
Enhanced L-Leucine Production in Escherichia coli via CRISPR-Associated Transposase Genome Engineering

 ,
,
Abstract
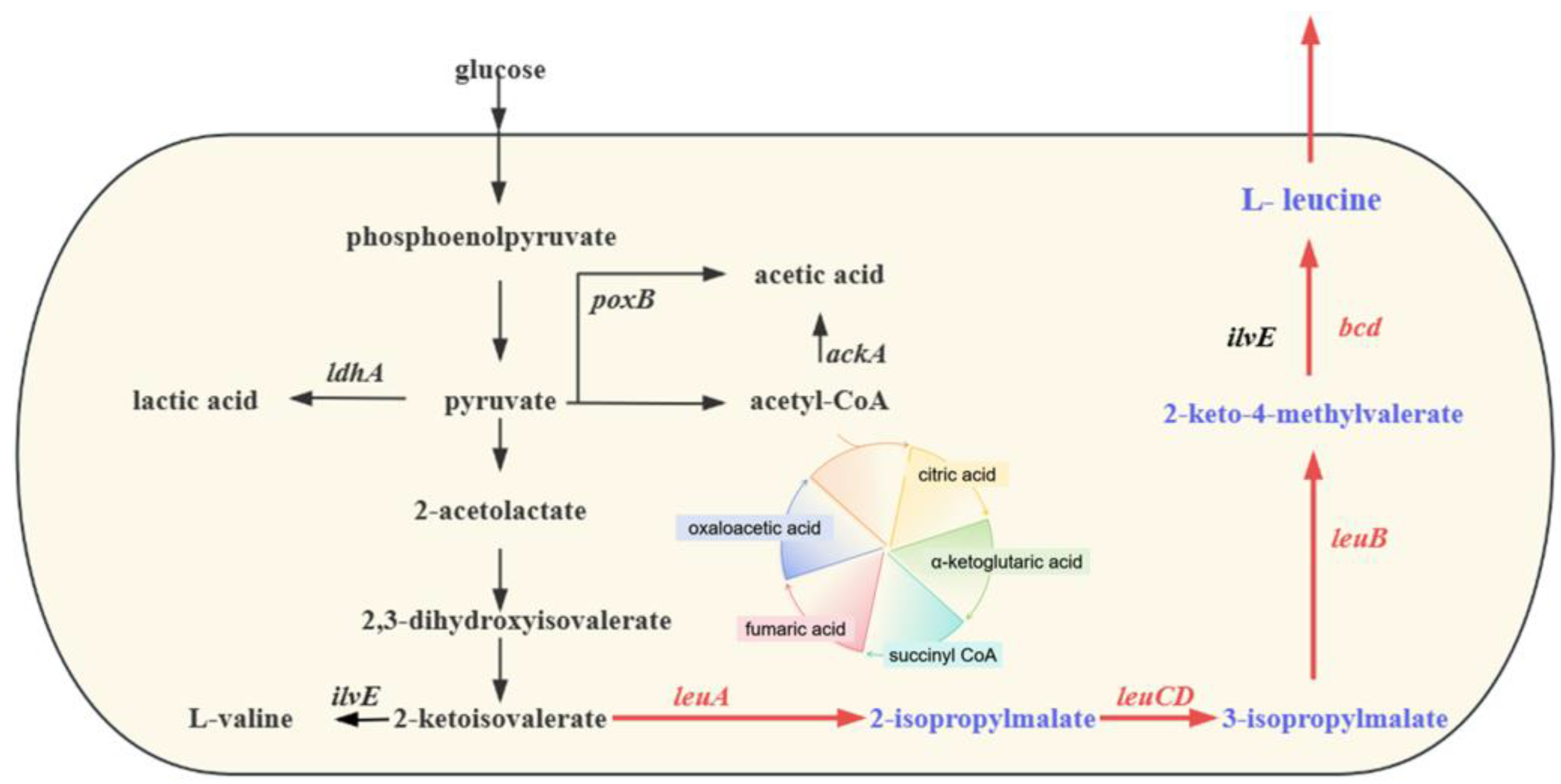
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Culture Media
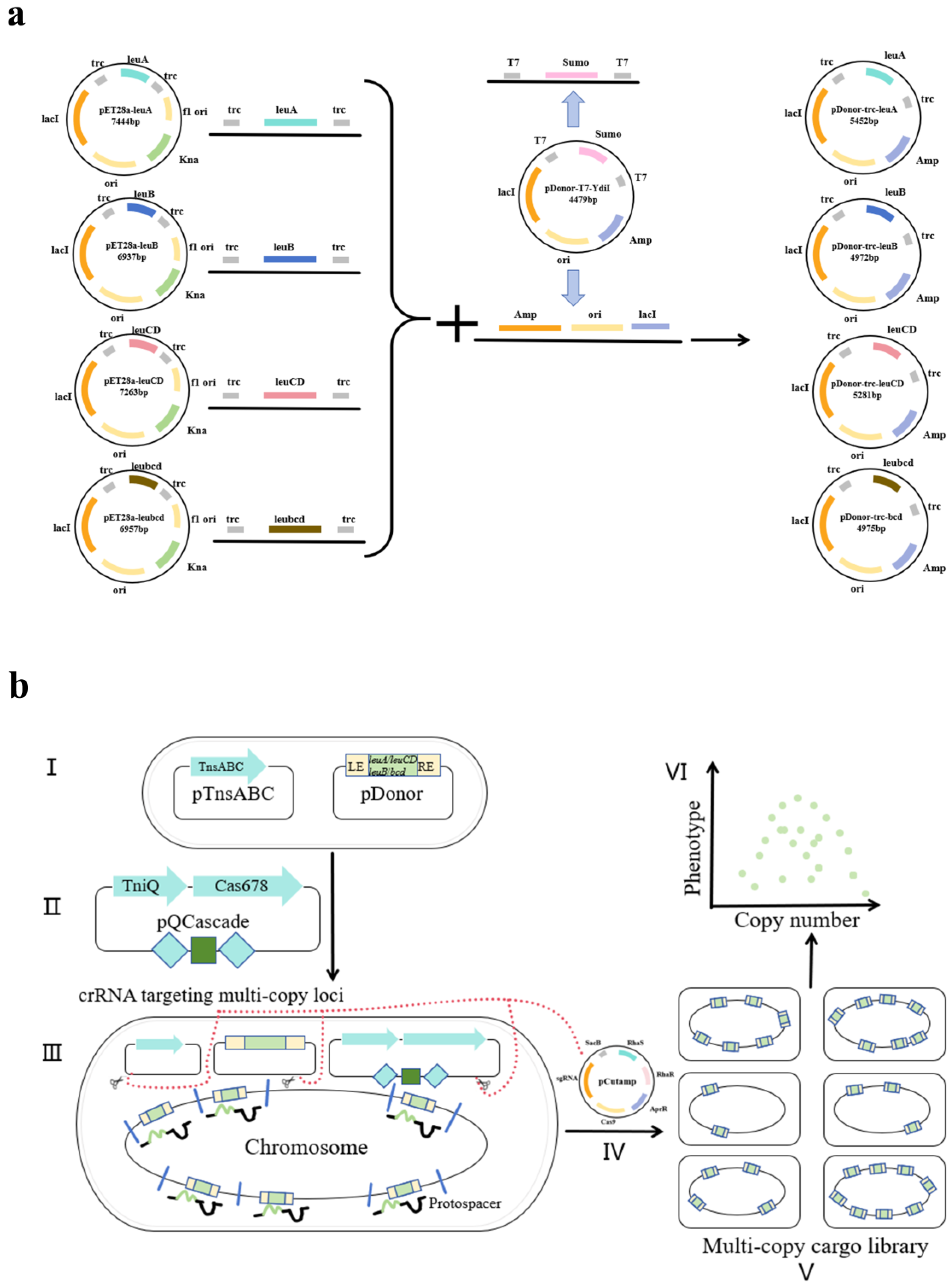
2.2. CRISPR-Associated Transposase Genome Integration
2.2.1. Plasmid Construction and Transformation
2.2.2. Induction
2.2.3. Plasmid Knockout
2.3. Plasmid Overexpression Enhancement
2.4. qPCR Analysis of Transposition Efficiency to Verify Gene Copy Number
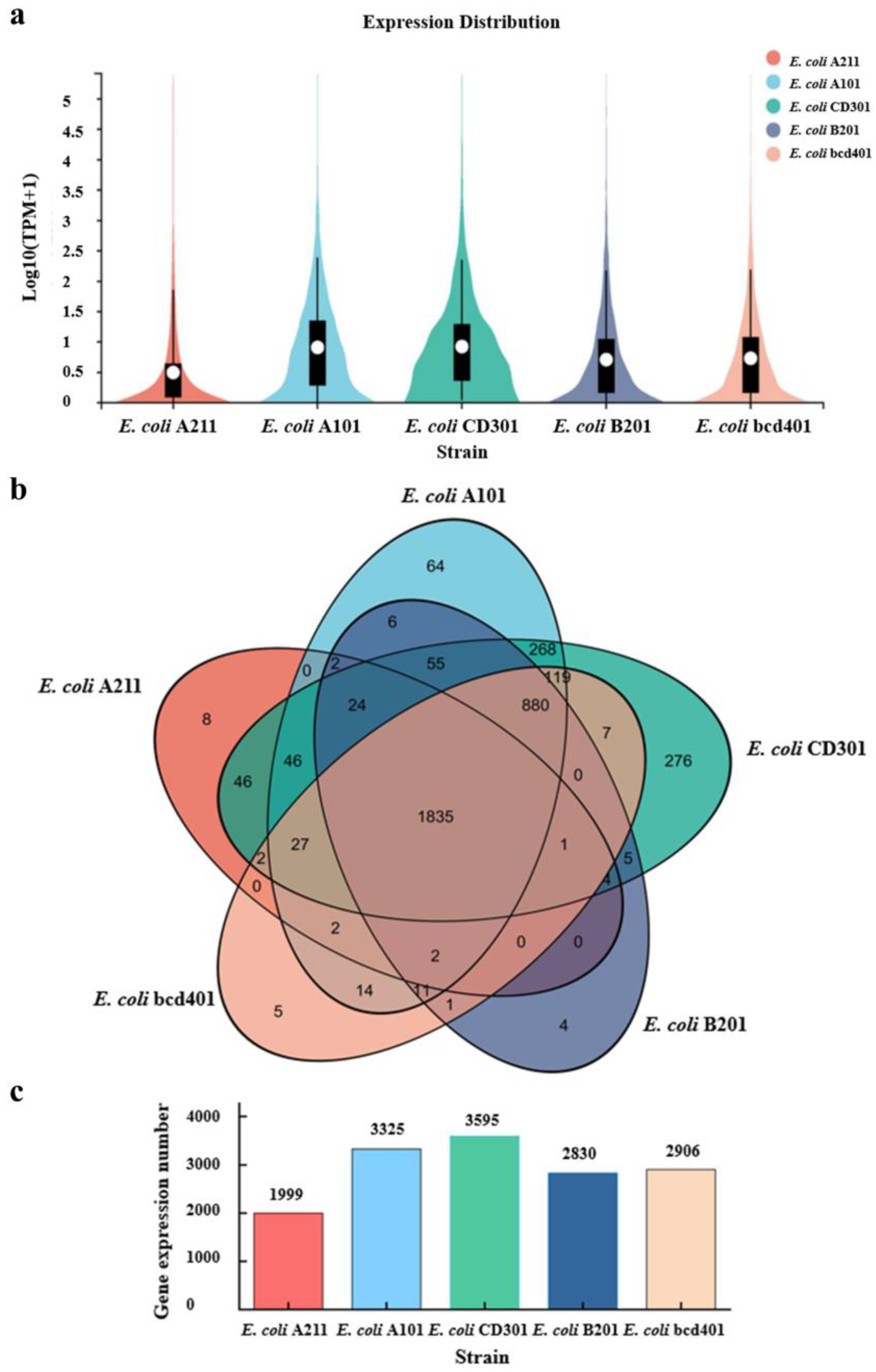
2.5. Transcriptomic Analysis of Engineering Bacterial Strains
2.6. Screening of L-Leucine-Producing Strains
2.7. Fermentation Optimization
2.8. Statistical Analytical
3. Results
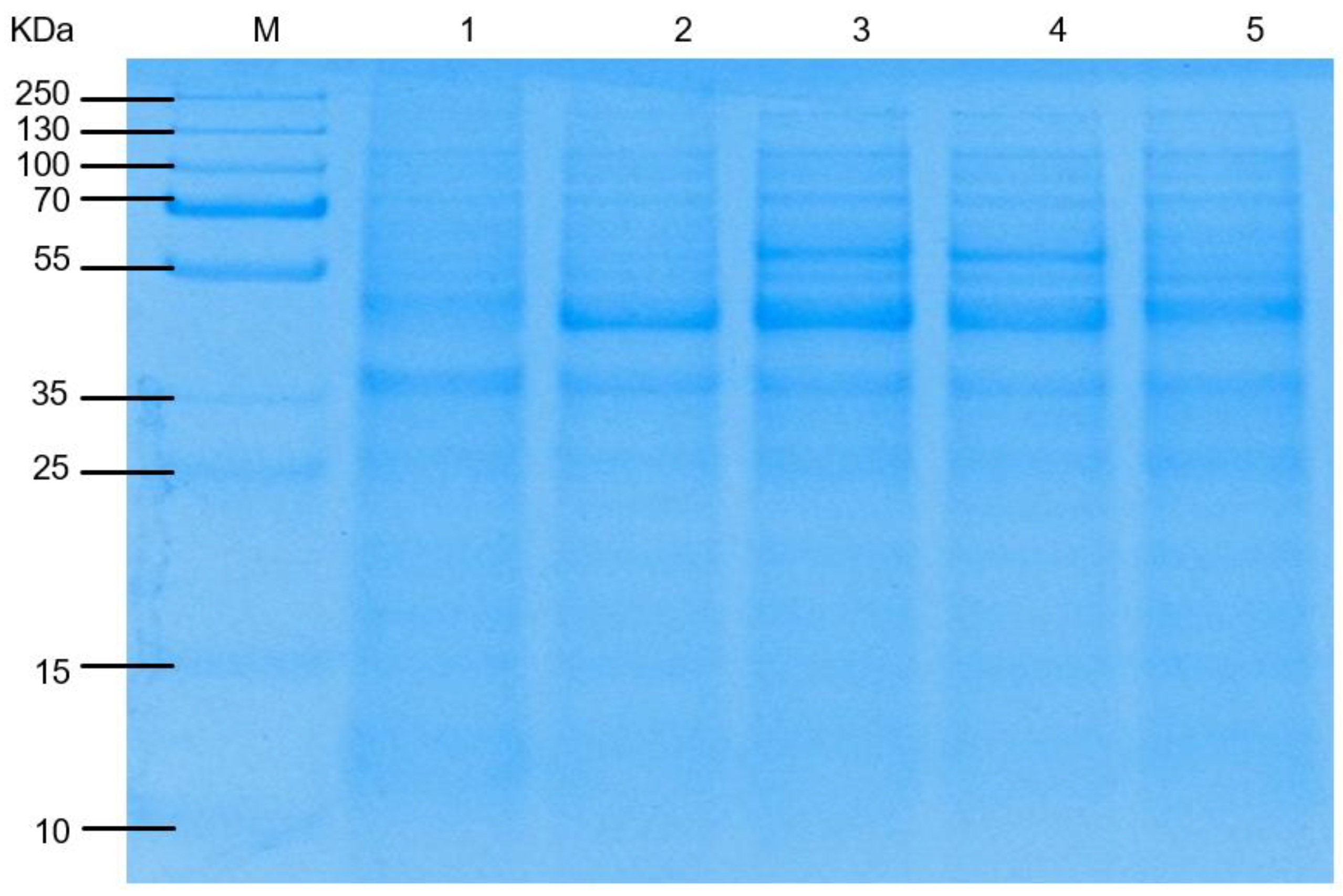
3.1. Construction of Engineered L-Leucine-Producing Strain
3.2. Estimation of Copy Number Using qPCR
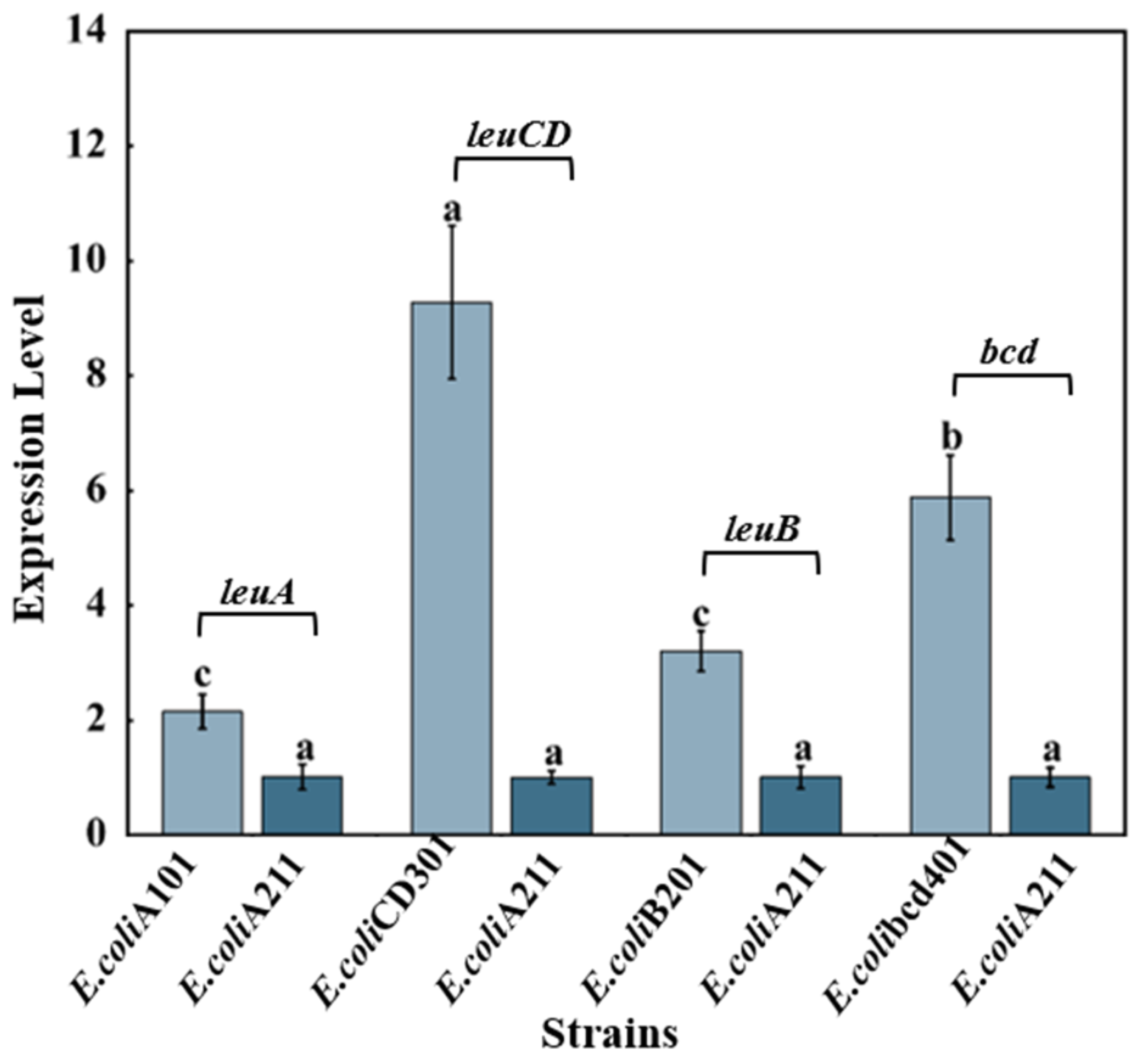
3.3. Analysis of Transcriptional Level of Engineering Bacterial Strains
3.4. Fermentation Screening to Identify Optimal Strains
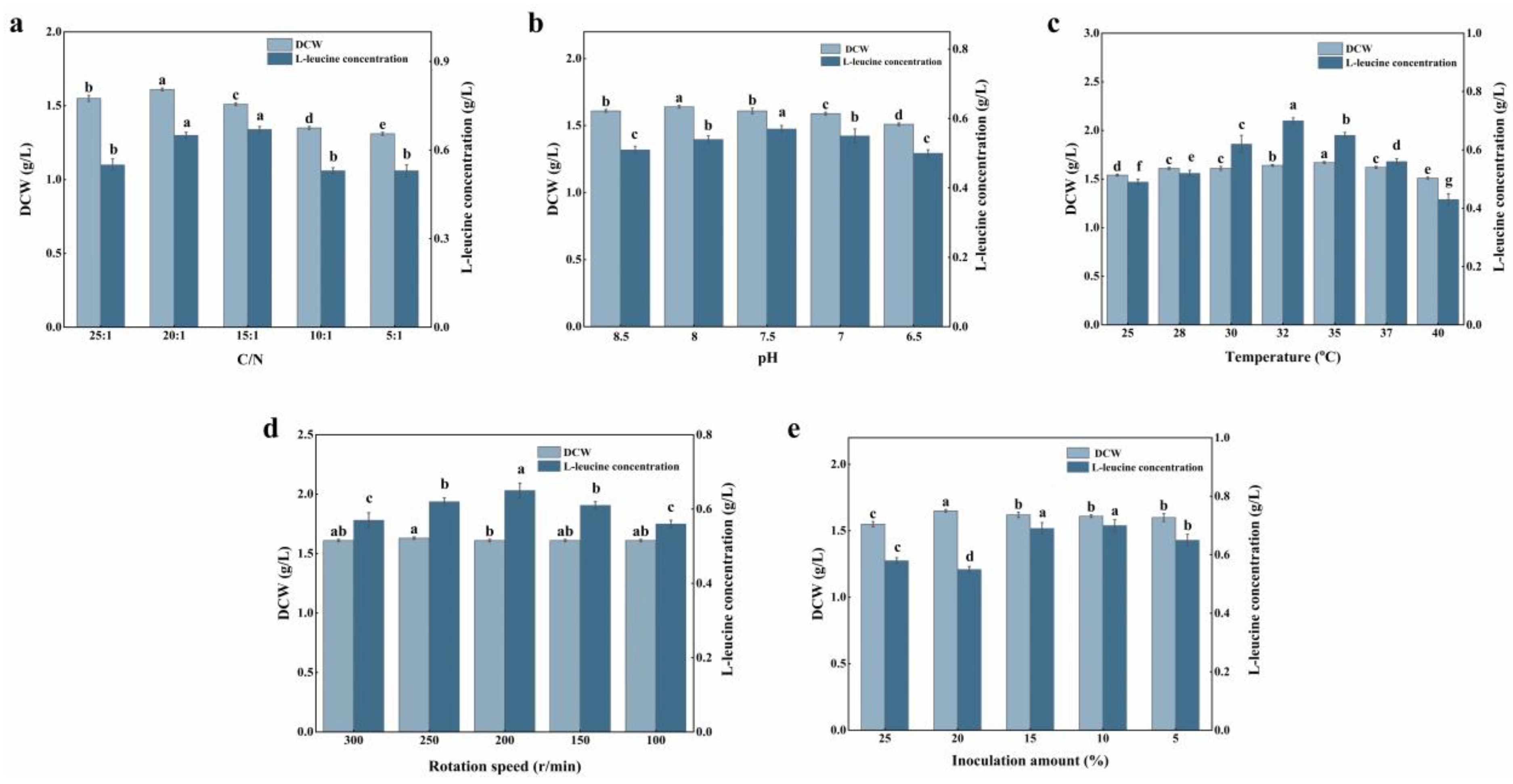
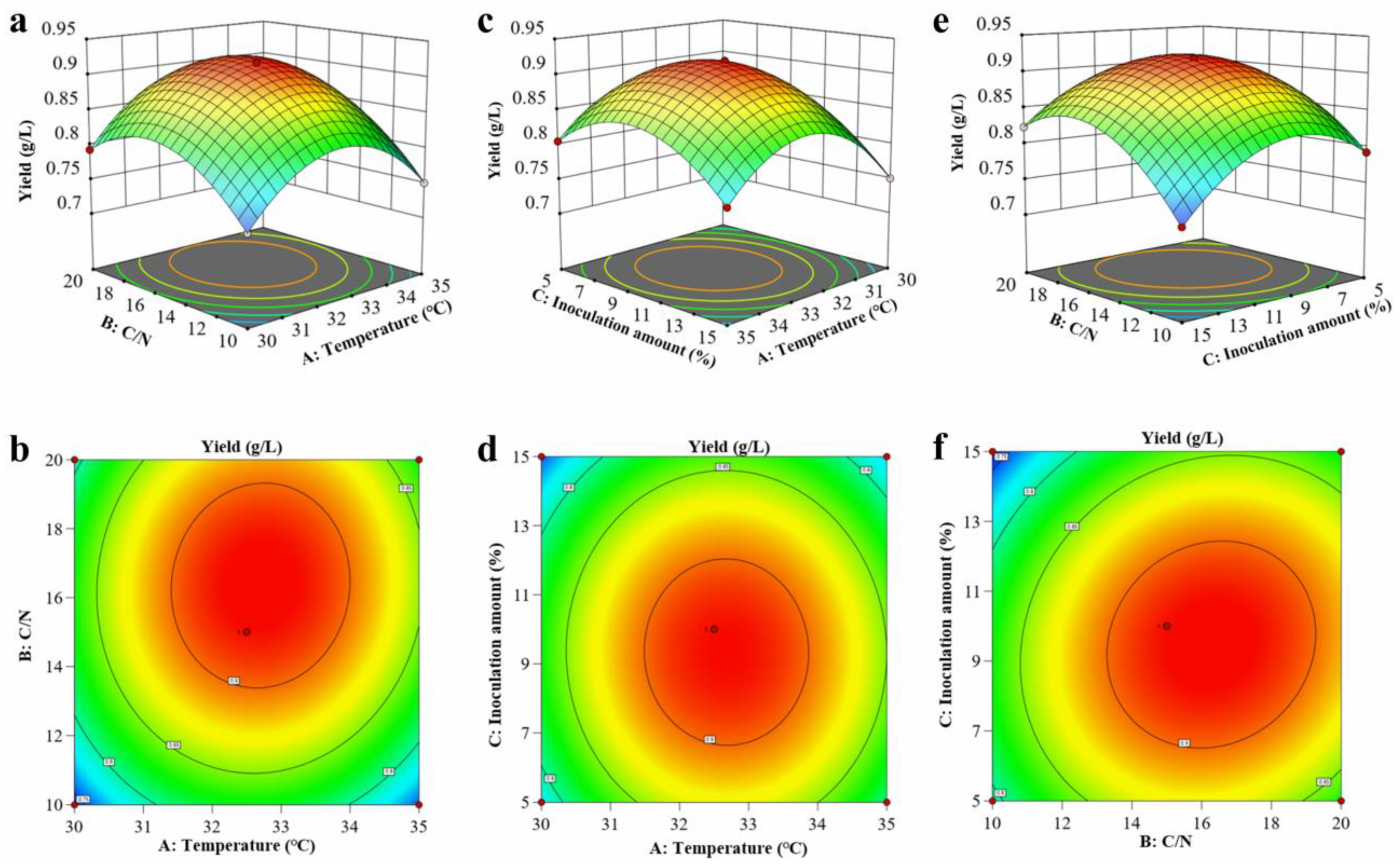
3.5. Control and Optimization of L-Leucine Fermentation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, J.H.; Lee, S.Y. Fermentative production of branched chain amino acids: A focus on metabolic engineering. Appl. Microbiol. Biotechnol. 