Abstract
Aspartic proteases are of significant importance for medicine and biotechnology. In spite of sufficient evidence that many non-Saccharomyces yeasts produce extracellular proteases, previous research has focused on the enzymes of Candida species because of their role as virulence factors. Nowadays, there is also increasing interest for their applications in industrial processes, mainly because of their activities at low pH values. Here, we report the features of new acid proteases isolated from wine-relevant yeasts Metschnikovia pulcherrima and Wickerhamomyces anomalus. To our knowledge, this is the first detailed description of such an enzyme derived from strains of W. anomalus. Deviating to most former studies, we could demonstrate that the yeasts produce these enzymes in a natural substrate (grape juice) during the active growth phase. The enzymes were purified from concentrated grape juice by preparative isoelectric focusing. Biochemical data (maximum activity at ≈ pH 3.0, inhibition by pepstatin A) classify them as aspartic proteases. For W. anomalus 227, this assumption was confirmed by the protein sequence of WaAPR1 determined by LC-MS/MS. The sequence revealed a signal peptide for secretion, as well as a peptidase A1 domain with two aspartate residues in the active site. The enzyme has a calculated molecular mass of 47 kDa and an isolelectric point of 4.11.
1. Introduction
The use of mixed starters of selected non-Saccharomyces yeasts and Saccharomyces cerevisiae is of increasing interest for production of novel wines with more complex organoleptic characteristics [1] and/or lower ethanol contents [2]. Non-Saccharomyces wine yeasts, also called “wild” yeasts, can enhance the analytical composition and aroma profile of wine by production of secondary metabolites and secretion of enzymes [3,4,5]. Yeasts of the genera Kloeckera, Candida, Debaryomyces, Rhodotorula, Pichia, Wickerhamomyces, Zygosaccharomyces, Hanseniaspora, Kluyveromyces and Metschnikowia produce hydrolytic exoenzymes (esterases, lipases, glycosidases, glucanases, pectinases, amylases, and proteases) that interact with grape compounds [6]. Particularly, glycoside hydrolases can release aroma active compounds in grape must from their odourless glycosidic precursors [7]. Others produce pectinolytic enzymes that can promote grape must clarification and may substitute for fungal enzymes, which are currently used for winemaking [8].
Recent research in wine biotechnology has focused on acid proteases to prevent formation of wine haze. A thermotolerant fungal protease (aspergilloglutamic peptidase) has already been approved for winemaking in Australia [9].
Degradation of haze forming-proteins by enzymes is an attractive alternative to bentonite fining because it would minimize losses of wine volume and aroma. Appropriate proteases must be active under harsh winemaking conditions, i.e., low pH (~3.5), low temperature (~15 °C), presence of ethanol (≥ 10% v/v), phenolic compounds and sulphites. Another problem is the intrinsic stability of haze forming proteins due to their high numbers of disulphide bonds as those present in lipid transfer proteins, chitinases and thaumatin-related proteins [9].
Yeasts producing acid proteases may offer an alternative or supplement to bentonite treatment for removal of undesirable wine proteins [10,11,12,13]. In contrast to the classical wine yeast S. cerevisiae, non-Saccharomyces yeasts are important sources of extracellular enzymes including proteases [6,14]. In the study of Fernández et al. [15], 53 of 141 isolates of “wild yeasts” hydrolyzed casein. The positive strains were identified as Metschnikowa pulcherrima and Pichia membranifaciens. In a similar study with 245 yeast isolates, 10 strains of Candida stellata, C. pulcherrima, Kloeckera apiculata and one strain of Debaromyces hansenii showed proteolytic activity [16]. Oenological isolates of Hanseniaspora [17], Metchnikowia pulcherrima and Candida apicola [18] produce extracellular proteases with potential applications in biotechnological processes.
Wine yeasts secreting proteolytic enzymes are of high biotechnological interest for protein haze prevention because they could be directly added as starter cultures to the grape must. Besides cost reductions, there are no administrative restrictions for their applications in must and wine, which should be considered with enzyme preparations.
In this study, we describe the purification and properties of extracellular acid proteases isolated from wine relevant yeasts strains of Wickerhamomyces anomalus and Metschnikovia pulcherrima, which are produced during cultivation in grape juice.
2. Materials and Methods
2.1. Yeast Strains and Cultures
Yeast strains investigated are deposited at the local culture collection of the Institute for Microbiology and Wine Research (IMW), Johannes Gutenberg University Mainz, Country. The identity of Wickerhamomyces anomalus strain 227 and Metschnikowia pulcherrima 446 have been verified by sequence analysis of the ITS (internal transcribed spacer region). The primers used for PCR amplification were ITS (F) GGAAGTAAAAGTCGTAACAAGG and ITS (R) TCCTCCGCTTATTGATATGC. Sequencing was performed by LCG Genomics (Berlin, Germany) and identification was accomplished by BLAST searches in public databases.
