
Temporal Comparison of Microbial Community Structure in an Australian Winery


Abstract
:1. Introduction
2. Materials and Methods
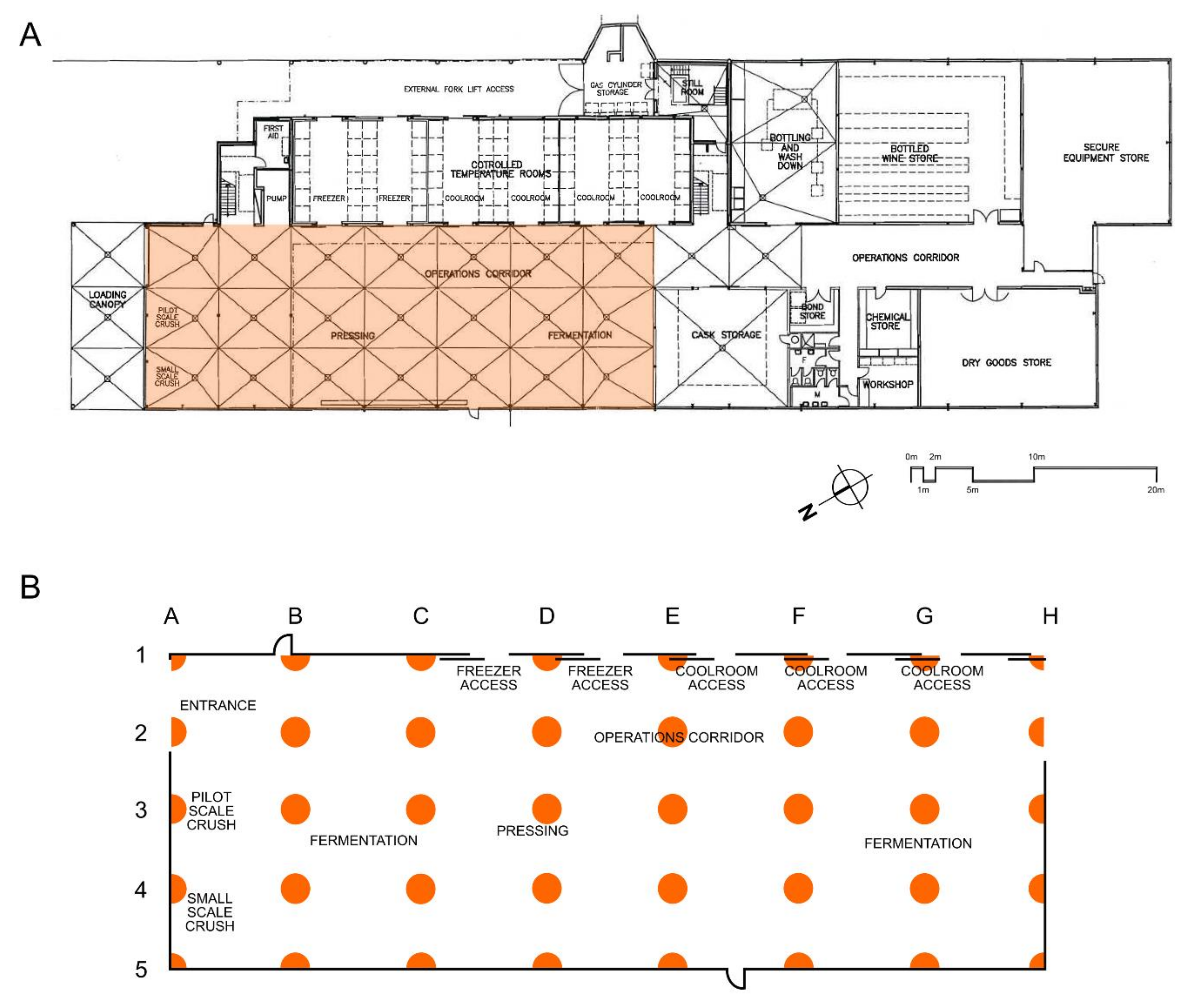
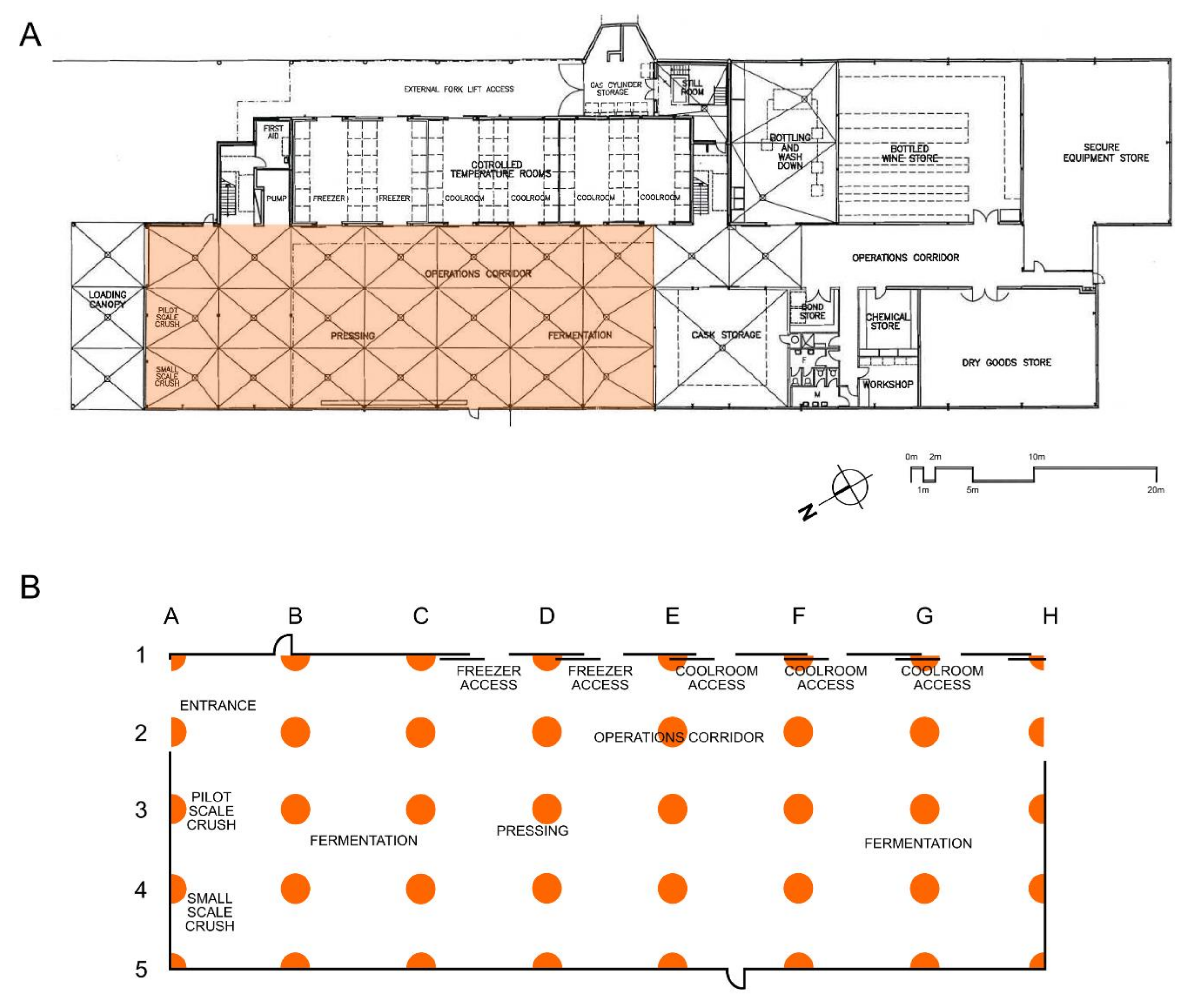
2.1. Winery Sampling
2.2. Determination of Microbial Populations
2.3. Data Analysis
3. Results
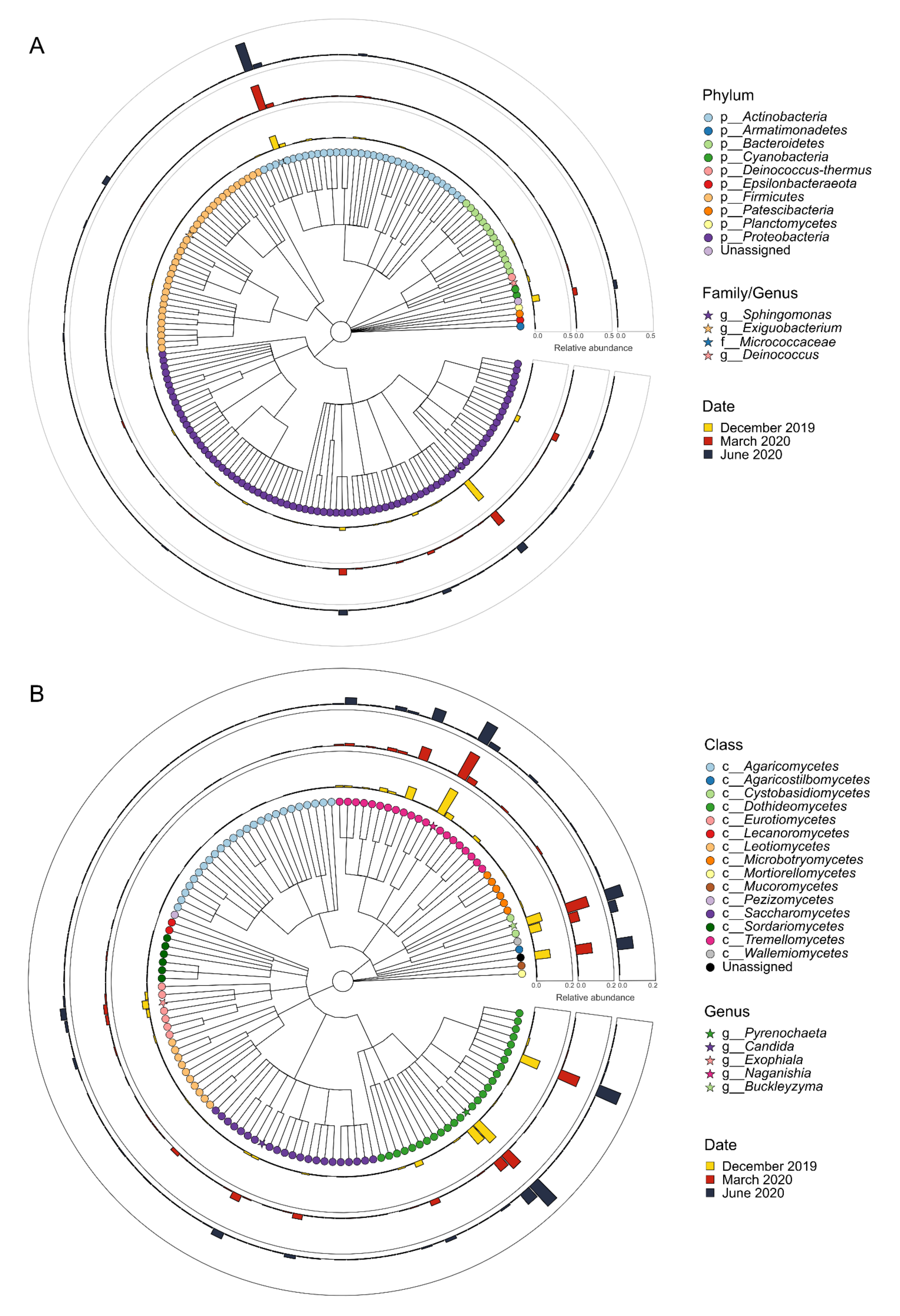
3.1. Microbial Populations in the Winery
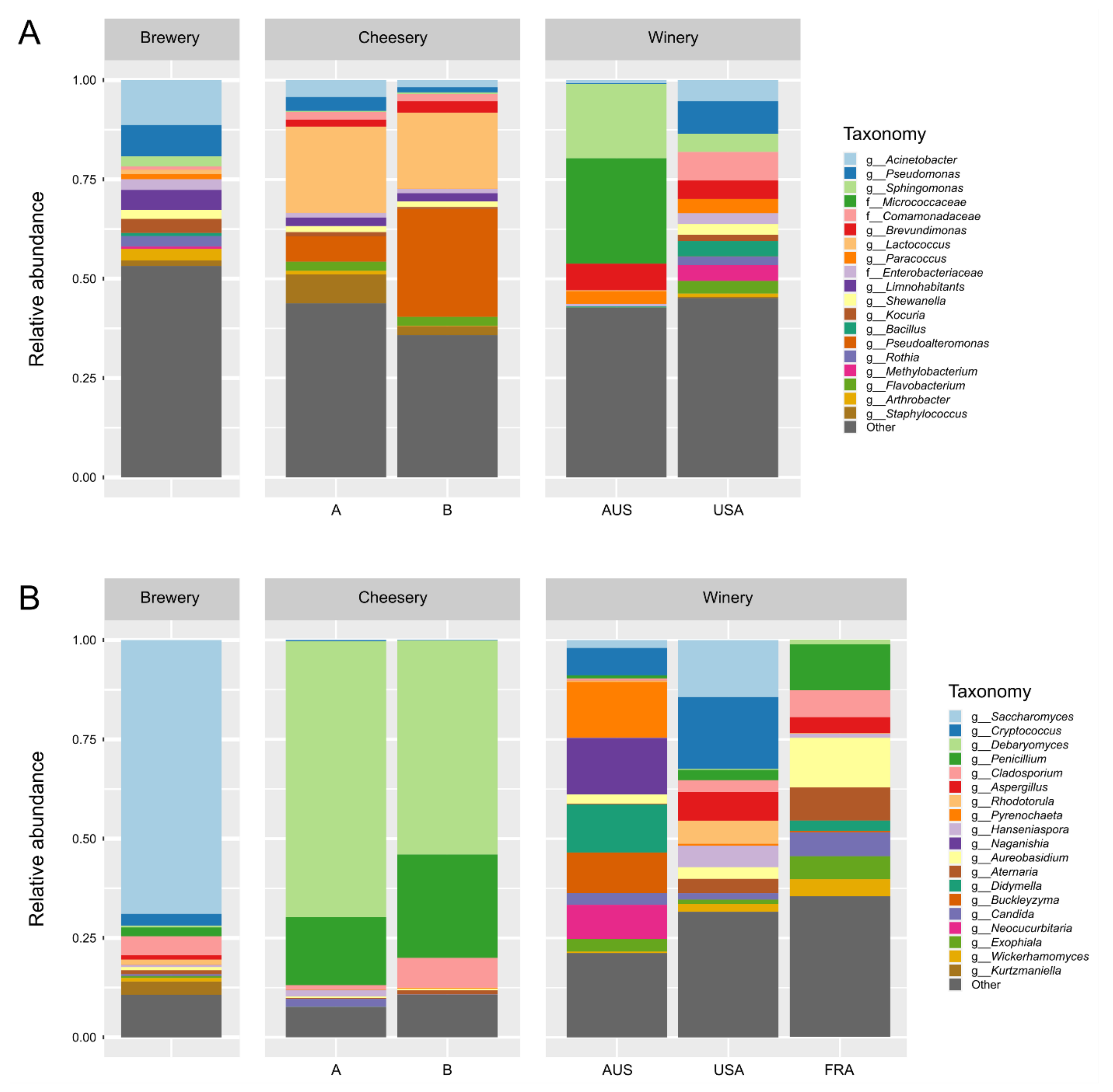
3.2. Community Structure Differs Between Food and Beverage Facilities
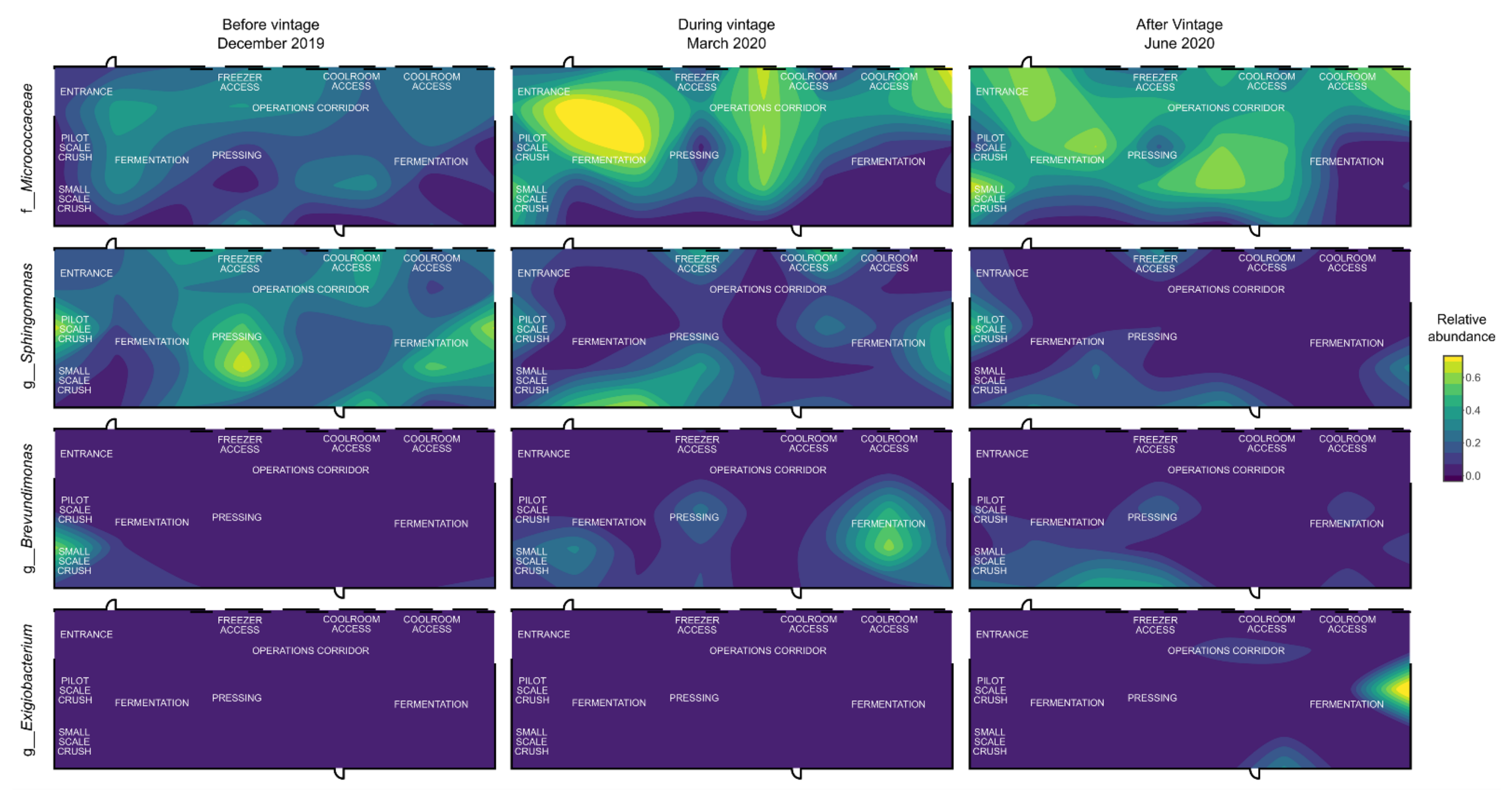
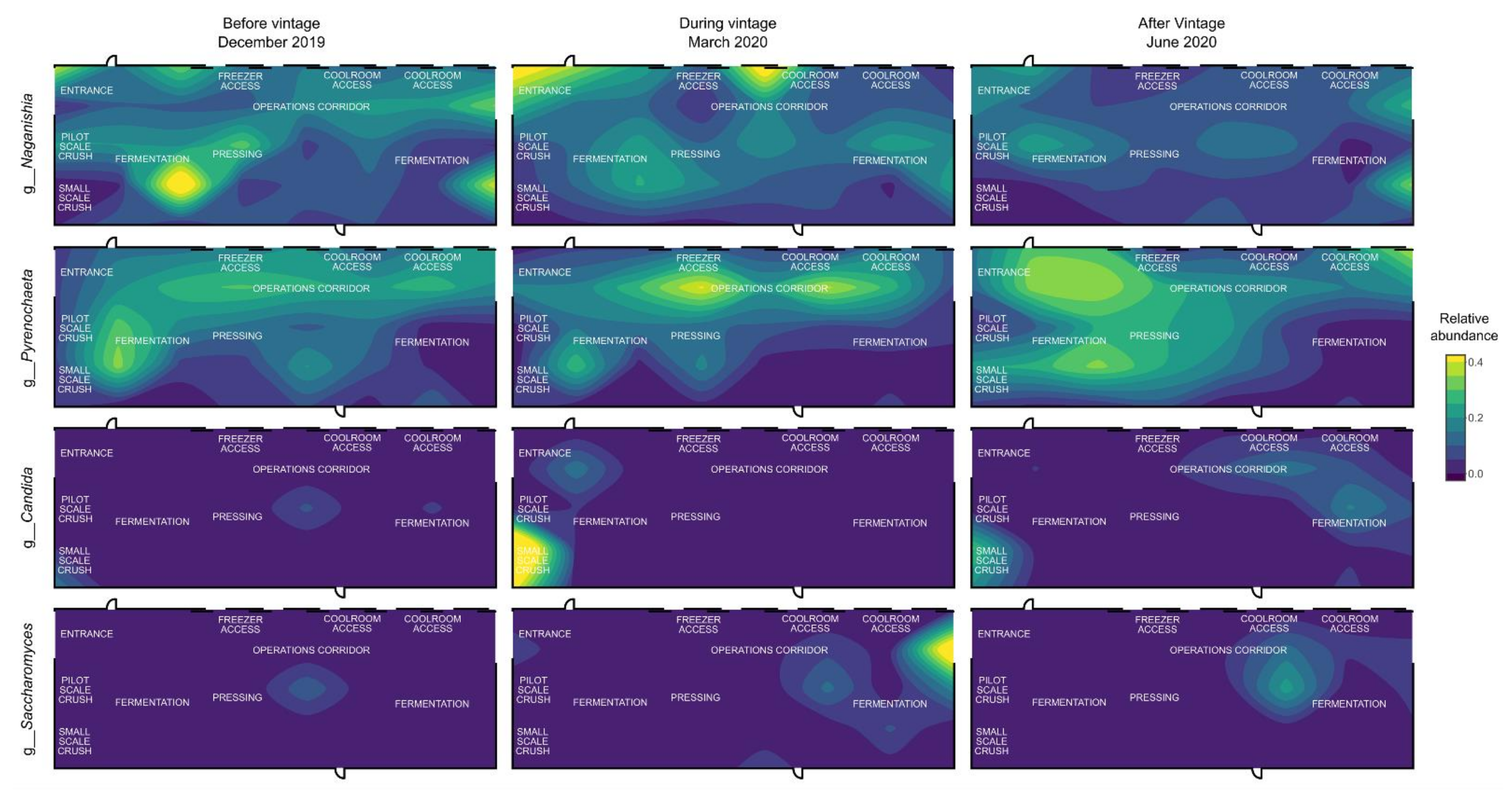
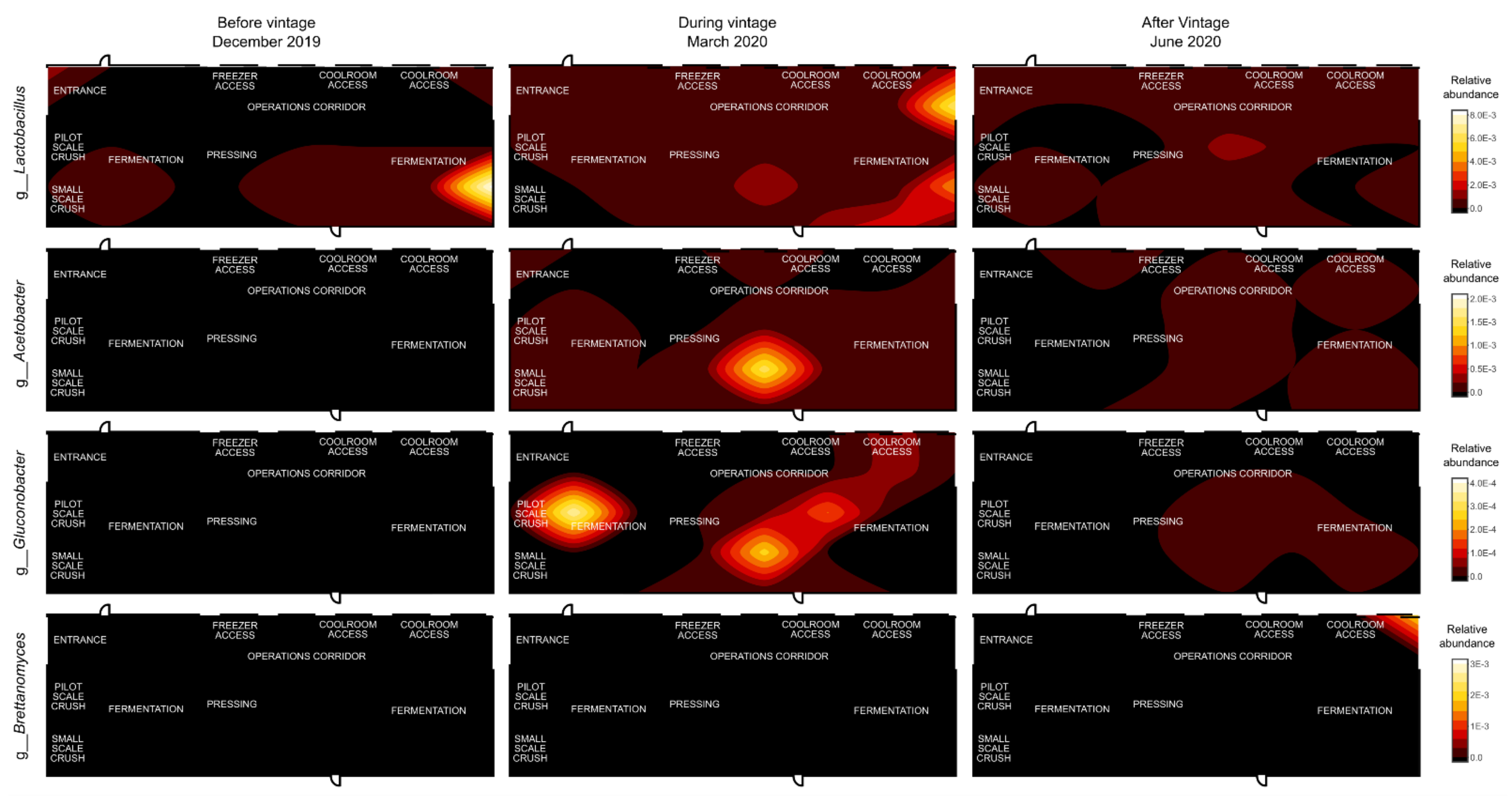
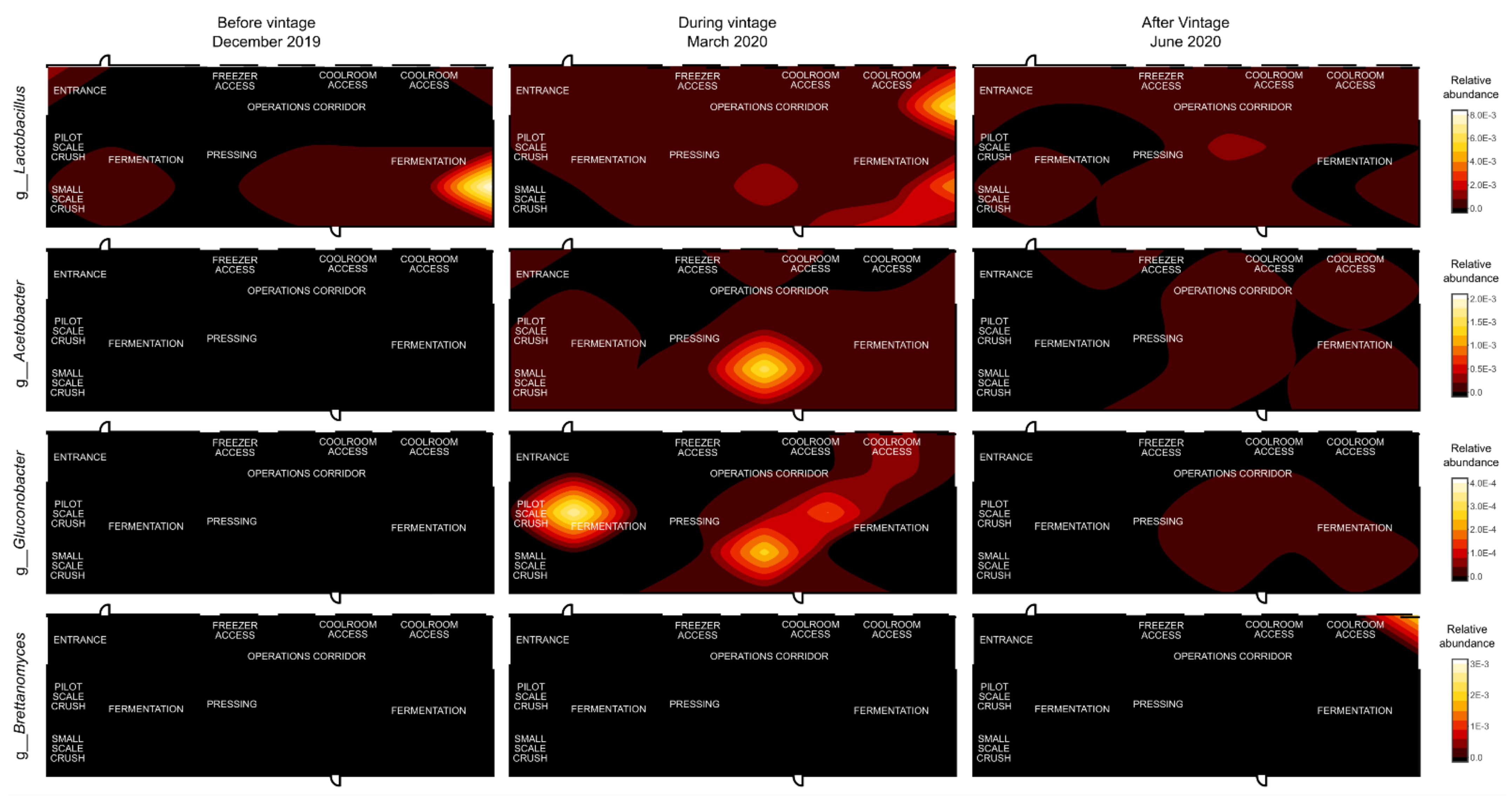
3.3. Mapping Microbial Abundance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGovern, P.E.; Zhang, J.; Tang, J.; Zhang, Z.; Hall, G.R.; Moreau, R.; Nunez, A.; Butrym, E.D.; Richards, M.P.; Wang, C.S.; et al. Fermented beverages of pre- and proto-historic China. Proc. Natl. Acad. Sci. USA 2004, 101, 17593–17598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamang, J.P.; Holzapfel, W.H.; Shin, D.H.; Felis, G.E. Microbiology of ethnic fermented foods and alcoholic beverages of the world. Front. Microbiol. 2017, 8, 1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, K.R.; Chaves, O.M.; Mallott, E.K.; Eppley, T.M.; Abreu, F.; Baden, A.L.; Barnett, A.A.; Bicca-Marques, J.C.; Boyle, S.A.; Campbell, C.J.; et al. Fermented food consumption in wild nonhuman primates and its ecological drivers. Am. J. Phys. Anthropol. 