
Monascus purpureus Fermented Product Ameliorates Learning and Memory Impairment in the Amyloid Precursor Protein Transgenic J20 Mouse Model of Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. J20 APP Transgenic Mice
2.2. In Vivo Drug Treatments
2.3. Morris Water Maze Test
2.4. Passive Avoidance Test
2.5. Measurement of Aβ Levels
2.6. Measurement of Phosphorylated Tau, TNF-α, and Acetylcholinesterase Activity
2.7. Gene Expression Analysis by Quantitative Polymerase Chain Reaction
2.8. Immunohistochemistry
2.8.1. Aβ Immunohistochemical Staining
2.8.2. APP IHC Stain
2.9. Statistical Analysis
3. Results
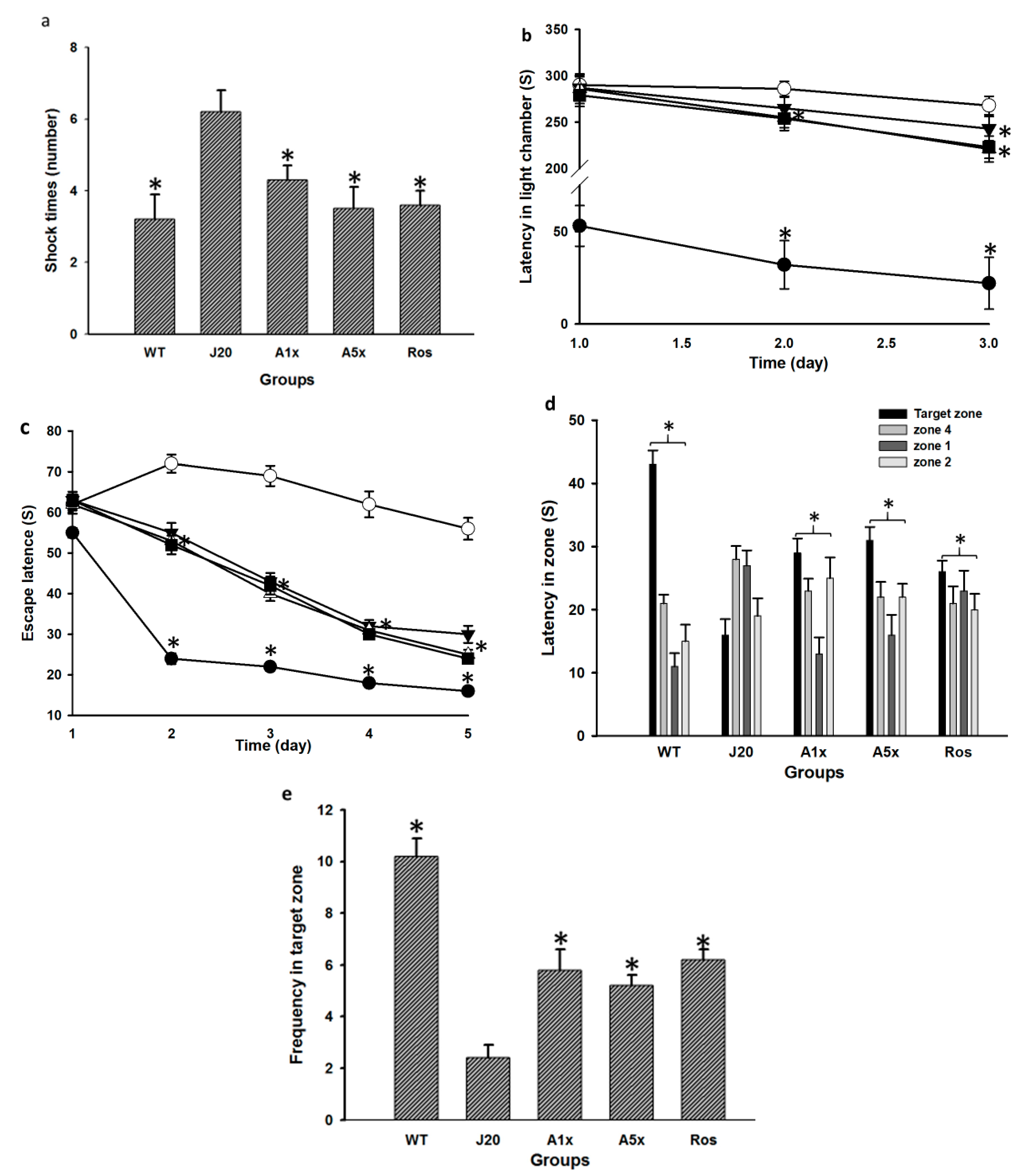
3.1. MPFP Significantly Improved Cognitive Behavior, Learning, and Memory Ability
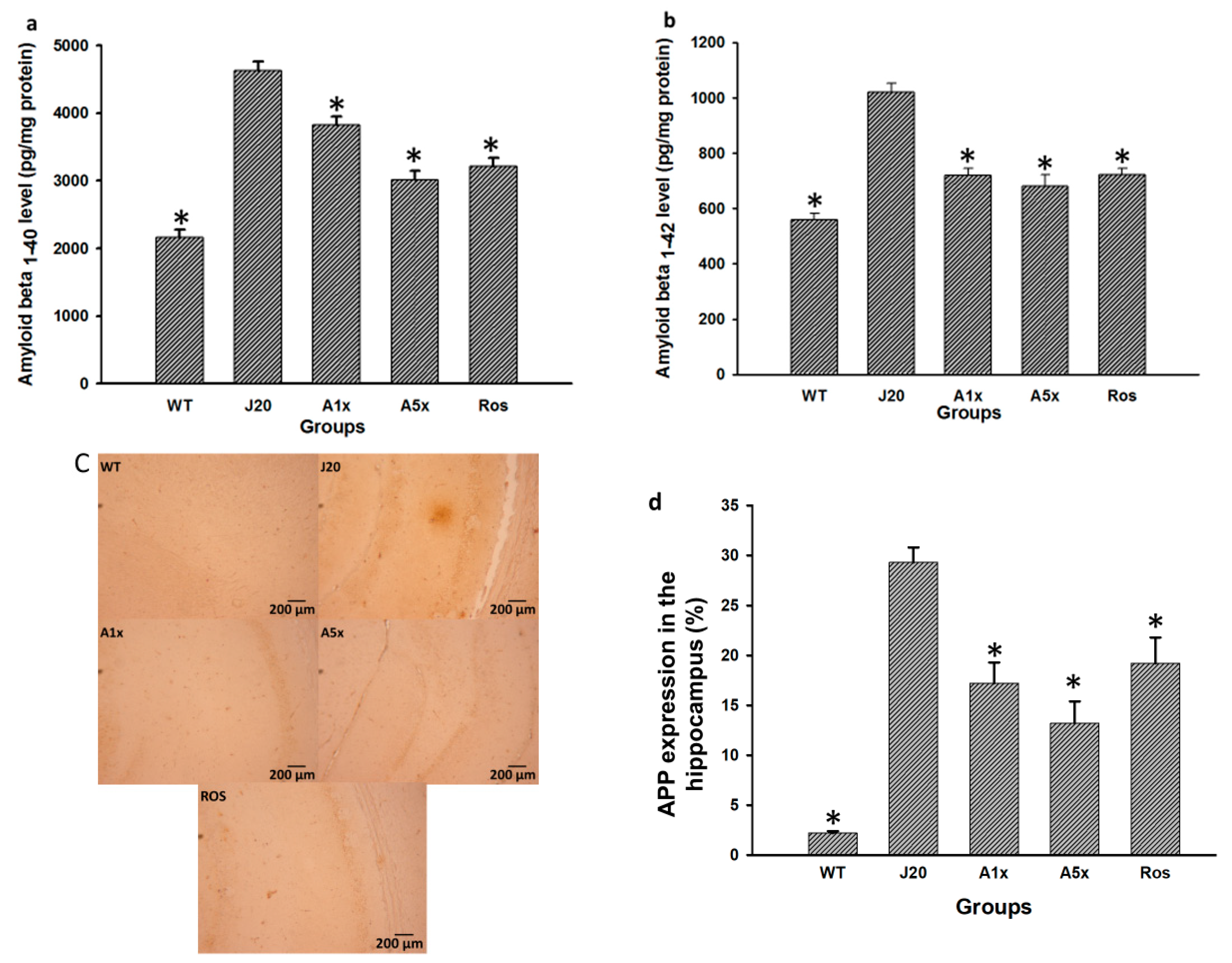
3.2. MPFP Treatment Reduced Brain Aβ Levels
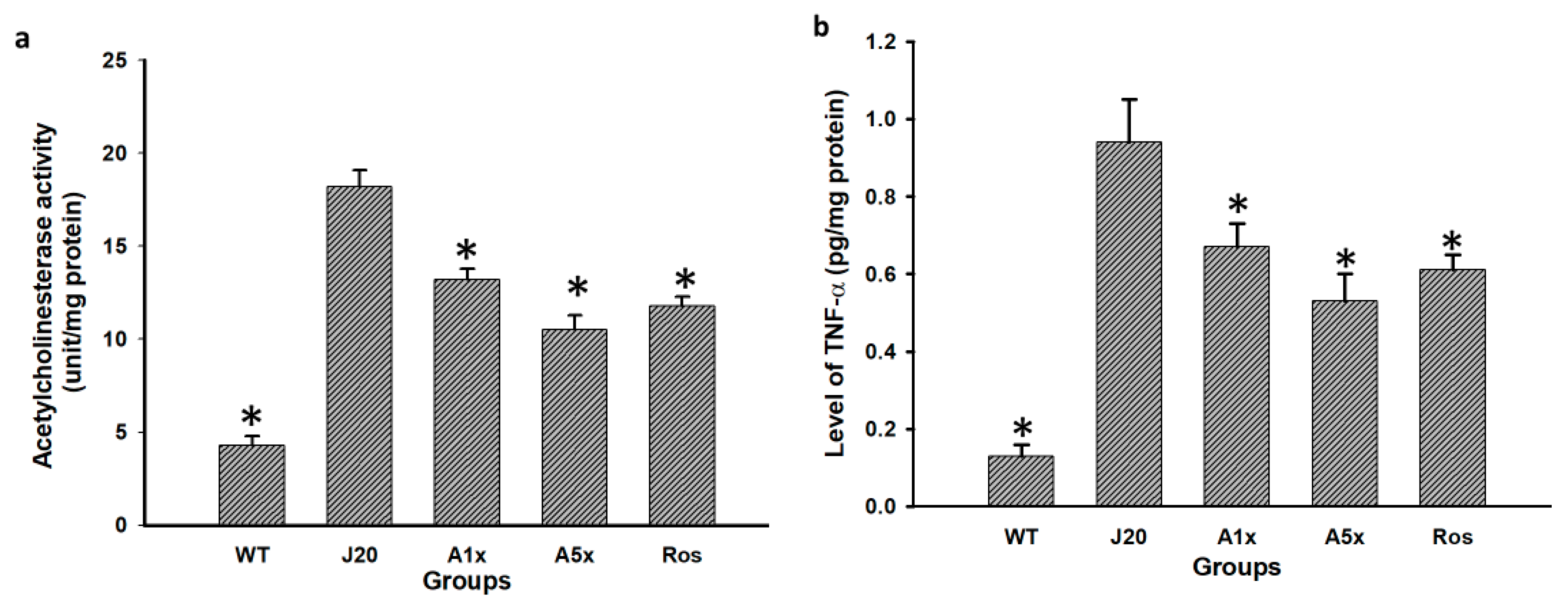
3.3. MPFP Enhanced Brain Cholinergic Activity in J20 Mice
3.4. Effect of MPFP on TNF-α and Neuroinflammation
3.5. MPFP Improved AD-Related Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Filippo, M.; Sarchielli, P.; Picconi, B.; Calabresi, P. Neuroinflammation and synaptic plasticity: Theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol. Sci. 2008, 29, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [Green Version]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Tosh, J.L.; Rickman, M.; Rhymes, E.; Norona, F.E.; Clayton, E.; Mucke, L.; Isaacs, A.M.; Elizabeth, M.C.; Fisher, E.C.M.; Wiseman, F.K. The integration site of the APP transgene in the J20 mouse model of Alzheimer’s disease. Wellcome Open Res. 2018, 2, 84. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.L.; Wang, J.J.; Pan, T.M. Red mold rice extract represses amyloid beta peptide-induced neurotoxicity via potent synergism of anti-inflammatory and antioxidative effect. Appl. Microbiol. Biotechnol. 2008, 79, 829–841. [Google Scholar] [CrossRef]