Yeasts were maintained on GYP medium (20 g·L−1 glucose, 10 g·L−l yeast extract, meat peptone 20 g·L−l). Solid media were prepared with 15 g·L−1 agar. For cultivation and protease production, a white grape juice (Lindavia®, Niehoffs-Vaihinger Fruchtsaft GmbH, Lauterecken, Germany) was diluted to 50% (v/v) with deionized water and steam-sterilized for 10 min at 100 °C. Volumes of 100 mL or 300 mL of this medium in Erlenmeyer flasks were inoculated with 1% (v/v) of a washed yeast preculture grown in GYP medium. The cultures were incubated on a rotary shaker (100 rpm) at 20 °C for 7 days. On each day, 2 mL samples were taken to monitor cell growth (OD600nm) and proteolytic activities in the supernatants.
2.2. Buffer Solutions
Tri-sodium phosphate (20.0 g·L−1, pH 12); sodium phosphate (100 mM, pH 7.0: 17.8 g·L−1 NaHPO4 X H2O adjusted with 0.1 M HCl); sodium acetate (100 mM: 6.0 g·L−1 glacial acetic acid adjusted to pH 4.5 with 0.1 M NaOH); sodium tartrate (5.0 g·L−1 tartaric acid adjusted to pH 3.5 with 0.1 M NaOH); and universal buffer (tris 6.5 g·L−1, maleic acid 2.32 g·L−1, boric acid 1.26 g·L−1; the pH desired was adjusted with 0.1 M NaOH).
2.3. Endo-Protease Activity of Secreted Yeast Enzymes
Endoprotease activities were measured with Megazyme® protease substrate AZCL-collagen® (Megazyme International, Wicklow, Ireland). This compound is prepared by dying and crosslinking collagen to the intensively blue-coloured copper protein azurine to produce a material, which hydrates in water but is still water insoluble. Hydrolysis by proteases produces water-soluble dyed fragments, and the rate of release of these (increase in absorbance at 590nm) is directly related to enzyme activity. The standard test was performed as follows: suspensions of AZCL-collagen® were prepared in appropriate buffers depending on the individual experiment. Afterwards, 100 µL of these suspensions were incubated with 100 µL sample for 24 h at 40 °C under vigorous shaking (750 rpm) in a thermo mixer (Eppendorf, Hamburg, Germany). The reaction was stopped by adding 1.0 mL tri-sodium phosphate-buffer (pH 12) and the tubes centrifuged at 16,000 g for 30 min. The absorbance of the supernatants were measured at 590nm in a spectrophotometer (Shimadzu UV-2450, Duisburg, Germany). The control solutions were prepared as above, but test samples were heat-treated (30 min, 100 °C) to inactivate any protease activity. Experiments were performed in triplicate.
2.4. Protease Inhibition Studies
Effects of selected inhibitors on proteolytic activities were tested in triplicate by incubation of the samples (1 h) with phenylmethylsulfonylfluoride (PMSF, 2 mM), pepstatin A (20 µM) or Na-EDTA (10 mM) before starting the reaction at 40 °C.
2.5. pH Optimum
The pH optimum of yeast proteases was determined in triplicate with the AZCL-collagen® substrate suspended in universal buffer between pH 3.0 and 8.0. Data presented are mean values of triplicate determinations.
2.6. Temperature Optimum
The temperature optimum of yeast proteases was determined in triplicate with the standard AZCL-collagen® assay at pH 3.5 between 20 and 60 °C.
2.7. Real-Time Monitoring of Proteolytic Yeast Enzymes
A test introduced by Chasseriaud et al. [13] was used with some modifications to monitor proteolytic activity by yeasts directly during growth in grape must. In brief, white grape juice (50% v/v) was steam-sterilized for 10 min at 100 °C. A stock solution (20 mg·mL−1) of the chromogenic protease substrate azocasein (Sigma-Aldrich, Munich, Germany) was prepared in 0.1 M NaOH and added to the grape juice at a final concentration of 2.0 mg·mL−1.
At regular intervals, 600 µL samples of the yeast cultures were withdrawn and mixed with 90 µL trichloroacetic acid (20% w/v). Samples were centrifuged at 16,000 g for 30 min. In addition, 500 µL NaOH (1 M) were added to the supernatants and colour release from azocasein was measured in a spectrophotometer (Shimadzu UV-2450, Duisburg, Germany) at 500nm. Non-inoculated culture media served as controls. Experiments were performed in duplicate.
