
Bacillus- and Lactobacillus-Based Dietary Synbiotics Are Associated with Shifts in the Oropharyngeal, Proximal Colonic, and Vaginal Microbiomes of Korean Native Black Pigs


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Animals, Experimental Design, and Housing
2.2. Sampling
2.3. DNA Isolation
2.4. 16S rRNA Gene Amplification and Sequencing
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
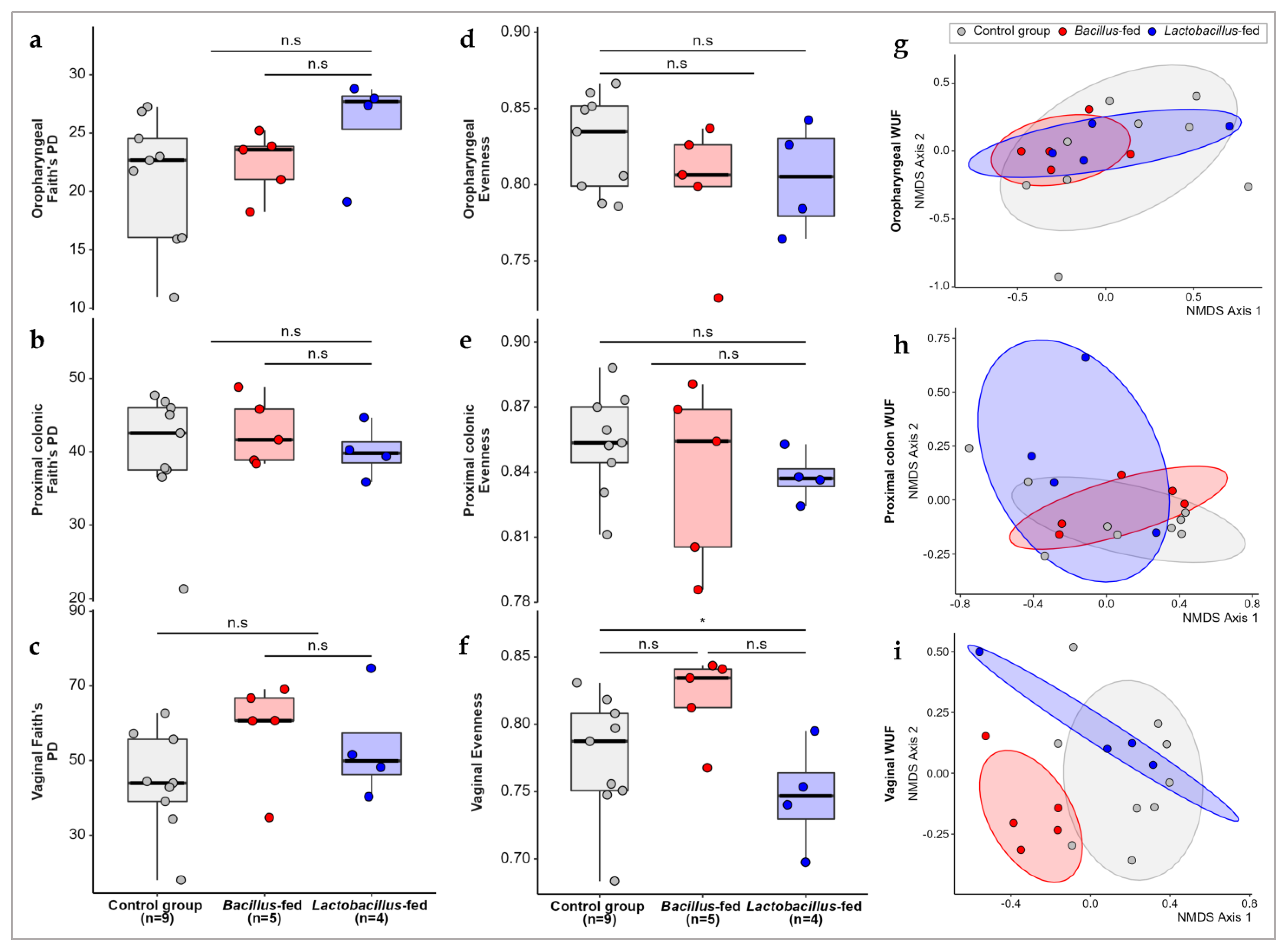
3.1. Synbiotic Impact on Microbial Diversity
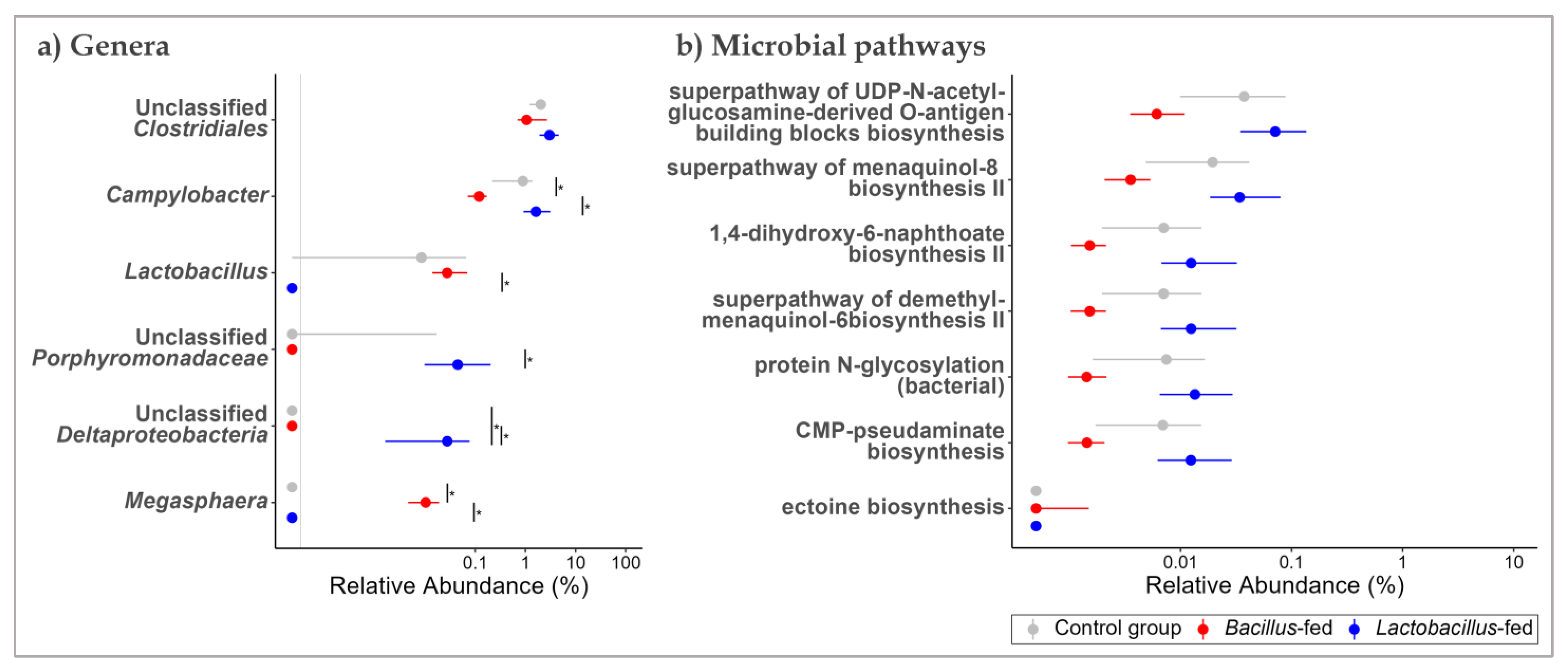
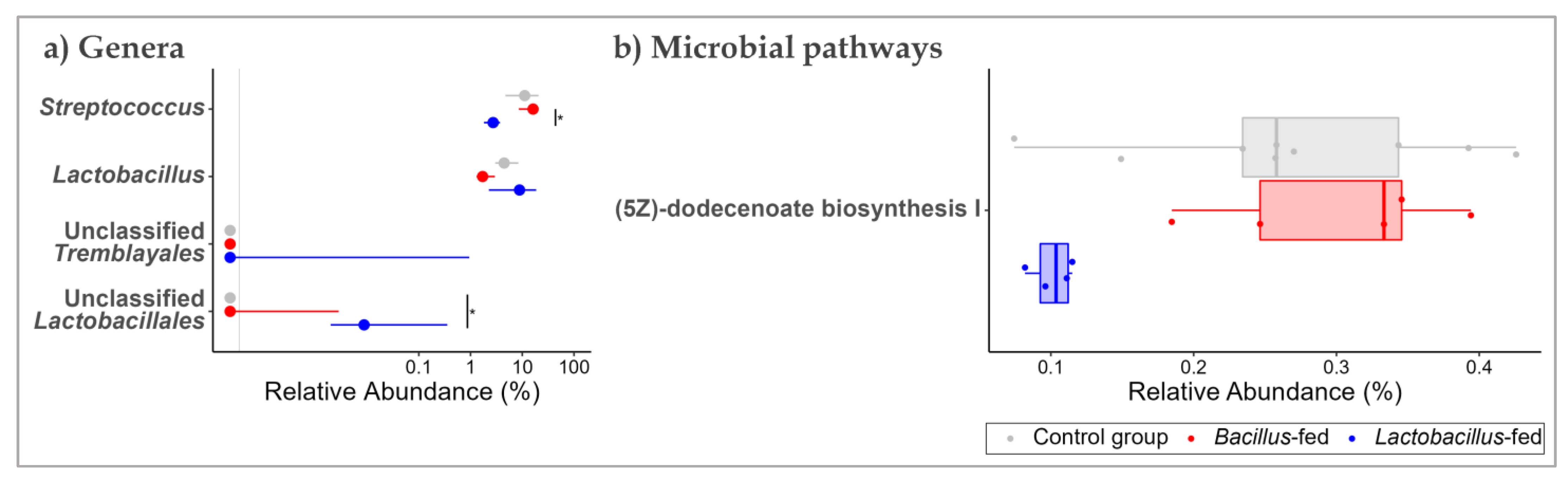
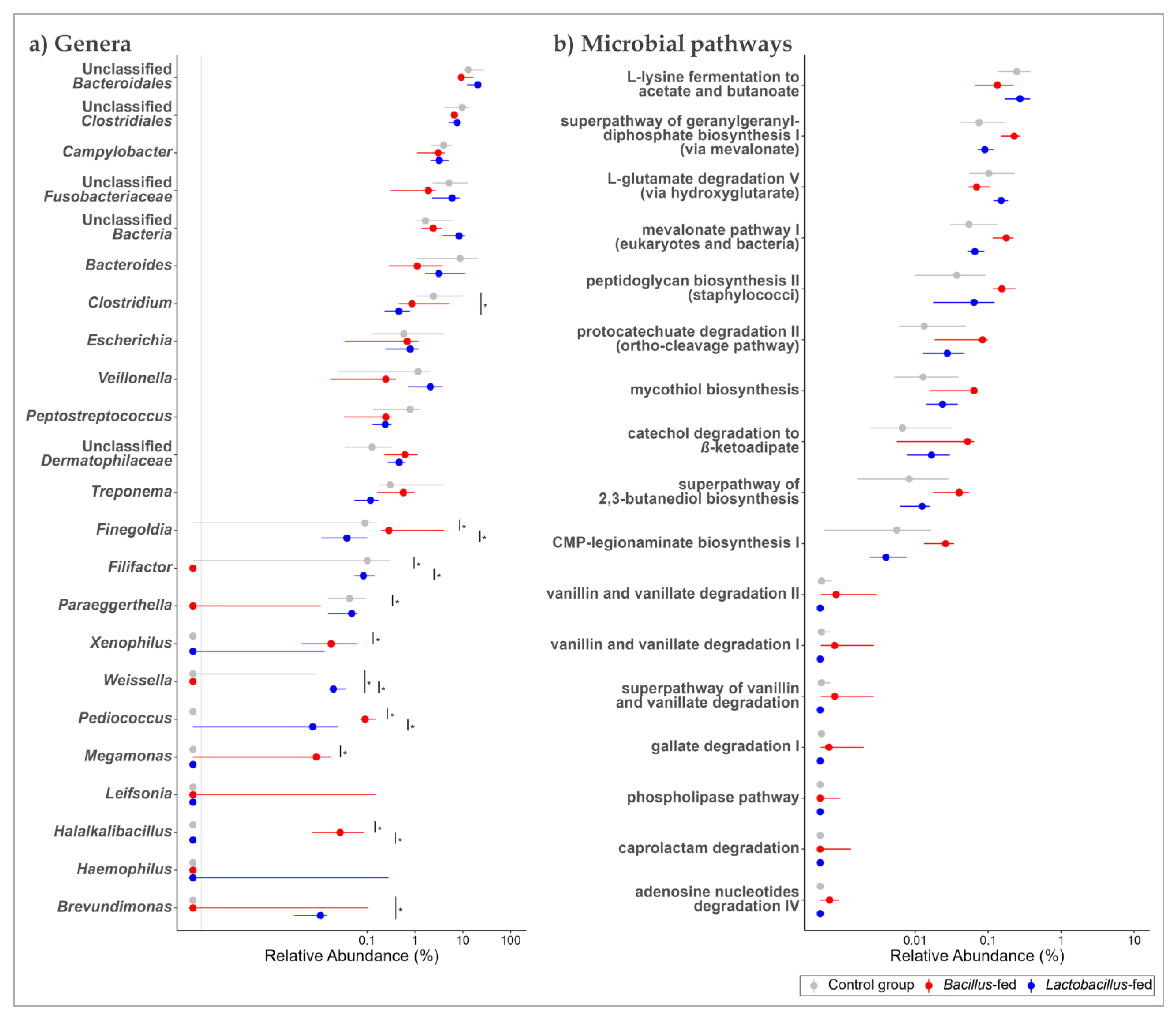
3.2. Synbiotic Impacts on Taxonomic Composition
3.3. Synbiotic Effect on Predicted Microbial Functionality across the Body Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenny, M.; Smidt, H.; Mengheri, E.; Miller, B. Probiotics–do they have a role in the pig industry? Animal 2011, 5, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Maron, D.F.; Smith, T.J.; Nachman, K.E. Restrictions on antimicrobial use in food animal production: An international regulatory and economic survey. Glob. Health 2013, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Lusk, J.L.; Norwood, F.B.; Pruitt, J.R. Consumer demand for a ban on antibiotic drug use in pork production. Am. J. Agric. Econ. 2006, 88, 1015–1033. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Markowiak, P.; Śliżewska, K. The role of probiotics, prebiotics and synbiotics in animal nutrition. Gut Pathog. 2018, 10, 21. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Kim, Y.-H.; Rhee, M.-H.; Song, J.-C.; Lee, K.-W.; Kim, K.-S.; Lee, S.-P.; Lee, I.-S.; Park, S.-C. Selection of Lactobacillus sp. PSC101 that produces active dietary enzymes such as amylase, lipase, phytase and protease in pigs. J. Gen. Appl. Microbiol. 2007, 53, 111–117. [Google Scholar] [CrossRef]
- Konstantinov, S.R.; Smidt, H.; Akkermans, A.D.; Casini, L.; Trevisi, P.; Mazzoni, M.; De Filippi, S.; Bosi, P.; De Vos, W.M. Feeding of Lactobacillus sobrius reduces Escherichia coli F4 levels in the gut and promotes growth of infected piglets. FEMS Microbiol. Ecol. 2008, 66, 599–607. [Google Scholar] [CrossRef]
- Alakomi, H.-L.; Skytta, E.; Saarela, M.; Mattila-Sandholm, T.; Latva-Kala, K.; Helander, I. Lactic acid permeabilizes gram-negative bacteria by disrupting the outer membrane. Appl. Environ. Microbiol. 2000, 66, 2001–2005. [Google Scholar] [CrossRef]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef]
- He, Y.; Jinno, C.; Kim, K.; Wu, Z.; Tan, B.; Li, X.; Whelan, R.; Liu, Y. Dietary Bacillus spp. enhanced growth and disease resistance of weaned pigs by modulating intestinal microbiota and systemic immunity. J. Anim. Sci. Biotechnol. 2020, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.A.; Duc, L.H.; Cutting, S.M. The use of bacterial spore formers as probiotics. FEMS Microbiol. Rev. 2005, 29, 813–835. [Google Scholar] [CrossRef] [PubMed]
- Urdaci, M.; Pinchuk, I. Antimicrobial activity of Bacillus probiotics. In Bacterial Spore Formers–Probiotics Emerging Applications; Horizon Bioscience: Norfolk, UK, 2004; pp. 171–182. [Google Scholar]