2010, 85, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zheng, B.; Chen, Z.; Huo, X.Y. Metabolic engineering of Corynebacterium glutamicum for producing branched chain amino acids. Microb. Cell Factories 2021, 20, 230. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.; Meininger, C.J.; Wu, G. L-Leucine and NO-mediated cardiovascular function. Amino Acids 2015, 47, 435–447. [Google Scholar] [CrossRef]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic andphysiological functions of branched chain aminoacids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Quinn, P.J. Production of L-valinefrom metabolically engineered Corynebacterium glu-tamicum. Appl. Microbiol. Biotechnol. 2018, 102, 4319–4330. [Google Scholar] [CrossRef]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Xu, J.Z.; Zhang, W.G. Metabolic engineering of L-leucine production in Escherichia coli and Corynebacterium glutamicum: A review. Crit. Rev. Biotechnol. 2019, 39, 633–647. [Google Scholar] [CrossRef]
- Hao, Y.; Pan, X.; You, J.; Li, G.; Xu, M.; Rao, Z. Microbial production of branched chain amino acids: Advances and perspectives. Bioresour. Technol. 2024, 397, 130502. [Google Scholar] [CrossRef]
- Choi, K.R.; Jiao, S.; Lee, S.Y. Metabolic engineering strategies toward production of biofuels. Curr. Opin. Chem. Biol. 2020, 59, 1–14. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Sun, Z.; Luan, Y.; Li, Y.; Wang, J.; Liang, Q.; Qi, Q. Engineering an in vivo EP-bifido pathway in Escherichia coli for high-yield acetyl-CoA generation with low CO2 emission. Metab. Eng. 2019, 51, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Eun, H.; Prabowo, C.P.S.; Cho, S.; Lee, S.Y. Metabolic and cellular engineering for the production of natural products. Curr. Opin. Biotechnol. 2022, 77, 102760. [Google Scholar] [CrossRef]
- Gusyatiner, M.M.; Lunts, M.G.; Kozlov, Y.I.; Vanovskaya, L.V.; Voroshilova, E.B. DNA coding for mutant isopropylmalate synthase L-leucine-producing microorganism and method for producing L-leucine. U.S. Patent. 2002, 403, 342. [Google Scholar]
- Vogt, M.; Haas, S.; Klaffl, S.; Polen, T.; Eggeling, L.; van Ooyen, J.; Bott, M. Pushing product formation to its limit: Metabolic engineering of Corynebacterium glutamicum for L-leucine overproduction. Metab. Eng. 2014, 22, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Jin, Z.; Xia, X.; Zhang, W. Improvement of acetyl-CoA supply and glucose utilization increases l-leucine production in Corynebacterium glutamicum. Biotechnol. J. 2022, 17, 2100349. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G., III; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Rybarski, J.R.; Hu, K.; Hill, A.M.; Wilke, C.O.; Finkelstein, I.J. Metagenomic discovery of CRISPR-associated transposons. Proc. Natl. Acad. Sci. USA 2021, 118, e2112279118. [Google Scholar] [CrossRef]