2.8. Preparative Isoelectric Focusing (pIEF)
Purification of the extracellular proteases was achieved by preparative IEF using the Rotofor® Preparative IEF-Cell (BioRad, Munich, Germany). Yeasts were grown for 6 days in 100 mL grape juice on a shaker (100 rpm) at 20 °C. After centrifugation (10,000 g for 10 min), supernatants were dialyzed (cut-off: 10 kDa) and lyophilized. Concentrated culture supernatants (3.0–5.0 mL) were filled up to 50 mL with double-deionized water and mixed with 1.0 mL of 40% (w/v) ampholyte solution pH 3.0 to 5.0 (Rotilyte®, Roth, Karlsruhe, Germany). The electrode buffers were 0.5 M acetic acid (anode) and 0.25 M HEPES (cathode), respectively. The separation was performed under a preset power of 15 W until constant voltage was reached after 4 to 5 h at 10 °C. The pH of the 20 fractions obtained was measured, their protein content checked by SDS-PAGE, and protease activities tested with AZCL-collagen®. The protease positive fractions were concentrated in 50 mL spin columns (cut-off: 10 kDa, Vivaspin®, Sartorius AG, Goettingen, Germany), washed three times with deionized water and stored at −20 °C.
2.9. Gel Electrophoretic Methods
Protein compositions in the culture filtrates and fractions of pIEF were checked by SDS-PAGE. Samples were separated in 12.5% (w/v) SDS gels (10 cm × 10 cm) with a 4% (w/v) stacking gel at room temperature for 1 h. A prestained protein ladder (Pink Color Protein Standard II®; Serva, Heidelberg, Germany) served as the molecular mass standard. Protein staining was performed with Quick Coomassie Stain® (Serva, Heidelberg, Germany).
2.10. Identification of Yeast Exoenzymes
The concentrated and dialyzed culture filtrates were separated by SDS-PAGE and gels treated with Quick Coomassie Stain®. W. anomalus 227 delivered two well separated protein bands after 24 h incubation, which were excised and sliced into small pieces. After destaining and drying, proteins were reduced with 2 mM DTT at 55 °C and alkylated with 15 mM iodacetamide at room temperature in the dark for 1 h each. After washing and drying, trypsin digests were done at 37 °C overnight (0.5 µg of trypsin per gel slice). Tryptic peptides were transferred into an autosampler vial for peptide analysis via LC-MS/MS. Nanoscale liquid chromatography of tryptic peptides was performed with a Waters NanoAcquity UPLC system (Eschborn, Germany) equipped with a 75 µm × 250 mm HSS-T3 reversed phase column and a 2.6 µL PEEKSIL-sample loop (SGE, Darmstadt, Germany) as described before (PMID: 23265486). Online mass spectrometry analysis of tryptic peptides was performed using a Waters Synapt G2-S QTOF mass spectrometer, operated at a resolving power of R = 20.000. All analyses were performed using positive mode ESI using a NanoLockSpray source as described (PMID: 23265486). Resulting liquid chromatography tandem MS (LC-MS/MS) data were processed and searched by using PROTEINLYNX GLOBAL SERVER, Ver. 3.0.2. (Waters, Eschborn, Germany). Protein identifications were assigned by searching a custom compiled database containing open reading frames obtained from transcriptome sequencing of W. anomalus [19] for gel bands derived from W. anomalus and all available (212.809) RefSeq database entries from the order of Saccharomycetales for gel bands derived from Metschnikowia pulcherrima. Sequence databases were supplemented by known possible contaminants (trypsin, human keratins) based on the precursor and fragmentation data afforded by the LC-MS/MS acquisition method as described before [20]. The false discovery rate (FDR) for peptide and protein identification was assessed searching a reverse database generated automatically in PLGS. FDR was set to 0.01 for database search.
2.11. In Silico Analysis
Protein sequence was analysed by public databases and tools (BLAST; ScanPROSITE; PROTPARAM; SIGNAL P, NetNGlyc) offered by the ExPASY Bioinformatics Portal.
2.12. Degradation Experiments
Proteins from a German Riesling wine (vintage 2012) were prepared by dialysis and lyophilisation as recently described by Jaeckels et al. [20]. The proteolytic activity against isolated wine proteins and bovine serum albumin (Sigma-Aldrich, Munich, Germany) were tested after incubation at 20 °C in sodium tartrate buffer (pH 3.5) and subsequently in an actual German Riesling wine (vintage 2014, 11.5% v/v alcohol). The specific reaction conditions are indicated in the corresponding results section. Degradation of the model proteins was checked by SDS-PAGE.
3. Results
In a previous screening study with 102 yeasts from the internal strain collection (IMW), W. anomalus 227 and M. pulcherrima 446 were identified as potential protease producers as evidenced by clearing zones on turbid milk powder agar plates (unpublished). In the present study, both strains grew equally well in 50% grape juice and reached a maximum cell density after six days (Figure 1).