- Hosoi, T.; Ametani, A.; Kiuchi, K.; Kaminogawa, S. Improved growth and viability of lactobacilli in the presence of Bacillus subtilis (natto), catalase, or subtilisin. Can. J. Microbiol. 2000, 46, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Olveira, G.; González-Molero, I. An update on probiotics, prebiotics and symbiotics in clinical nutrition. Endocrinol. Y Nutr. 2016, 63, 482–494. [Google Scholar] [CrossRef]
- Cho, I.-C.; Han, S.-H.; Fang, M.; Lee, S.-S.; Ko, M.-S.; Lee, H.; Lim, H.-T.; Yoo, C.-K.; Lee, J.-H.; Jeon, J.-T. The robust phylogeny of Korean wild boar (Sus scrofa coreanus) using partial D-loop sequence of mtDNA. Mol. Cells 2009, 28, 423–430. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Bugenyi, A.W.; Lee, M.-R.; Choi, Y.-J.; Song, K.-D.; Lee, H.-K.; Son, Y.-O.; Lee, D.-S.; Lee, S.-C.; Son, Y.-J.; Heo, J. Oropharyngeal, proximal colonic, and vaginal microbiomes of healthy Korean native black pig gilts. BMC Microbiol. 2023, 23, 3. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, B.D.; Bokulich, N.A.; McDonald, D.; Knight, R.; Caporaso, J.G.; Huttley, G.A. Species abundance information improves sequence taxonomy classification accuracy. Nat. Commun. 2019, 10, 4643. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. In Breakthroughs in Statistics; Springer: Berlin/Heidelberg, Germany, 1992; pp. 196–202. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wickham, H. Data Analysis. In ggplot2; Springer: Berlin/Heidelberg, Germany, 2016; pp. 189–201. [Google Scholar]
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 2008, 9, 321–332. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.L.; Kjeldsen, K.U.; Rattei, T.; Pester, M.; Loy, A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi) sulfite reductases. ISME J. 2015, 9, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Sen, N. Functional and molecular insights of hydrogen sulfide signaling and protein sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef]
- Li, L.; Moore, P. Putative biological roles of hydrogen sulfide in health and disease: A breath of not so fresh air? Trends Pharmacol. Sci. 2008, 29, 84–90. [Google Scholar] [CrossRef]
- Singh, S.B.; Lin, H.C. Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Vieira, R.d.S.; Castoldi, A.; Basso, P.J.; Hiyane, M.I.; Câmara, N.O.S.; Almeida, R.R. Butyrate attenuates lung inflammation by negatively modulating Th9 cells. Front. Immunol. 2019, 10, 67. [Google Scholar] [CrossRef]
- Ni, Y.-F.; Wang, J.; Yan, X.-L.; Tian, F.; Zhao, J.-B.; Wang, Y.-J.; Jiang, T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir. Res. 2010, 11, 33. [Google Scholar] [CrossRef]
- Man, S.M. The clinical importance of emerging Campylobacter species. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 669–685. [Google Scholar] [CrossRef]