- Lee, C.L.; Kuo, T.F.; Wang, J.J.; Pan, T.M. Red mold rice ameliorates impairment of memory and learning ability in intracerebroventricular amyloid beta-infused rat by repressing amyloid beta accumulation. J. Neurosci. Res. 2007, 85, 3171–3182. [Google Scholar] [CrossRef]
- Lee, C.L.; Wang, J.J.; Kuo, S.L.; Pan, T.M. Monascus fermentation of dioscorea for increasing the production of cholesterol-lowering agent—Monacolin K and antiinflammation agent—Monascin. Appl. Microbiol. Biotechnol. 2006, 72, 1254–1262. [Google Scholar] [CrossRef]
- Lee, B.H.; Hsu, W.H.; Huang, T.; Chang, Y.Y.; Hsu, Y.W.; Pan, T.M. Monascin improves diabetes and dyslipidemia by regulating PPARgamma and inhibiting lipogenesis in fructose-rich diet-induced C57BL/6 mice. Food Funct. 2013, 4, 950–959. [Google Scholar] [CrossRef]
- Hsu, W.H.; Lee, B.H.; Chang, Y.Y.; Hsu, Y.W.; Pan, T.M. A novel natural Nrf2 activator with PPARγ-agonist (monascin) attenuates the toxicity of methylglyoxal and hyperglycemia. Toxicol. Appl. Pharmacol. 2013, 272, 842–851. [Google Scholar] [PubMed]
- Hsu, W.H.; Lee, B.H.; Liao, T.H.; Hsu, Y.W.; Pan, T.M. Monascus-fermented metabolite monascin suppresses inflammation via PPAR-gamma regulation and JNK inactivation in THP-1 monocytes. Food Chem. Toxicol. 