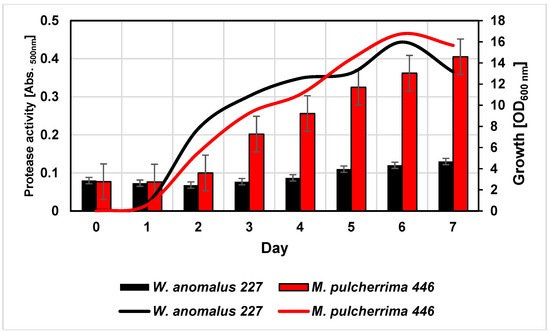
Figure 1.
Cell densities (lines) and proteolytic activities (columns) of Wickerhamoyces anomalus 227 and Metschnikovia pulcherrima 446 during cultivation in a white grape juice at 20 °C. Proteolytic activity was directly detected in the cultures by hydrolysis of supplemented azocasein.
In the course of cultivation, yeasts exhibited proteolytic activities as indicated by cleavage of azocasein supplemented to the medium, although at quite different levels. In order to characterize these activities in more detail, the cultures were harvested after six days and the supernatants were dialyzed and subsequently concentrated by lyophilisation. As detected by SDS-PAGE (Figure 2), only a few distinct proteins were present in the culture concentrates but not in the juice controls (not shown). The protein bands were cut out of the gel and tryptic fragment patterns were analysed by LC-MS/MS. By database research, two extracellular proteins of W. anomalus 227 could be identified. The band at ca. 30 kDa corresponded with 100% identity to an exo-β-1,3-glucanase of W. anomalus AS1 described in our recent study [21].
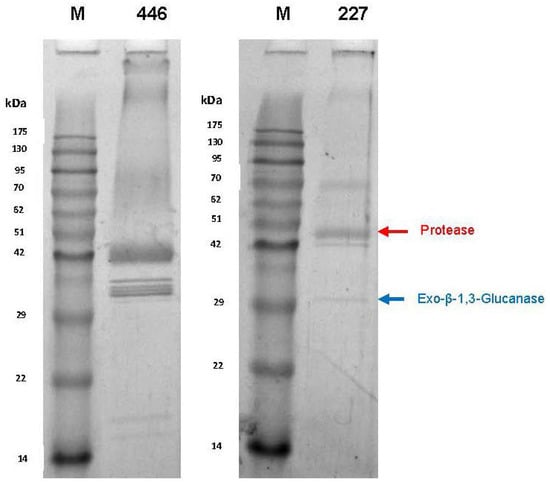
Figure 2.
SDS-PAGE (12.5%) of dialyzed and lyophilized culture supernatants after six days of yeast growth in 50% (v/v) grape juice. Strains: M. pulcherrima 446 and W. anomalus 227. M: Molecular mass standard. Arrows indicate proteins identified by LC-MS/MS.
The higher molecular mass protein delivered 100% identity with a hypothetic protein (NCBI Acc. No. XP_019036036) annotated in the genome of W. anomalus NRRL Y-366-8. It could be attributed to peptidase family A1 (Figure 3). The sequence contains two aspartyl residues (D79 and D291) as typically found in the active site of acid proteases. In addition, a putative signal peptide for secretion, possible cysteine bridges and a single N-glycosylation site were detected. Due to the lack of reference sequence data, the extracellular proteins of M. pulcherrima 446 were not identified by database searches.

Figure 3.
Sequence coverage (peptides identified by LC-MS/MS) and annotated putative protein sequence of the extracellular protease WaAPR1 of W. anomalus 227. Amino acids are given in the short code.
Above: Peptides identified by LC-MS/MS are highlighted according to their type (see legend). Total sequence coverage was 49.4%.
Below: Annotated putative protein sequence obtained from the W. anomalus transcriptome database. Number of amino acids: 447; Molecular mass: 47328 Da (PROTPARAM); Theoretical pI: 4.11 (PROTPARAM). A possible signal peptide of 19 amino acids (SIGNAL P) is shown in red and underlined. The sequence contains a peptidase family A1 domain (amino acids61-395) with aspartyl residues D79 and D291 (green) in the active site (ScanPROSITE) and a single N-glycoyslation signature (blue; NetGlyc 1.0 server). Cysteine residues C329 and C260 (yellow) may build a disulfide bridge (ScanPROSITE).
The extracellular proteases of both yeasts were purified by preparative IEF. The proteolytic activity of W. anomalus 227 focused at ca. pH 3.4, whereas two activity peaks at pH 3.96 and 4.61 were found in fractions of M. pulcherrima 446 (Figure 4).