- De Vrese, M.; Winkler, P.; Rautenberg, P.; Harder, T.; Noah, C.; Laue, C.; Ott, S.; Hampe, J.; Schreiber, S.; Heller, K. Effect of Lactobacillus gasseri PA 16/8, Bifidobacterium longum SP 07/3, B. bifidum MF 20/5 on common cold episodes: A double blind, randomized, controlled trial. Clin. Nutr. 2005, 24, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Berggren, A.; Lazou Ahrén, I.; Larsson, N.; Önning, G. Randomised, double-blind and placebo-controlled study using new probiotic lactobacilli for strengthening the body immune defence against viral infections. Eur. J. Nutr. 2011, 50, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Yitbarek, A.; Taha-Abdelaziz, K.; Hodgins, D.C.; Read, L.; Nagy, É.; Weese, J.S.; Caswell, J.L.; Parkinson, J.; Sharif, S. Gut microbiota-mediated protection against influenza virus subtype H9N2 in chickens is associated with modulation of the innate responses. Sci. Rep. 2018, 8, 13189. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Dang, A.T.; Marsland, B.J. Microbes, metabolites, and the gut–lung axis. Mucosal Immunol. 2019, 12, 843–850. [Google Scholar] [CrossRef]
- Von Dohlen, C.D.; Kohler, S.; Alsop, S.T.; McManus, W.R. Mealybug β-proteobacterial endosymbionts contain γ-proteobacterial symbionts. Nature 2001, 412, 433–436. [Google Scholar] [CrossRef]
- Thao, M.L.; Gullan, P.J.; Baumann, P. Secondary (γ-Proteobacteria) endosymbionts infect the primary (β-Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Appl. Environ. Microbiol. 2002, 68, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2012, 10, 13–26. [Google Scholar] [CrossRef]
- Oh, J.K.; Chae, J.P.; Pajarillo, E.A.B.; Kim, S.H.; Kwak, M.J.; Eun, J.S.; Chee, S.W.; Whang, K.Y.; Kim, S.H.; Kang, D.K. Association between the body weight of growing pigs and the functional capacity of their gut microbiota. Anim. Sci. J. 2020, 91, e13418. [Google Scholar] [CrossRef]
- Makarova, K.S.; Koonin, E.V. Evolutionary genomics of lactic acid bacteria. J. Bacteriol. 2007, 189, 1199–1208. [Google Scholar] [CrossRef]
- Fayol-Messaoudi, D.; Berger, C.N.; Coconnier-Polter, M.-H.; Lievin-Le Moal, V.; Servin, A.L. pH-, Lactic acid-, and non-lactic acid-dependent activities of probiotic Lactobacilli against Salmonella enterica Serovar Typhimurium. Appl. Environ. Microbiol. 2005, 71, 6008–6013. [Google Scholar] [CrossRef] [PubMed]
- Twomey, D.; Ross, R.; Ryan, M.; Meaney, B.; Hill, C. Lantibiotics produced by lactic acid bacteria: Structure, function and applications. Antonie Van Leeuwenhoek 2002, 82, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Yu, H.; Gao, X.; Li, X.; Qiao, S. Influence of Lactobacillus fermentum I5007 on the intestinal and systemic immune responses of healthy and E. coli challenged piglets. Antonie Van Leeuwenhoek 2009, 96, 89–98. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; O’Regan, P.; Fanning, Á.; O’Mahony, C.; MacSharry, J.; Lyons, A.; Bienenstock, J.; O’Mahony, L.; Shanahan, F. Functional modulation of human intestinal epithelial cell responses by Bifidobacterium infantis and Lactobacillus salivarius. Immunology 2006, 118, 202–215. [Google Scholar] [CrossRef]
- Ohashi, Y.; Tokunaga, M.; Taketomo, N.; Ushida, K. Stimulation of indigenous lactobacilli by fermented milk prepared with probiotic bacterium, Lactobacillus delbrueckii subsp. bulgaricus strain 2038, in the pigs. J. Nutr. Sci. Vitaminol. 2007, 53, 82–86. [Google Scholar] [CrossRef]
- Konstantinov, S.R.; Awati, A.; Smidt, H.; Williams, B.A.; Akkermans, A.D.; De Vos, W.M. Specific response of a novel and abundant Lactobacillus amylovorus-like phylotype to dietary prebiotics in the guts of weaning piglets. Appl. Environ. Microbiol. 2004, 70, 3821–3830. [Google Scholar] [CrossRef]