2012, 50, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPAR gamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano, L.; Simon, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; Lopez de Maturana, R.; Garcia-Osta, A.; Ricobaraza, A.; Perez-Mediavilla, A.; Del Rio, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, I.H.; Scearce-Levie, K.; Legleiter, J.; Palop, J.J.; Gerstein, H.; Bien-Ly, N.; Puolivali, J.; Lesne, S.; Ashe, K.H.; Muchowski, P.J.; et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 2007, 282, 23818–23828. [Google Scholar] [CrossRef] [Green Version]
- Ratner, M. FDA pharmacogenomics guidance sends clear message to industry. Nat. Rev. Drug Discov. 2005, 4, 359. [Google Scholar] [CrossRef]
- Crawley, J.N. What’s Wrong with My Mouse? Behavioral Phyenotyping of Transgenic and Knockout Mice, 2nd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007. [Google Scholar]
- Arendt, T.; Bigl, V.; Tennstedt, A.; Arendt, A. Correlation between cortical plaque count and neuronal loss in the nucleus basalis in Alzheimer’s disease. Neurosci. Lett. 1984, 48, 81–85. [Google Scholar]
- Aucoin, J.S.; Jiang, P.; Aznavour, N.; Tong, X.K.; Buttini, M.; Descarries, L.; Hamel, E. Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer’s disease. Neuroscience 2005, 132, 73–86. [Google Scholar]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Bandeira-Echtler, E.; Bergerhoff, K.; Clar, C.; Ebrahim, S.H. Rosiglitazone for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2007, 18, CD006063. [Google Scholar]
- Scott, H.D.; Laake, K. Statins for the prevention of Alzheimer’s disease. Cochrane Database Syst. Rev. 2001, 3, CD003160. [Google Scholar]