2021, 175, 513–530. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Mills, D.A. Facility-specific “house” microbiome drives microbial landscapes of artisan cheesemaking plants. Appl. Environ. Microbiol. 2013, 79, 5214–5223. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Bergsveinson, J.; Ziola, B.; Mills, D.A. Mapping microbial ecosystems and spoilage-gene flow in breweries highlights patterns of contamination and resistance. elife 2015, 4, e04634. [Google Scholar] [CrossRef]
- Doyle, C.J.; O’Toole, P.W.; Cotter, P.D. Metagenome-based surveillance and diagnostic approaches to studying the microbial ecology of food production and processing environments. Environ. Microbiol. 2017, 19, 4382–4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdo, H.; Catacchio, C.R.; Ventura, M.; D’Addabbo, P.; Alexandre, H.; Guilloux-Benatier, M.; Rousseaux, S. The establishment of a fungal consortium in a new winery. Sci. Rep. 2020, 10, 7962. [Google Scholar] [CrossRef]
- Einson, J.E.; Rani, A.; You, X.M.; Rodriguez, A.A.; Randell, C.L.; Barnaba, T.; Mammel, M.K.; Kotewicz, M.L.; Elkins, C.A.; Sela, D.A. A vegetable fermentation facility hosts distinct microbiomes reflecting the production environment. Appl. Environ. Microbiol. 