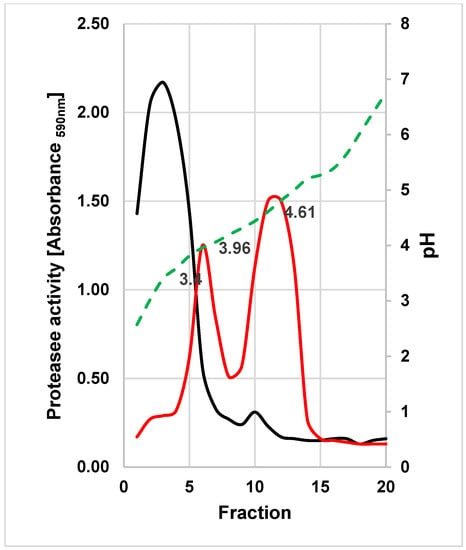
Figure 4.
Preparative IEF of concentrated culture supernatants of W. anomalus 227 (black line) and M. pulcherrima 446 (red line). Collected fractions were examined for protease activities under the standard assay conditions. The pH gradient is illustrated as the broken green line.
The protease-active fractions were pooled, concentrated and subjected to SDS-PAGE and sensitive protein staining (Figure 5). One single band appeared in the fractions collected from the main activity peak of W. anomalus 227 (at ≈ pH 3.4) and in the second peak of M. pulcherrima 446 (at ≈ pH 4.61).
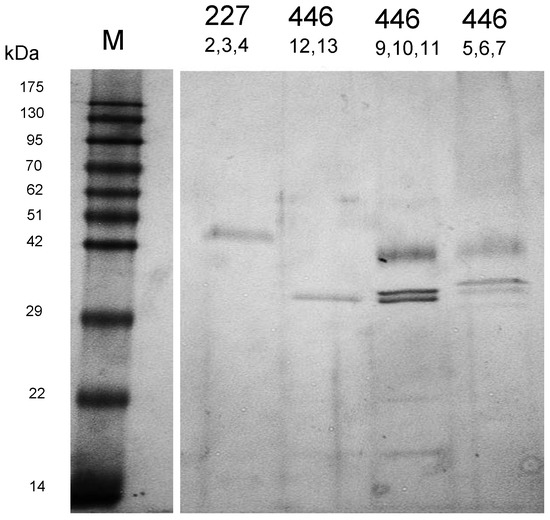
Figure 5.
SDS-PAGE (12.5%) of protease-active fractions from W. anomalus 227 and M. pulcherrima 446 as obtained by preparative IEF.
The isoelectric point and the apparent molecular mass of the purified protease WaAPR1 from W. anomalus 227 correspond fairly well with the theoretical values (pI 4.11, MW 47 kDa) derived from the amino acid sequence (Figure 3). The protein purified from the culture supernatant (in fractions 12 and 13) of M. pulcherrima 446 is probably one of several protease isoenzymes secreted by this strain. This can be concluded from the appearance of at least five protein bands in SDS-PAGE (Figure 2) and by two activity peaks displayed in preparative IEF. The protein concentrations and enzymatic activities at different stages of purification are listed in Table 1.

Table 1.
Protein concentrations and proteolytic activities at different stages of purification.
In accordance with the sequence data, inhibitor studies confirmed that both yeasts secrete acid proteases: activities were most significantly impaired by low concentrations of pepstatin A, which specifically inhibits aspartic proteases (Table 2).
Table 2.
Effect of inhibitors on protease activities of W. anomalus 227 and M. pulcherrima 446.
Inhibitors for serine proteases (PMFS) and metalloproteinase (EDTA) were far less effective even at high concentrations. In addition, enzymatic activity on azurine collagen was highest at pH 3–4 and dropped at higher pH values (Figure 6).
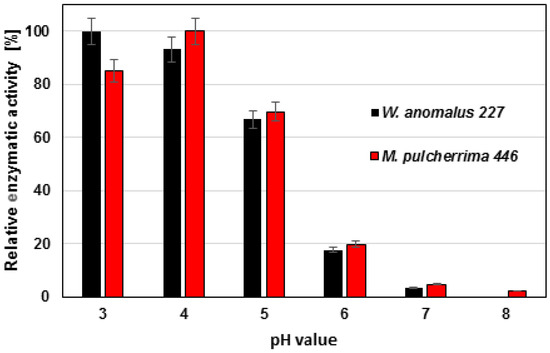
Figure 6.
Effect of pH on protease activities of W. anomalus 227 and M. pulcherrima 446.
The temperature maximum of proteolytic activity for both yeast strains was found at 40 °C, although significant activities could be determined at lower and higher temperatures (Figure 7).
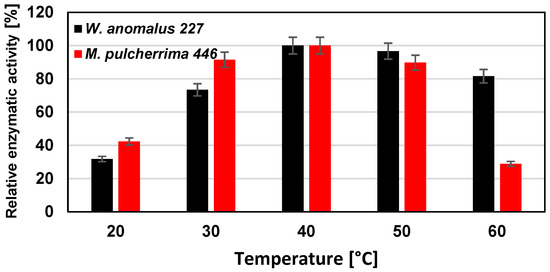
Figure 7.
Effect of temperature on protease activities of W. anomalus 227 and M. pulcherrima 446.
