response to drought stress. Plant Physiol. 2010, 154, 1232–1243. [Google Scholar] [CrossRef]
- Zou, J.J.; Li, X.D.; Ratnasekera, D.; Wang, C.; Liu, W.X.; Song, L.F.; Zhang, W.Z.; Wu, W.H. Arabidopsis CALCIUM-DEPENDENT PROTEIN KINASE8 and CATALASE3 Function in Abscisic Acid-Mediated Signaling and H2O2 Homeostasis in Stomatal Guard Cells under Drought Stress. Plant Cell 2015, 27, 1445–1460. [Google Scholar] [CrossRef]
- Curran, A.; Chang, I.F.; Chang, C.L.; Garg, S.; Miguel, R.M.; Barron, Y.D.; Li, Y.; Romanowsky, S.; Cushman, J.C.; Gribskov, M.; et al. Calcium-dependent protein kinases from Arabidopsis show substrate specificity differences in an analysis of 103 substrates. Front. Plant Sci. 2011, 2, 36. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Seq_1 | Seq_2 | Ka | Ks | Ka_Ks | Duplication Type |
|---|---|---|---|---|---|
| MeMADS1 | MeMADS25 | 0.0332 | 0.2781 | 0.1195 | WGD or Segmental |
| MeMADS2 | MeMADS26 | 0.0922 | 0.2898 | 0.3181 | WGD or Segmental |
| MeMADS3 | MeMADS24 | 0.0666 | 0.3600 | 0.1851 | WGD or Segmental |
| MeMADS4 | MeMADS11 | 0.0305 | 0.3127 | 0.0976 | WGD or Segmental |
| MeMADS5 | MeMADS12 | 0.0921 | 0.3774 | 0.2441 | WGD or Segmental |
| MeMADS6 | MeMADS13 | 0.0730 | 0.4825 | 0.1513 | WGD or Segmental |
| MeMADS7 | MeMADS14 | 0.0638 | 0.3022 | 0.2111 | WGD or Segmental |
| MeMADS9 | MeMADS23 | 0.0640 | 0.3485 | 0.1835 | WGD or Segmental |
| MeMADS15 | MeMADS82 | 0.1066 | 0.4907 | 0.2173 | WGD or Segmental |
| MeMADS17 | MeMADS83 | 0.0727 | 0.3083 | 0.2359 | WGD or Segmental |
| MeMADS18 | MeMADS86 | 0.1087 | 0.3209 | 0.3387 | WGD or Segmental |
| MeMADS32 | MeMADS72 | 0.0719 | 0.3576 | 0.2010 | WGD or Segmental |
| MeMADS33 | MeMADS71 | 0.0700 | 0.2197 | 0.3186 | WGD or Segmental |
| MeMADS39 | MeMADS48 | 0.0564 | 0.2575 | 0.2191 | WGD or Segmental |
| MeMADS58 | MeMADS62 | 0.0272 | 0.2605 | 0.1042 | WGD or Segmental |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Li, Y.; Geng, S.; Liu, Q.; Zhou, Y.; Shen, S.; Shen, Z.; Ma, D.; Xiao, M.; Luo, X.; et al. Genome-Wide Identification and Expression Analysis of the MADS-Box Gene Family in Cassava (Manihot esculenta). Horticulturae 2024, 10, 1073. https://doi.org/10.3390/horticulturae10101073
Zhang Q, Li Y, Geng S, Liu Q, Zhou Y, Shen S, Shen Z, Ma D, Xiao M, Luo X, et al. Genome-Wide Identification and Expression Analysis of the MADS-Box Gene Family in Cassava (Manihot esculenta). Horticulturae. 2024; 10(10):1073. https://doi.org/10.3390/horticulturae10101073
Chicago/Turabian StyleZhang, Qin, Yanan Li, Sha Geng, Qian Liu, Yingchun Zhou, Shaobin Shen, Zhengsong Shen, Dongxiao Ma, Mingkun Xiao, Xin Luo, and et al. 2024. "Genome-Wide Identification and Expression Analysis of the MADS-Box Gene Family in Cassava (Manihot esculenta)" Horticulturae 10, no. 10: 1073. https://doi.org/10.3390/horticulturae10101073
APA StyleZhang, Q., Li, Y., Geng, S., Liu, Q., Zhou, Y., Shen, S., Shen, Z., Ma, D., Xiao, M., Luo, X., Che, B., Li, K., & Yan, W. (2024). Genome-Wide Identification and Expression Analysis of the MADS-Box Gene Family in Cassava (Manihot esculenta). Horticulturae, 10(10), 1073. https://doi.org/10.3390/horticulturae10101073