2018, 84, e01680-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwirzitz, B.; Wetzels, S.U.; Dixon, E.D.; Stessl, B.; Zaiser, A.; Rabanser, I.; Thalguter, S.; Pinior, B.; Roch, F.F.; Strachan, C.; et al. The sources and transmission routes of microbial populations throughout a meat processing facility. Npj Biofilms Microbiomes 2020, 6, 26. [Google Scholar] [CrossRef]
- Chacon-Vargas, K.; Torres, J.; Giles-Gomez, M.; Escalante, A.; Gibbons, J.G. Genomic profiling of bacterial and fungal communities and their predictive functionality during pulque fermentation by whole-genome shotgun sequencing. Sci. Rep. 2020, 10, 15115. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.J.; Nam, Y.D.; Roh, S.W.; Bae, J.W. Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol. 2012, 30, 112–123. [Google Scholar] [CrossRef]
- Varela, C.; Sundstrom, J.; Cuijvers, K.; Jiranek, V.; Borneman, A. Discovering the indigenous microbial communities associated with the natural fermentation of sap from the cider gum Eucalyptus gunnii. Sci. Rep. 2020, 10, 14716. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Bamforth, C.W.; Mills, D.A. Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PLoS ONE 2012, 7, e35507. [Google Scholar] [CrossRef] [PubMed]
- Spitaels, F.; Wieme, A.D.; Janssens, M.; Aerts, M.; Daniel, H.-M.; Van Landschoot, A.; De Vuyst, L.; Vandamme, P. The microbial diversity of traditional spontaneously fermented lambic beer. PLoS ONE 2014, 9, e95384. [Google Scholar] [CrossRef] [PubMed]