- Willems, A.; Amat-Marco, M.; Collins, M.D. Phylogenetic analysis of Butyrivibrio strains reveals three distinct groups of species within the Clostridium subphylum of the gram-positive bacteria. Int. J. Syst. 1996, 46, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Li, M.; Lei, H.; Jiang, X.; Tu, W.; Lu, Y.; Xia, D. Butyric acid regulates progesterone and estradiol secretion via cAMP signaling pathway in porcine granulosa cells. J. Steroid Biochem. Mol. Biol. 2017, 172, 89–97. [Google Scholar] [CrossRef]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef]
- Taneike, T.; Narita, T.; Kitazawa, T.; Bando, S.; Teraoka, H.; Ohga, A. Binding and functional characterization of alpha-2 adrenoceptors in isolated swine myometrium. J. Auton. Pharmacol. 1995, 15, 93–105. [Google Scholar] [CrossRef]
- Kitazawa, T.; Nakagoshi, K.; Teraoka, H.; Taneike, T. 5-HT7 receptor and β2-adrenoceptor share in the inhibition of porcine uterine contractility in a muscle layer-dependent manner. Eur. J. Pharmacol. 2001, 433, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Fanchin, R.; Righini, C.; Olivennes, F.; Taylor, S.; de Ziegler, D.; Frydman, R. Uterine contractions at the time of embryo transfer alter pregnancy rates after in-vitro fertilization. Hum. Reprod. 1998, 13, 1968–1974. [Google Scholar] [CrossRef] [PubMed]
- Echigo, A.; Fukushima, T.; Mizuki, T.; Kamekura, M.; Usami, R. Halalkalibacillus halophilus gen. nov., sp. nov., a novel moderately halophilic and alkaliphilic bacterium isolated from a non-saline soil sample in Japan. Int. J. Syst. Evol. Microbiol. 2007, 57, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.T.; Fontana, C.; Pasteris, S.E.; Bassi, D.; Cocconcelli, P.S.; Otero, M.C. Vaginal bacterial diversity from healthy gilts and pregnant sows subjected to natural mating or artificial insemination. Res. Vet. Sci. 2021, 140, 26–37. [Google Scholar] [CrossRef]
- Sanglard, L.; Schmitz-Esser, S.; Gray, K.; Linhares, D.; Yeoman, C.; Dekkers, J.; Niederwerder, M.; Serão, N. Vaginal microbiota diverges in sows with low and high reproductive performance after porcine reproductive and respiratory syndrome vaccination. Sci. Rep. 2020, 10, 3046. [Google Scholar] [CrossRef]
- Jeon, S.J.; Vieira-Neto, A.; Gobikrushanth, M.; Daetz, R.; Mingoti, R.D.; Parize, A.C.B.; de Freitas, S.L.; da Costa, A.N.L.; Bicalho, R.C.; Lima, S. Uterine microbiota progression from calving until establishment of metritis in dairy cows. Appl. Environ. Microbiol. 2015, 81, 6324–6332. [Google Scholar] [CrossRef]
- Rogosa, M. THE GENUS VEILLONELLA I: General Cultural, Ecological, and Biochemical Considerations. Carbohydr. Res. 1964, 87, 162–170. [Google Scholar]
- Rogosa, M.; Bishop, F.S. THE GENUS VEILLONELLA II: Nutritional Studies. J. Bacteriol. 1964, 87, 574–580. [Google Scholar] [CrossRef]
- Kiefer, Z.E.; Koester, L.R.; Studer, J.M.; Chipman, A.L.; Mainquist-Whigham, C.; Keating, A.F.; Schmitz-Esser, S.; Ross, J.W. Vaginal microbiota differences associated with pelvic organ prolapse risk during late gestation in commercial sows. Biol. Reprod. 2021, 105, 1545–1561. [Google Scholar] [CrossRef]