With respect to a potential application for protein haze reduction, we finally assessed the activity at wine-relevant conditions. Bovine serum albumin was completely degraded after 24 h in a white wine. However, thaumatin-like wine proteins were resistant to enzymatic hydrolysis (Figure 8).
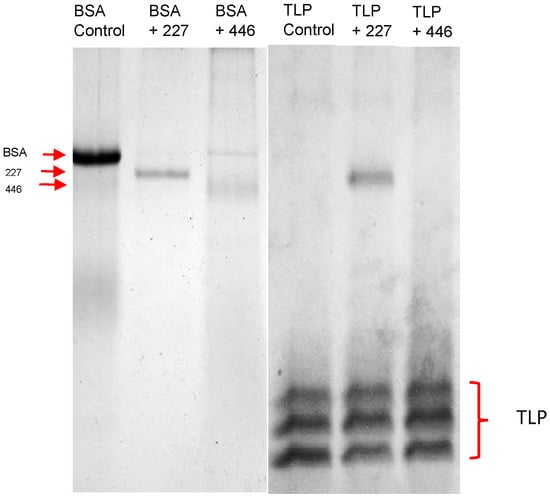
Figure 8.
Detection of proteolytic activity of yeasts by SDS-PAGE (12.5%). Bovine serum albumine (BSA, left) and thaumatin-like wine proteins (TLP, right) were added to a white wine at final concentrations of 100 μg·mL−1 and incubated for 24 h at 20 °C with concentrated culture supernatants of W. anomalus 227 and M. pulcherrima 448.
4. Discussion
Aspartic proteases are ubiquitously distributed proteolytic enzymes that are active in acidic environments [22]. Several microorganisms secrete such proteases as virulence factors and/or in order to break down proteins, thereby deliberating sources of nitrogen.
Extracellular proteinases secreted by yeasts have been isolated and examined by several researchers, especially with respect to their enzymatic properties and the physiology of their induction and secretion. Most of these studies focus on the acid aspartic proteases of Candida albicans because of their involvement in human diseases. Nowadays, there is growing interest on yeast acid proteases for industrial applications [10].
Although simple screening procedures with solid casein agar plates have repeatedly demonstrated proteolytic activities in various non-Saccharomyces species, only limited knowledge exists on their cellular location, substrate preferences, catalytic properties or molecular structure [11,15,16]. In contrast to some previous studies using synthetic culture media and proteins as inducers [18,23], we demonstrated the secretion of proteases by two wine associated yeasts directly in a natural substrate, i.e., grape juice. Remarkably, they were the only dominant extracellular enzymes produced during the active growth phase. Recent studies have already demonstrated protease activities in Wickerhamomyces isolates from enological ecosystems [24]. However, the activities detected were relatively low and not expressed in all strains examined [25]. The extracellular protease WaAPR1 of W. anomalus 227 with a molecular mass of 47 kDa and an isoelectric point of 4.11 is in the typical range of microbial aspartic proteases [10,22]. As only a single N-glycosylation site is present in the protein sequence, any covalent glycosylation would be difficult to demonstrate. A similar situation has been reported for the aspartic protease from M. pulcherrima IWBT Y1123 [18]. The bioinformatic tool ScanPROSITE predicts the position of the two essential aspartyl residues in the active site at D79 and D291. Results of a BLAST search reveals 100% identity with a hypothetic protein (NCBI Acc. No. XP_019036036) in the genome of W. anomalus NRRL Y-366-8 and ca. 35% identity with putative acid proteases of other yeast species belonging to the genera Wickerhamomyces, Saccharomyces, Zygosaccharomyces, Metschnikovia, Yarrowia, Torulaspora and Candida.
The amino acid sequence of the M. pulcherrima 446 protease could not be identified in this study. This is attributed to a lack of reference sequence data. In addition, the identification attempts reported in our study were performed with a non-purified concentrate (Figure 2), which probably contained a mixture of several protease isoenzymes. The MALDI analysis will be repeated with a purified sample. Nevertheless, biochemical data (pH optimum, inhibition by pepstatin A) and results of other researchers give strong hints for an aspartic protease. Reid et al. [18] reported on genetic and biochemical levels of the existence of an extracellular aspartic protease in M. pulcherrima strain IWBT Y1123. Gotoh et al. [23] purified a protease from the culture supernatant from strain KSY 188-5. The latter had an apparent molecular mass of 37 kDa, an isolelectric point of 4.7, and an optimum activity around 45 °C at pH 3.0. These parameters are very close to the data obtained in the present study for the protease of M. pulcherrima 446.