- Li, C.; Zou, Y.; Jiang, T.; Zhang, J.; Yan, Y.; Li, C.; Zou, Y.; Jiang, T.; Zhang, J.; Yan, Y. Harnessing plasmid replication mechanism to enable dynamic control of gene copy in bacteria. Metab. Eng. 2022, 70, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, X.; Wang, Q.; Xu, J.; Dong, F.; Yang, S.; Yang, J.; Zhang, Z.; Qian, Y.; Chen, J.; et al. Multicopy chromosomal integration using CRISPR-associated transposases. ACS Synth. Biol. 2020, 9, 1998–2008. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chen, A.; Dong, Y.; Jiang, H.; Yang, S.; Zhang, J.; Wei, D. Identification and analysis of the key genes for Escherichia coli heterologous protein expression by transcriptomic profiling. Mol. Biol. Rep. 2024, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Li, G.; Xie, H.; Yang, W.; Xu, H.; Han, S.; Wang, J.; Meng, Y.; Xu, Q.; Li, Y.; et al. Sustainable production of 4-hydroxyisoleucine with minimised carbon loss by simultaneously utilising glucose and xylose in engineered Escherichia coli. Bioresour. Technol. 2022, 354, 127196. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Oh, J.E.; Lee, K.H.; Kim, J.Y.; Lee, S.Y. Rational design of Escherichia coli for Lisoleucine production. ACS Synth. Biol. 2012, 1, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaw, G.B. Leucine biosynthesis in fungi: Entering metabolism through the back door. Microbiol. Mol. Biol. Rev. 2003, 67, 1–5. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, H.U.; Kim, T.Y.; Park, J.S.; Kim, S.S.; Lee, S.Y. Metabolic engineering of Corynebacterium glutamicum for L-Arginine production. Nat. Commun. 2014, 5, 4618. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Ding, X.; Yang, W.; Du, X.; Chen, N.; Xu, Q.; Wei, M.; Zhang, C. High-level and high-yield production of L-leucine in Escherichia coli using multistep metabolic engineering. Metab. Eng. 2023, 78, 128–136. [Google Scholar] [CrossRef]
- Pournejati, R.; Karbalaei-Heidari, H.R. Optimization of Fermentation Conditions to Enhance Cytotoxic Metabolites Production by Bacillus velezensis Strain RP137 from the Persian Gulf. Avicenna J. Med. Biotechnol. 2020, 12, 116–123. [Google Scholar]
- Yu, J.; Liu, Q.; Liu, Q.; Liu, X.; Sun, Q.; Yan, J.; Qi, X.; Fan, S. Effect of liquid culture requirements on antifungal antibiotic production by Streptomyces rimosus MY02. Bioresour. Technol. 2008, 99, 2087–2091. [Google Scholar] [CrossRef]
- Wichmann; Wandreg, C.; Große-Wiesmann, J. Continuous microbial production of l-leucine with cell retention. Appl. Microbiol. Biotechnol. 1990, 32, 373–379. [Google Scholar] [CrossRef]
- Putra, M.A.D.; Rukmi, M.I.; Pujiyanto, S.; Mulyani, N.S. Produksi Protease Alkalis Termostabil Dari Aspergillus flavus DUCC- K225Dengan Ammonium Sulfat Sebagai Sumber Nitrogen. Bioma Berk. Ilm. Biologi. 2019, 21, 60–64. [Google Scholar]
- Usuda, Y.; Nishio, Y.; Iwatani, S.; Van Dien, S.J.; Imaizumi, A.; Shimbo, K.; Kageyama, N.; Iwahata, D.; Miyano, H.; Matsui, K. Dynamic modeling of Escherichia coli metabolic and regulatory systems for amino-acid production. J. Biotechnol. 