- Calasso, M.; Ercolini, D.; Mancini, L.; Stellato, G.; Minervini, F.; Di Cagno, R.; De Angelis, M.; Gobbetti, M. Relationships among house, rind and core microbiotas during manufacture of traditional Italian cheeses at the same dairy plant. Food Microbiol. 2016, 54, 115–126. [Google Scholar] [CrossRef]
- Lortal, S.; Di Blasi, A.; Madec, M.N.; Pediliggieri, C.; Tuminello, L.; Tanguy, G.; Fauquant, J.; Lecuona, Y.; Campo, P.; Carpino, S.; et al. Tina wooden vat biofilm: A safe and highly efficient lactic acid bacteria delivering system in PDO Ragusano cheese making. Int. J. Food Microbiol. 2009, 132, 1–8. [Google Scholar] [CrossRef]
- Hultman, J.; Rahkila, R.; Ali, J.; Rousu, J.; Björkroth, K.J. Meat processing plant microbiome and contamination patterns of cold-tolerant bacteria causing food safety and spoilage risks in the manufacture of vacuum-packaged cooked sausages. Appl. Environ. Microbiol. 2015, 81, 7088–7097. [Google Scholar] [CrossRef] [Green Version]
- Spescha, C.; Stephan, R.; Zweifel, C. Microbiological contamination of pig carcasses at different stages of slaughter in two European Union-approved abattoirs. J. Food Prot. 2006, 69, 2568–2575. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Ohta, M.; Richardson, P.M.; Mills, D.A. Monitoring seasonal changes in winery-resident microbiota. PLoS ONE 2013, 8, e66437. [Google Scholar] [CrossRef]