- Fink, D.L.; Geme, J.W. The Genus Haemophilus. In The Prokaryotes: A Handbook on the Biology of Bacteria Volume 6: Proteobacteria: Gamma Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 1034–1061. [Google Scholar]
- Messing, M.; Sottnek, F.O.; Biddle, J.W.; Schlater, L.K.; Kramer, M.A.; Kraus, S.J. Isolation of Haemophilus species from the genital tract. Sex. Transm. Dis. 1983, 10, 56–61. [Google Scholar] [CrossRef]
- Kwiecien, J.M.; Little, P.B. Haemophilus somnus and reproductive disease in the cow: A review. Can. Vet. J. 1991, 32, 595. [Google Scholar] [PubMed]
- Schoenhofen, I.C.; McNally, D.J.; Brisson, J.-R.; Logan, S.M. Elucidation of the CMP-pseudaminic acid pathway in Helicobacter pylori: Synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction. Glycobiology 2006, 16, 8C–14C. [Google Scholar] [CrossRef] [PubMed]
- Samuel, G.; Reeves, P. Biosynthesis of O-antigens: Genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 2003, 338, 2503–2519. [Google Scholar] [CrossRef]
- Bentley, R.; Meganathan, R. Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiol. Rev. 1982, 46, 241–280. [Google Scholar] [CrossRef]
- Pastor, J.M.; Salvador, M.; Argandoña, M.; Bernal, V.; Reina-Bueno, M.; Csonka, L.N.; Iborra, J.L.; Vargas, C.; Nieto, J.J.; Cánovas, M. Ectoines in cell stress protection: Uses and biotechnological production. Biotechnol. Adv. 2010, 28, 782–801. [Google Scholar] [CrossRef] [PubMed]
- Seelbinder, B.; Chen, J.; Brunke, S.; Vazquez-Uribe, R.; Santhaman, R.; Meyer, A.-C.; de Oliveira Lino, F.S.; Chan, K.-F.; Loos, D.; Imamovic, L. Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome 2020, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-S.; Keum, Y.-S.; Li, Q.X. Bacterial degradation of aromatic compounds. Int. J. Environ. Res. Public Health 2009, 6, 278–309. [Google Scholar] [CrossRef]
- Yurgel, S.N.; Nearing, J.T.; Douglas, G.M.; Langille, M.G. Metagenomic functional shifts to plant induced environmental changes. Front. Microbiol. 2019, 10, 1682. [Google Scholar] [CrossRef]
- Ong, C.T.; Ross, E.M.; Boe-Hansen, G.; Turni, C.; Hayes, B.J.; Fordyce, G.; Tabor, A.E. Adaptive sampling during sequencing reveals the origins of the bovine reproductive tract microbiome across reproductive stages and sexes. Sci. Rep. 2022, 12, 15075. [Google Scholar] [CrossRef]
- Kazunga, C.; Aitken, M.D. Products from the incomplete metabolism of pyrene by polycyclic aromatic hydrocarbon-degrading bacteria. Appl. Environ. Microbiol. 2000, 66, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Smanski, M.J.; Peterson, R.M.; Huang, S.-X.; Shen, B. Bacterial diterpene synthases: New opportunities for mechanistic enzymology and engineered biosynthesis. Curr. Opin. Chem. Biol. 2012, 16, 132–141. [Google Scholar] [CrossRef]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 2017, 25, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Tyo, K.; Carlsen, S.; Mucha, O.; Phon, T.H.; Stephanopoulos, G. Terpenoids: Opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol. Pharm. 2008, 5, 167–190. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.O.; Russell, J.B. An rRNA approach for assessing the role of obligate amino acid-fermenting bacteria in ruminal amino acid deamination. Appl. Environ. Microbiol. 1996, 62, 815–821. [Google Scholar] [CrossRef]