2010, 147, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N. Stimulatory effect of NH4+ on the transport of leucine and glucose in an anaerobic alkaliphile. Eur. J. Biochem. 1993, 217, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Membré, J.M.; Leporq, B.; Vialette, M.; Mettler, E.; Perrier, L.; Thuault, D.; Zwietering, M. Temperature effect on bacterial growth rate: Quantitative microbiology approach including cardinal values and variability estimates to perform growth simulations on/in food. Int. J. Food Microbiol. 2005, 100, 179–186. [Google Scholar] [CrossRef]
- Dai, Y.; Xu, Z.; Xia, X. Effects of fermentation temperature on bacterial community, physicochemical properties and volatile flavor in fermented soy whey and its coagulated tofu. LWT 2023, 173, 114355. [Google Scholar] [CrossRef]
- Wang, Y.H.; Feng, J.T.; Zhang, Q.; Zhang, X. Optimization of fermentation condition for antibiotic production by Xenorhabdus nematophila with response surface methodology. J. Appl. Microbiol. 2008, 104, 735–744. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Name | Characteristics | Source |
|---|---|---|---|
| Bacterial strain | E. coli A211 | Original engineering strain | This study |
| E. coli DH5α | Clone | Vazyme | |
| Plasmid | pET28a-leuA | Carrying the target gene leuA | This study |
| pET28a-leuB | Carrying the target gene leuB | This study | |
| pET28a-leuCD | Carrying the target gene leuCD | This study | |
| pET28a-bcd | Carrying the target gene bcd | This study | |