- Morrison-Whittle, P.; Goddard, M.R. Quantifying the relative roles of selective and neutral processes in defining eukaryotic microbial communities. ISME J. 2015, 9, 2003–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.W.; Tsai, P.; Anfang, N.; Ross, H.A.; Goddard, M.R. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 2014, 16, 2848–2858. [Google Scholar] [CrossRef] [Green Version]
- Grangeteau, C.; Gerhards, D.; von Wallbrunn, C.; Alexandre, H.; Rousseaux, S.; Guilloux-Benatier, M. Persistence of two non-Saccharomyces yeasts (Hanseniaspora and Starmerella) in the cellar. Front. Microbiol. 2016, 7, 268. [Google Scholar] [CrossRef] [PubMed]
- Ocon, E.; Gutierrez, A.R.; Garijo, P.; Lopez, R.; Santamaria, P. Presence of non-Saccharomyces yeasts in cellar equipment and grape juice during harvest time. Food Microbiol. 2010, 27, 1023–1027. [Google Scholar] [CrossRef]
- Pérez-Martín, F.; Seseña, S.; Fernández-González, M.; Arévalo, M.; Palop, M.L. Microbial communities in air and wine of a winery at two consecutive vintages. Int. J. Food Microbiol. 2014, 190, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Sabate, J.; Cano, J.; Esteve-Zarzoso, B.; Guillamon, J.M. Isolation and identification of yeasts associated with vineyard and winery by RFLP analysis of ribosomal genes and mitochondrial DNA. Microbiol. Res. 2002, 157, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Varela, C.; Borneman, A.R. Yeasts found in vineyards and wineries. Yeast 2017, 34, 111–128. [Google Scholar] [CrossRef]