- Buckel, W.; Barker, H.A. Two pathways of glutamate fermentation by anaerobic bacteria. J. Bacteriol. 1974, 117, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Lehtoranta, L.; Ala-Jaakkola, R.; Laitila, A.; Maukonen, J. Healthy vaginal microbiota and influence of probiotics across the female life span. Front. Microbiol. 2022, 13, 787. [Google Scholar] [CrossRef]
- Buggio, L.; Somigliana, E.; Borghi, A.; Vercellini, P. Probiotics and vaginal microecology: Fact or fancy? BMC Women’s Health 2019, 19, 25. [Google Scholar] [CrossRef]
- Pang, X.; Hua, X.; Yang, Q.; Ding, D.; Che, C.; Cui, L.; Jia, W.; Bucheli, P.; Zhao, L. Inter-species transplantation of gut microbiota from human to pigs. ISME J. 2007, 1, 156–162. [Google Scholar] [CrossRef]
- Chen, C.; Hao, L.; Zhang, Z.; Tian, L.; Zhang, X.; Zhu, J.; Jie, Z.; Tong, X.; Xiao, L.; Zhang, T. Cervicovaginal microbiome dynamics after taking oral probiotics. J. Genet. Genom. 2021, 48, 716–726. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S. Lactobacillus reuteri induces gut intraepithelial CD4+ CD8αα+ T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, T.; Li, Y.; Zhang, T.; Wang, Q.; He, J.; Wang, L.; Li, L.; Yang, N.; Fang, Y. Probiotics for the treatment of women with bacterial vaginosis: A systematic review and meta-analysis of randomized clinical trials. Eur. J. Pharmacol. 2019, 864, 172660. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, A.; Luisa, F.M.; Dellino, M.; Erica, S.; Loizzi, V.; Bocciolone, L.; Rabaiotti, E.; Cioffi, R.; Sabetta, G.; Cormio, G. Fertility sparing surgery in sex-cord stromal tumors: Oncological and reproductive outcomes. Int. J. Gynecol. Cancer 2022, 32, 1063–1070. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body Site | Synbiotic | Genus | logFC 1 | logCPM 1 | LR 1 | p-Value | FDR 1 |
|---|---|---|---|---|---|---|---|
| oropharyngeal cavity | Bacillus-based synbiotic supplementation | Campylobacter | −3.19 | 13.17 | 19.98 | 7.83 × 10−6 | 1.02 × 10−3 |
| Megasphaera | 6.09 | 6.16 | 12.29 | 4.55 × 10−4 | 2.95 × 10−2 | ||
| Unclassified Porphyromonadaceae | −7.59 | 8.32 | 9.19 | 2.43 × 10−3 | 1.05 × 10−1 | ||
| Unclassified Clostridiales | −1.46 | 13.78 | 8.42 | 3.71 × 10−3 | 1.21 × 10−1 | ||
| oropharyngeal cavity | Lactobacillus-based synbiotic supplementation | Unclassified Deltaproteobacteria | 7.39 | 6.79 | 14.56 | 1.36 × 10−4 | 1.76 × 10−2 |
| Lactobacillus | −7.28 | 8.12 | 9.84 | 1.71 × 10−3 | 1.11 × 10−1 | ||
| Proximal colon | Lactobacillus-based synbiotic supplementation | Unclassified Tremblayales | 10.44 | 9.76 | 24.38 | 7.89 × 10−7 | 8.13 × 10−5 |
| Unclassified Lactobacillales | 7.11 | 7.74 | 11.99 | 5.35 × 10−4 | 2.76 × 10−2 | ||
| Lactobacillus | 1.69 | 15.84 | 8.46 | 3.62 × 10−3 | 1.24 × 10−1 | ||
| Streptococcus | −1.92 | 16.63 | 7.85 | 5.09 × 10−3 | 1.31 × 10−1 | ||
| Vaginal canal | Bacillus-based synbiotic supplementation | Filifactor | −10.41 | 10.76 | 28.22 | 1.08 × 10−7 | 2.85 × 10−5 |