| pET22b-leuAB | Carrying genes leuA, leuB | This study | |
| pET22b-leuCD-bcd | Carrying genes leuCD, bcd | This study | |
| pDonor-T7-YdiI | Key enzyme expression vector | This study | |
| pQCascade-IS1 | Tool enzyme expression | This study | |
| pTnsABC | Tool enzyme expression | This study | |
| pCutamp | Tool enzyme expression | This study |
| Sequence | Time (min) | Mobile Phase A (%) | Mobile Phase B (%) | Notes |
|---|---|---|---|---|
| 1 | 0 | 16 | 84 | Initial state |
| 2 | 0.18 | 16 | 84 | |
| 3 | 2.4 | 30 | 70 | |
| 4 | 4.2 | 34 | 66 | |
| 5 | 7.2 | 43 | 57 | |
| 6 | 13.3 | 55 | 45 | |
| 7 | 15 | 55 | 45 | |
| 8 | 20.4 | 98 | 2 | |
| 9 | 21.3 | 16 | 84 | |
| 10 | 30 | 16 | 84 | Rebalance the system and restore its initial state |
| Factor | Code | Level | ||
|---|---|---|---|---|
| −1 | 0 | 1 | ||
| Temperature (°C) | A | 30 | 32.5 | 35 |
| C/N | B | 10 | 15 | 20 |
| Inoculation amount (%) | C | 5 | 10 | 15 |
| Sequence | Temperature (°C) | C/N | Inoculation Amount (%) |
|---|---|---|---|
| 1 | −1 | −1 | 0 |
| 2 | 1 | −1 | 0 |
| 3 | −1 | 1 | 0 |
| 4 | 1 | 1 | 0 |
| 5 | −1 | 0 | −1 |
| 6 | 1 | 0 | −1 |
| 7 | −1 | 0 | 1 |
| 8 | 1 | 0 | 1 |
| 9 | 0 | −1 | −1 |
| 10 | 0 | 1 | −1 |
| 11 | 0 | −1 | 1 |
| 12 | 0 | 1 | 1 |
| 13 | 0 | 0 | 0 |
| 14 | 0 | 0 | 0 |
| 15 | 0 | 0 | 0 |
| 16 | 0 | 0 | 0 |
| 17 | 0 | 0 | 0 |
| Fermentation Conditions | Culture Medium C/N | Initial pH of the Culture Medium | Fermentation Temperature (°C) | Shaking Speed (r/min) | Strain Inoculation Amount (%) |