- Curtin, C.; Varela, C.; Borneman, A. Harnessing improved understanding of Brettanomyces bruxellensis biology to mitigate the risk of wine spoilage. Aust. J. Grape Wine Res. 2015, 21, 680–692. [Google Scholar] [CrossRef]
- Loureiro, V.; Malfeito-Ferreira, M. Spoilage yeasts in the wine industry. Int. J. Food Microbiol. 2003, 86, 23–50. [Google Scholar] [CrossRef]
- Ribéreau-Gayón, P.; Dubourdieu Donéche, D.B.; Lonvaud, A. Red winemaking. In Handbook of Enology; Ribéreau-Gayón, P., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Alston, J.M.; Arvik, T.; Hart, J.; Lapsley, J.T. Brettanomics I: The cost of Brettanomyces in California wine production. J. Wine Econ. 2021, 16, 4–31. [Google Scholar] [CrossRef]
- Parker, M.; Onetto, C.; Hixson, J.; Bilogrevic, E.; Schueth, L.; Pisaniello, L.; Borneman, A.; Herderich, M.; de Barros Lopes, M.; Francis, L. Factors contributing to interindividual variation in retronasal odor perception from aroma glycosides: The tole of odorant sensory detection threshold, oral microbiota, and hydrolysis in saliva. J. Agric. Food Chem. 2019, 68, 10299–10309. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Mills, D.A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 2013, 79, 2519–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 1. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Mahe, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. Peer J. 2014, 2, e593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternes, P.R.; Lee, D.; Kutyna, D.R.; Borneman, A.R. A combined meta-barcoding and shotgun metagenomic analysis of spontaneous wine fermentation. bioRxiv 2017, 6, gix040. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Lahti, L.; Shetty, S. Tools for Microbiome Analysis in R. Version 1.9.1. Available online: http://microbiome.github.com/microbiome (accessed on 30 June 2019).
- Oksanen, J.; Blanchet, G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5.4. Available online: https://CRAN.R-project.org/package=vegan (accessed on 30 June 2019).
- Li, C.; Yu, G.; Zhu, C. microbiomeviz—An R Package for Visualizing Microbiome Data. Available online: https://github.com/lch14forever/microbiomeViz (accessed on 30 June 2019).
- Dash Enterprise, Collaborative Data Science. Available online: https://plot.ly (accessed on 30 June 2019).
- Yu, G.; Smith, D.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods. Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Xu, S.; Yu, G. ggtreeExtra: An R Package to Add Geom Layers on Circular or Other Layout Tree of “ggtree”. Available online: https://github.com/YuLab-SMU/ggtreeExtra/ (accessed on 30 June 2019).
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Eady Plots. R Package Version 0.2. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 30 June 2019).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Matplotlib. Default Color Maps from ‘Matplotlib’. Available online: https://github.com/sjmgarnier/viridis (accessed on 30 November 2020).