| Clostridium | 6.27 | 6.05 | 20.52 | 5.91 × 10−6 | 7.77 × 10−4 | ||
| Halalkalibacillus | 7.12 | 6.59 | 17.94 | 2.28 × 10−5 | 2.00 × 10−3 | ||
| Veillonella | −4.24 | 14.03 | 11.77 | 6.02 × 10−4 | 3.96 × 10−2 | ||
| Leifsonia | 7.46 | 6.84 | 10.80 | 1.01 × 10−3 | 5.33 × 10−2 | ||
| Unclassified Fusobacteriaceae | −3.36 | 16.06 | 10.02 | 1.55 × 10−3 | 5.89 × 10−2 | ||
| Unclassified Clostridiales | −2.64 | 16.00 | 10.00 | 1.57 × 10−3 | 5.89 × 10−2 | ||
| Bacteroides | −3.40 | 16.48 | 9.08 | 2.58 × 10−3 | 8.48 × 10−2 | ||
| Paraeggerthella | −5.06 | 9.24 | 8.62 | 3.32 × 10−3 | 9.17 × 10−2 | ||
| Clostridium | −5.27 | 10.71 | 8.53 | 3.49 × 10−3 | 9.17 × 10−2 | ||
| Unclassified Bacteroidales | −2.05 | 17.53 | 7.92 | 4.90 × 10−3 | 1.17 × 10−1 | ||
| Peptostreptococcus | −2.96 | 12.62 | 7.65 | 5.66 × 10−3 | 1.24 × 10−1 | ||
| Unclassified Bacteria | −2.41 | 15.66 | 7.23 | 7.18 × 10−3 | 1.38 × 10−1 | ||
| Megamonas | 4.84 | 5.39 | 7.18 | 7.36 × 10−3 | 1.38 × 10−1 | ||
| Finegoldia | 2.71 | 11.79 | 7.02 | 8.07 × 10−3 | 1.41 × 10−1 | ||
| Weissella | −5.82 | 6.60 | 6.87 | 8.74 × 10−3 | 1.43 × 10−1 | ||
| Unclassified Dermatophilaceae | 5.31 | 6.93 | 6.77 | 9.25 × 10−3 | 1.43 × 10−1 | ||
| Campylobacter | −1.93 | 15.48 | 6.61 | 1.01 × 10−2 | 1.48 × 10−1 | ||
| Vaginal canal | Lactobacillus-based synbiotic supplementation | Haemophilus | 10.11 | 8.82 | 20.36 | 6.42 × 10−6 | 1.69 × 10−3 |
| Oribacterium | 7.74 | 6.77 | 10.23 | 1.38 × 10−3 | 1.75 × 10−1 |
| Body Site | Synbiotic | Superpathway Class | Pathway Name | Log FC * | Log CPM * | LR * | p-Value | FDR * |
|---|---|---|---|---|---|---|---|---|
| oropharyngeal cavity | Bacillus-based synbiotic | Carbohydrate Biosynthesis | superpathway of UDP-N-acetylglucosamine-derived O-antigen building blocks biosynthesis | −3.12 | 8.61 | 28.72 | 8.37 × 10−8 | 2.84 × 10−5 |
| Cofactor, Carrier, and Vitamin Biosynthesis | superpathway of menaquinol-8 biosynthesis II | −3.11 | 7.65 | 24.24 | 8.51 × 10−7 | 1.44 × 10−4 | ||
| Carbohydrate Biosynthesis | CMP-pseudaminate biosynthesis | −3.30 | 6.13 | 17.80 | 2.46 × 10−5 | 1.50 × 10−3 | ||
| Carbohydrate Biosynthesis | protein N-glycosylation (bacterial) | −3.36 | 6.21 | 18.32 | 1.86 × 10−5 | 1.50 × 10−3 | ||
| Cofactor, Carrier, and Vitamin Biosynthesis | 1,4-dihydroxy-6-naphthoate biosynthesis II | −3.22 | 6.19 | 17.65 | 2.66 × 10−5 | 1.50 × 10−3 | ||
| Cofactor, Carrier, and Vitamin Biosynthesis | superpathway of demethylmenaquinol-6 biosynthesis II | −3.22 | 6.19 | 17.68 | 2.62 × 10−5 | 1.50 × 10−3 | ||
| Amine and Polyamine Biosynthesis | ectoine biosynthesis | 7.29 | −0.15 | 15.64 | 7.65 × 10−5 | 3.70 × 10−3 | ||
| Proximal colon | Lactobacillus-based synbiotic | Fatty Acid and Lipid Biosynthesis | (5Z)-dodecenoate biosynthesis I | −1.47 | 11.22 | 22.44 | 2.17 × 10−6 | 7.39 × 10−4 |
| Vaginal canal | Bacillus-based synbiotic | Degradation/Utilization/Assimilation-Other | caprolactam degradation | 7.24 | −0.16 | 17.29 | 3.20 × 10−5 | 1.56 × 10−3 |
| Nucleosides and Nucleotides Degradation | adenosine nucleotides degradation IV | 6.84 | −0.42 | 20.11 | 7.33 × 10−6 | 5.73 × 10−4 | ||
| Fatty Acid and Lipids Degradation | phospholipase pathway | 6.28 | −0.77 | 15.00 | 1.08 × 10−4 | 2.81 × 10−3 | ||