- Shi, Y.C.; Pan, T.M. Beneficial effects of Monascus purpureus NTU 568-fermented products: A review. Appl. Microbiol. Biotechnol. 2011, 90, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano, L.; Simon, A.M.; Perez-Mediavilla, A.; Salazar-Colocho, P.; Del Rio, J.; Frechilla, D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer’s disease mouse model. Biochem. Biophys. Res. Commun. 2009, 379, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, W.A.; McMillan, P.J.; Kulstad, J.J.; Leverenz, J.B.; Craft, S.; Haynatzki, G.R. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 2006, 199, 265–273. [Google Scholar] [CrossRef]
- Toledo, E.M.; Inestrosa, N.C. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol. Psychiatry 2010, 15, 272–285. [Google Scholar] [CrossRef]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G.; et al. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.; Sawmiller, D.; Tan, J. Restoring soluble amyloid precursor protein α functions as a potential treatment for Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 973–991. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.L.; Kuo, T.F.; Wu, C.L.; Wang, J.J.; Pan, T.M. Red mold rice promotes neuroprotective sAPPα secretion instead of Alzheimer’s risk factors and amyloid beta expression in hyperlipidemic Aβ40-infused rats. J. Agric. Food Chem. 2010, 58, 2230–2238. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Godoy, J.A.; Quintanilla, R.A.; Koenig, C.S.; Bronfman, M. Peroxisome proliferator-activated receptor γ is expressed in hippocampal neurons and its activation prevents β-amyloid neurodegeneration: Role of Wnt signaling. Exp. Cell Res. 2005, 304, 91–104. [Google Scholar] [CrossRef]