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 30 June 2019).
- Shetty, S.; Lahti, L. Microbiomeutilities: An R Package with Utility Functions for The Microbiome R Package. Available online: https://github.com/microsud/microbiomeutilities-shiny (accessed on 30 June 2019).
- Abdo, H.; Catacchio, C.R.; Ventura, M.; D’Addabbo, P.; Calabrese, F.M.; Laurent, J.; David-Vaizant, V.; Alexandre, H.; Guilloux-Benatier, M.; Rousseaux, S. Colonization of wild Saccharomyces cerevisiae strains in a new winery. Beverages 2020, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleet, G.H.; Heard, G.M. Yeasts: Growth during fermentation. In Wine Microbiology and Biotechnology; Fleet, G.H., Ed.; Harwood Academic: Chur, Switzerland, 1993; pp. 27–54. [Google Scholar]
- Liu, X.Z.; Wang, Q.M.; Göker, M.; Groenewald, M.; Kachalkin, A.V.; Lumbsch, H.T.; Millanes, A.M.; Wedin, M.; Yurkov, A.M.; Boekhout, T.; et al. Towards an integrated phylogenetic classification of the Tremellomycete. Stud. Mycol. 2015, 81, 85–147. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.K.; Vimercati, L.; Darcy, J.L.; Arán, P.; Gendron, E.M.S.; Solon, A.J.; Porazinska, D.; Dorador, C.A. Naganishia in high places: Functioning populations or dormant cells from the atmosphere? Mycology 2017, 8, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Infantino, A.; Aragona, M.; Brunetti, A.; Lahoz, E.; Oliva, A.; Porta-Puglia, A. Molecular and physiological characterization of Italian isolates of Pyrenochaeta lycopersici. Mycol. Res. 2003, 107, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Lević, J.; Stanković, S.; Krnjaja, V.; Bočarov-Stančić, A. Simple and efficient method for detection and identification of Pyrenochaeta terrestris on maize root. Crop. Prot. 2012, 38, 66–71. [Google Scholar] [CrossRef]
- Keinath, A.P. From native plants in central Europe to cultivated crops worldwide: The emergence of Didymella bryoniae as a cucurbit pathogen. HortScience 2011, 46, 532. [Google Scholar] [CrossRef] [Green Version]
- De Roos, J.; Van der Veken, D.; De Vuyst, L. The interior surfaces of wooden barrels are an additional microbial inoculation source for lambic beer production. Appl. Environ. Microbiol. 2019, 85, e02226-18. [Google Scholar] [CrossRef] [Green Version]
- Quigley, L.; O’Sullivan, D.J.; Daly, D.; O’Sullivan, O.; Burdikova, Z.; Vana, R.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; McSweeney, P.L.H.; et al. Thermus and the pink discoloration defect in cheese. mSystems 2016, 1, e00023-16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Podtburg, T.C.; Zhao, P.; Chen, D.; Baker, D.A.; Cords, B.; Doyle, M.P. Reduction by competitive bacteria of Listeria monocytogenes in biofilms and Listeria bacteria in floor drains in a ready-to-eat poultry processing plant. J. Food Prot. 2013, 76, 601–607. [Google Scholar] [CrossRef]
- Santamaría, P.; Garijo, P.; López, R.; Tenorio, C.; Rosa Gutiérrez, A. Analysis of yeast population during spontaneous alcoholic fermentation: Effect of the age of the cellar and the practice of inoculation. Int J. Food Microbiol. 2005, 103, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Theisinger, S.M.; de Smidt, O. Bioaerosols in the food and beverage industry. In Ideas and Applications toward Sample Preparation for Food and Beverage Analysis; InTech: Rijeka, Croatia, 2017. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Date | |||
|---|---|---|---|
| Before Vintage December 2019 | During Vintage March 2020 | After Vintage June 2020 | |
| Bacterial communities | |||
| Richness | 549.90 a | 476.05 b | 494.03 b |
| Diversity 1 | 4.04 a | 3.50 b | 3.33 b |
| Dominance | 0.19 a | 0.26 b | 0.32 b |
| Evenness | 0.64 a | 0.57 b | 0.54 b |
| Fungal communities | |||
| Richness | 322.88 a | 292.63 b | 358.78 a |
| Diversity 1 | 2.84 a | 2.75 a | 2.86 a |
| Dominance | 0.27 a | 0.29 a | 0.29 a |
| Evenness | 0.49 a | 0.49 a | 0.49 a |
| Bacterial Communities | |||||
|---|---|---|---|---|---|
| Df | SS | F Model | p Value | Significance | |
| Location | 39 | 16.865 | 2.872 | 0.001 | *** |
| Sampling date | 2 | 2.638 | 8.762 | 0.001 | *** |
| Residuals | 78 | 11.743 | |||
| Fungal Communities | |||||
| Df | SS | F Model | p Value | Significance | |
| Location | 39 | 14.990 | 4.006 | 0.001 | *** |
| Sampling date | 2 | 0.608 | 3.169 | 0.001 | *** |
| Residuals | 73 | 7.004 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varela, C.; Cuijvers, K.; Borneman, A. Temporal Comparison of Microbial Community Structure in an Australian Winery. Fermentation 2021, 7, 134. https://doi.org/10.3390/fermentation7030134
Varela C, Cuijvers K, Borneman A. Temporal Comparison of Microbial Community Structure in an Australian Winery. Fermentation. 2021; 7(3):134. https://doi.org/10.3390/fermentation7030134
Chicago/Turabian StyleVarela, Cristian, Kathleen Cuijvers, and Anthony Borneman. 2021. "Temporal Comparison of Microbial Community Structure in an Australian Winery" Fermentation 7, no. 3: 134. https://doi.org/10.3390/fermentation7030134
APA StyleVarela, C., Cuijvers, K., & Borneman, A. (2021). Temporal Comparison of Microbial Community Structure in an Australian Winery. Fermentation, 7(3), 134. https://doi.org/10.3390/fermentation7030134