| Aromatic Compounds Degradation | gallate degradation I | 4.36 | 0.81 | 11.17 | 8.32 × 10−4 | 9.30 × 10−3 | ||
| Aromatic Compounds Degradation | vanillin and vanillate degradation I | 4.20 | 1.36 | 9.40 | 2.17 × 10−3 | 1.70 × 10−2 | ||
| Aromatic Compounds Degradation | superpathway of vanillin and vanillate degradation | 4.20 | 1.36 | 9.45 | 2.11 × 10−3 | 1.70 × 10−2 | ||
| Aromatic Compounds Degradation | vanillin and vanillate degradation II | 4.20 | 1.51 | 9.24 | 2.37 × 10−3 | 1.82 × 10−2 | ||
| Carbohydrates Biosynthesis | CMP-legionaminate biosynthesis I | 2.05 | 6.61 | 7.77 | 5.31 × 10−3 | 3.26 × 10−2 | ||
| Aromatic Compounds Degradation | catechol degradation to β-ketoadipate | 1.66 | 7.67 | 7.34 | 6.75 × 10−3 | 3.88 × 10−2 | ||
| Aromatic Compounds Degradation | protocatechuate degradation II (ortho-cleavage pathway) | 1.63 | 8.39 | 9.81 | 1.74 × 10−3 | 1.48 × 10−2 | ||
| Cell Structures Biosynthesis | peptidoglycan biosynthesis II (staphylococci) | 1.61 | 9.61 | 10.88 | 9.70 × 10−4 | 9.65 × 10−3 | ||
| Superpathways | superpathway of 2,3-butanediol biosynthesis | 1.57 | 7.46 | 8.42 | 3.71 × 10−3 | 2.57 × 10−2 | ||
| Cofactors, Prosthetic Groups, Electron Carriers Biosynthesis | mycothiol biosynthesis | 1.39 | 8.07 | 7.86 | 5.05 × 10−3 | 3.23 × 10−2 | ||
| Secondary Metabolite Biosynthesis | mevalonate pathway I (eukaryotes and bacteria) | 1.19 | 9.90 | 11.49 | 6.99 × 10−4 | 8.54 × 10−3 | ||
| Secondary Metabolite Biosynthesis | superpathway of geranylgeranyldiphosphate biosynthesis I (via mevalonate) | 1.11 | 10.30 | 12.44 | 4.21 × 10−4 | 6.58 × 10−3 | ||
| Amino Acid Degradation | L-lysine fermentation to acetate and butanoate | −1.03 | 11.14 | 11.54 | 6.79 × 10−4 | 8.54 × 10−3 | ||
| Amino Acid Degradation | L-glutamate degradation V (via hydroxyglutarate) | −1.06 | 10.24 | 8.19 | 4.21 × 10−3 | 2.79 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bugenyi, A.W.; Song, K.-D.; Lee, H.-K.; Heo, J. Bacillus- and Lactobacillus-Based Dietary Synbiotics Are Associated with Shifts in the Oropharyngeal, Proximal Colonic, and Vaginal Microbiomes of Korean Native Black Pigs. Fermentation 2023, 9, 359. https://doi.org/10.3390/fermentation9040359
Bugenyi AW, Song K-D, Lee H-K, Heo J. Bacillus- and Lactobacillus-Based Dietary Synbiotics Are Associated with Shifts in the Oropharyngeal, Proximal Colonic, and Vaginal Microbiomes of Korean Native Black Pigs. Fermentation. 2023; 9(4):359. https://doi.org/10.3390/fermentation9040359
Chicago/Turabian StyleBugenyi, Andrew Wange, Ki-Duk Song, Hak-Kyo Lee, and Jaeyoung Heo. 2023. "Bacillus- and Lactobacillus-Based Dietary Synbiotics Are Associated with Shifts in the Oropharyngeal, Proximal Colonic, and Vaginal Microbiomes of Korean Native Black Pigs" Fermentation 9, no. 4: 359. https://doi.org/10.3390/fermentation9040359
APA StyleBugenyi, A. W., Song, K.-D., Lee, H.-K., & Heo, J. (2023). Bacillus- and Lactobacillus-Based Dietary Synbiotics Are Associated with Shifts in the Oropharyngeal, Proximal Colonic, and Vaginal Microbiomes of Korean Native Black Pigs. Fermentation, 9(4), 359. https://doi.org/10.3390/fermentation9040359