Constitutive production in the natural substrate grape juice without need of external inducers combined with maximum activity at low pH suggests an essential physiological role of the secreted enzyme. The protease activity might help to gain nitrogen from grape proteins necessary for cell growth, especially considering the surplus of sugar carbon in nitrogen-limited milieus [26,27]. The secreted proteases may also act as a survival tool by degrading cell wall proteins of competitive microorganisms, analogous to its role as a virulence factor in C. albicans. The membrane-bound aspartic protease of S. cerevisiae, a member of the yapsin protease family, is involved in cell wall growth and maintenance. It is a challenge to get more insights in the natural role of secreted yeast aspartic proteases. On the other hand, their biotechnological potential needs to be further examined in future experiments. The use of aspartic proteases as alternatives or supplements to clarifying agents in various beverage industries is under intensive investigation, and the potential applications in the wine industry are thoroughly discussed [10]. Under wine-relevant conditions (pH 3.5, 20 °C), the proteases of both yeast strains were able to degrade bovine serum albumin but not wine specific thaumatin-like proteins (TLP). Nevertheless, it is well known that TLP are very inert to proteolytic hydrolysis and among the few protein classes that finally reach the bottled wine. One should keep in mind that even the actual used commercial fungal protease preparation is only effective at elevated (not wine-relevant) temperatures and cannot completely eliminate wine proteins. Other grape must proteins than TLP may be more susceptible to proteolytic hydrolysis by yeast enzymes. The fact that the proteolytic enzymes were produced during growth in grape juice offers the chance to lower the protein content by using our yeast strains directly for must fermentations. Recent studies have already shown that strains of Wickerhamomyces anomalus and Metschnikovia pulcherrima positively influence wine sensory properties [28]. Any reduction of grape must protein content would mean an advantage for wine-makers by lowering the effective bentonite dosages. These aspects are topics of our current research.
Acknowledgments
The authors would like to thank undergraduate student Hendrik Leibhan for conducting some of the experiments.
Author Contributions
Martina Schlander performed most of the experiments; Ute Distler performed HPLC-MS analysis; Stefan Tenzer supervised and evaluated the HPLC-MS/MS analysis; Eckhard Thines provided financial, laboratory and personnel capacities; Harald Claus conceived and designed the experiments, analyzed the data and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| TLP | Thaumatin-like Proteins |
| pIEF | Preparative Isoelectric Focusing |
References
- Padilla, B.; Gil, J.V.; Manzanares, P. Past and future of Non-Saccharomyces yeasts: From spoilage microorganisms to biotechnological tools for improving wine aroma complexity. Front. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ciani, M.; Morales, P.; Comitini, F.; Tronchoni, J.; Canonico, L.; Curiel, J.A.; Oro, L.; Rodrigues, A.J.; Gonzalez, R. Non-conventional yeast species for lowering ethanol content of wines. Front. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Maturano, Y.P.; Assof, M.; Fabani, M.P.; Nally, M.C.; Jofré, V.; Rodríguez Assaf, L.A.; Toro, M.E.; Castellanos de Figueroa, L.I.; Vazquez, F. Enzymatic activities produced by mixed Saccharomyces and non-Saccharomyces cultures: Relationship with wine voltile composition. Antonie van Leeuwenhoek 2015, 108, 1239–1256. [Google Scholar] [CrossRef] [PubMed]
- Suzzi, G.; Schirone, M.; Sergi, M.; Marianella, R.M.; Fasoli, G.; Aguzzi, I.; Tofalo, R. Multistarter from organic viticulture for red wine Montepulciano d´Abruzzo production. Front. Microbiol. 2012. [Google Scholar] [CrossRef]
- Tofalo, R.; Patrignani, F.; Lanciotti, R.; Perpetuini, G.; Schirone, M.; Di Gianvito, P.; Pizzoni, D.; Arfelli, G.; Suzzi, G. Aroma profile of Montepulciano d´Abruzzo wine fermented by single and co-culture starters of autochthonous Saccharomyces and non-Saccharomyces yeasts. 2016. [Google Scholar] [CrossRef]
- Claus, H. Exoenzymes of wine microoorganisms. In Biology of Microorganisms on Grapes, in Must and Wine; König, H., Unden, G., Fröhlich, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 259–271. [Google Scholar]
- Mateo, J.J.; Maicas, S. Application of Non-Saccharomyces yeasts to wine-making process. Fermentation 2016, 2, 14. [Google Scholar] [CrossRef]