|---|---|---|---|---|---|
| Before optimization | 25:1 | 7 | 37 | 200 | 5 |
| After optimization | 17:1 | 7.5 | 33 | 200 | 9.6 |
| Strain | Titer (g/L) | Sugar Consumption (g/L) | Yield (g/g) |
|---|---|---|---|
| E.coli A211 | 0.34 | 9.87 | 0.03 |
| E.coli CD301 | 0.89 | 8.83 | 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.; Li, N.; Li, Z.; Zhou, Y.; Lin, Z.; Du, P.; Xiao, J.; Guo, C.; Wang, J. Enhanced L-Leucine Production in Escherichia coli via CRISPR-Associated Transposase Genome Engineering. Fermentation 2025, 11, 314. https://doi.org/10.3390/fermentation11060314
Ren X, Li N, Li Z, Zhou Y, Lin Z, Du P, Xiao J, Guo C, Wang J. Enhanced L-Leucine Production in Escherichia coli via CRISPR-Associated Transposase Genome Engineering. Fermentation. 2025; 11(6):314. https://doi.org/10.3390/fermentation11060314
Chicago/Turabian StyleRen, Xiankun, Nan Li, Zhaoqi Li, Yangyi Zhou, Zerun Lin, Peng Du, Jing Xiao, Chuanzhuang Guo, and Jianbin Wang. 2025. "Enhanced L-Leucine Production in Escherichia coli via CRISPR-Associated Transposase Genome Engineering" Fermentation 11, no. 6: 314. https://doi.org/10.3390/fermentation11060314
APA StyleRen, X., Li, N., Li, Z., Zhou, Y., Lin, Z., Du, P., Xiao, J., Guo, C., & Wang, J. (2025). Enhanced L-Leucine Production in Escherichia coli via CRISPR-Associated Transposase Genome Engineering. Fermentation, 11(6), 314. https://doi.org/10.3390/fermentation11060314