- d’Abramo, C.; Ricciarelli, R.; Pronzato, M.A.; Davies, P. Troglitazone, a peroxisome proliferator-activated receptor-gamma agonist, decreases tau phosphorylation in CHOtau4R cells. J. Neurochem. 2006, 98, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Lee, E.J.; Kwon, K.J.; Shin, C.Y.; Song, K.; Park, J.; Jo, I.; Han, S. Troglitazone, a thiazolidinedione, decreases tau phosphorylation through the inhibition of cyclin-dependent kinase 5 activity in SH-SY5Y neuroblastoma cells and primary neurons. J. Neurochem. 2013, 126, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin mice. J. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, M.-C.; Cheng, I.H.-J.; Chen, C.-L. Monascus purpureus Fermented Product Ameliorates Learning and Memory Impairment in the Amyloid Precursor Protein Transgenic J20 Mouse Model of Alzheimer’s Disease. Fermentation 2022, 8, 193. https://doi.org/10.3390/fermentation8050193
Fang M-C, Cheng IH-J, Chen C-L. Monascus purpureus Fermented Product Ameliorates Learning and Memory Impairment in the Amyloid Precursor Protein Transgenic J20 Mouse Model of Alzheimer’s Disease. Fermentation. 2022; 8(5):193. https://doi.org/10.3390/fermentation8050193
Chicago/Turabian StyleFang, Ming-Chih, Irene Han-Juo Cheng, and Chien-Li Chen. 2022. "Monascus purpureus Fermented Product Ameliorates Learning and Memory Impairment in the Amyloid Precursor Protein Transgenic J20 Mouse Model of Alzheimer’s Disease" Fermentation 8, no. 5: 193. https://doi.org/10.3390/fermentation8050193
APA StyleFang, M.-C., Cheng, I. H.-J., & Chen, C.-L. (2022). Monascus purpureus Fermented Product Ameliorates Learning and Memory Impairment in the Amyloid Precursor Protein Transgenic J20 Mouse Model of Alzheimer’s Disease. Fermentation, 8(5), 193. https://doi.org/10.3390/fermentation8050193