- Ugliano, M. Enzymes in winemaking. In Wine Chemistry and Biochemistry; Moreana-Arribas, M.V., Polo, C., Eds.; Springer Science-Business Media: Adelaide, Australia, 2009; pp. 103–126. [Google Scholar]
- Van Sluyter, S.C.; McRae, J.M.; Falconer, R.J.; Smith, P.A.; Bacic, A.; Waters, E.J.; Marangon, M. Wine protein haze: Mechanisms of formation and advances in prevention. J. Agric. Food. Chem. 2015, 63, 4020–4030. [Google Scholar] [CrossRef] [PubMed]
- Theron, L.W.; Divol, B. Microbial aspartic proteases: Current and potential applications in industry. Appl. Microbiol. Biotechnol. 2014, 98, 8853–8863. [Google Scholar] [CrossRef] [PubMed]
- Rosi, I.; Costamagna, L.; Bertuccioli, M. Screening for extracellular protease(s) production by wine yeasts. J. Inst. Brew. 1987, 93, 322–324. [Google Scholar] [CrossRef]
- Lagace, L.S.; Bisson, L.F. Survey of yeast acid proteases for effectiveness of wine haze reduction. Am. J. Enol. Vitic. 1990, 41, 147–162. [Google Scholar]
- Chasseriaud, L.; Miot-Sertier, C.; Coulon, J.; Iturmendi, N.; Moine, V.; Albertin, W.; Bely, M. A new method for monitoring the extracellular proteolytic activity of wine yeasts during alcoholic fermentation of grape must. J. Microbiol. Methods 2015, 119, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Molnárova, J.; Vadkertiová, R.; Stratilová, E. Extracellular enzymatic activities and physiological profiles of yeasts colonizing fruit trees. J. Basic Microbiol. 2014, 51, S74–S84. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Úbeda, J.F.; Briones, A.I. Typing of non-Saccharomyces yeasts with enzymatic activities of interest in wine-making. Int. J. Food. Microbiol. 2000, 59, 29–36. [Google Scholar] [CrossRef]
- Strauss, M.L.A.; Jolly, N.P.; Lambrechts, M.G.; van Rensburg, P. Screening for the production of extracellular hydrolytic enzymes by non-Saccharomyces wine yeasts. J. Appl. Microbiol. 2001, 91, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.J.; Maicas, S.; Thießen, C. Biotechnological characterisation of extracellular proteases produced by enological Hanseniaspora isolates. Int. J. Food Sci. Technol. 2015, 50, 218–225. [Google Scholar] [CrossRef]
- Reid, V.J.; Theron, L.W.; du Toit, M.; Divol, B. Identification and partial characterization of extracellular aspartic protease genes from Metschnikowia pulcherrima IWBT Y1123 and Candida apicola IWBT Y1384. Appl. Environm. Microbiol. 2012, 78, 6838–6849. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Fupp, O.; Trost, S.; Jaenicke, S.; Passoth, V.; Goesmann, A.; Tauch, A.; Brinkrolf, K. Genome sequence of Wickerhamomyces anomalus DSM 6766 reveals genetic basis of biotechnologically important antimicrobial activities. FEMS Yeast Res. 2012, 12, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Jaeckels, N.; Tenzer, S.; Rosfa, S.; Schild, H.; Decker, H.; Wigand, P. Purification and structural characterization of lipid transfer protein from red wine and grapes. Food Chem. 2013, 138, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Schwentke, J.; Sabel, A.; Petri, A.; König, H.; Claus, H. The wine yeast Wickerhamomyces anomalus AS1 secretes a multifunctional exo-β-1,3 glucanase with implications for winemaking. Yeast 2014, 31, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Ogrydziak, D.M. Yeast extracellular proteases. Crit. Rev. Biotechnol. 1993, 13, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Kikuchi, K.; Kodama, K.; Konno, H.; Kakuta, T.; Koizumi, T.; Nojiro, K. Purification and properties of extracellular carboxyl proteinase secreted by Candida pulcherrima. Biosci. Biotech. Biochem. 1995, 59, 367–371. [Google Scholar] [CrossRef]
- Madrigal, T.; Maicas, S.; Mateo Tolosa, J.J. Glucose and ethanol tolerant enzymes produced by Pichia (Wickerhamomyces) isolates from enological ecosystems. Am. J. Enol. Vitic. 2013, 64, 126–133. [Google Scholar] [CrossRef]
- López, S.; Mateo, J.J.; Maicas, S. Screening of Hanseniaspora strains for production of enzymes with potential interest for winemaking. Fermentation 2016. [Google Scholar] [CrossRef]
- Christ, E.; Kowalczyk, M.; Zuchowska, M.; Claus, H.; Löwenstein, R.; Szopinska-Morawska, A.; Renaut, J.; König, H. An exemplary model study for overcoming stuck fermentation during spontaneous fermentation with the aid of a Saccharomyces triple hybrid. J. Agric. Sci. 2015, 7, 18–34. [Google Scholar] [CrossRef]
- Guo, Z.P.; Zhang, L.; Ding, Z.Y.; Wang, Z.X.; Shi, G.Y. Improving the performance of industrial ethanol-producing yeast by expressing the aspartyl protease on the cell surface. Yeast 2010, 27, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Varela, C. The impact of non-Saccharomyces yeasts in the production of alcoholic beverages. Appl. Microbiol. Biotechnol. 2016, 100, 9861–9874. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).