1. Introduction
Ensiling is a practical approach for ensuring the year-round availability of high-quality forage in livestock systems, especially in the mountainous regions of southwest China, where harsh winters and short growing seasons limit the fresh feed supply [
1,
2,
3]. As a forage crop of the
Pennisetum genus, king grass (
Pennisetum purpureum Schumacher ×
P. americanum), also known as hybrid giant Napier grass, is widely cultivated in the tropical and subtropical areas due to its high biomass yield and nutritive value [
4,
5,
6]. Introduced in southwest China in the 1990s, it has been adapted for local cultivation. However, its high fiber content and low water-soluble carbohydrate (WSC) levels limit its silage quality [
7]. Seasonal growth patterns further constrain its consistent availability, making ensiling a necessary strategy for feed preservation [
5].
The fermentation quality of silage is primarily determined by the structure and activity of its microbial community. Among these, lactic acid bacteria (LAB) are the key functional group responsible for rapidly converting water-soluble carbohydrates into lactic acid under anaerobic conditions, thereby lowering the pH and inhibiting spoilage organisms [
8]. Commercial inoculants based on LAB have been developed to enhance this process, including homofermentative species such as
Lactiplantibacillus plantarum and
Lacticaseibacillus casei, which mainly produce lactic acid, and obligate heterofermentative species like
Lentilactobacillus buchneri and
Lentilactobacillus hilgardii, which also generate acetic acid and improve aerobic stability. In addition, non-LAB species such as
Bacillus subtilis have been explored for their potential to complement LAB-driven fermentation through enzyme secretion or antagonistic effects against undesirable microbes [
9,
10]. The use of such additives can significantly influence fermentation outcomes, including acid production, dry matter loss, and microbial succession patterns. However, most current evaluations of these inoculants rely on laboratory-scale trials under controlled conditions, which may not reflect their actual performance under variable and often unpredictable farm environments [
11,
12,
13]. Furthermore, the efficacy of single-strain inoculants in complex plant–microbe interactions during silage fermentation is often limited, raising the need to explore multi-strain or consortia-based formulations that can adapt more effectively to fluctuating field conditions.
Although previous studies have characterized the microbial communities of silages made from various forages such as alfalfa, corn, and Napier grass [
14,
15,
16], relatively limited attention has been paid to king grass silage, particularly under temperate field conditions. The fermentation behavior of king grass silage may differ substantially due to its unique compositional characteristics and regional cultivation patterns. Moreover, while many studies have investigated the effects of microbial additives, most have focused on simplified systems using single strains or laboratory-scale trials. These approaches often fail to capture the complexity and variability of farm-scale silage production, where environmental fluctuations, native microbial backgrounds, and plant–microbe interactions can significantly influence the outcomes. Therefore, it remains unclear whether commercial multi-strain inoculants can successfully dominate microbial succession and consistently improve the fermentation performance in real-world conditions. A deeper understanding of how these additives affect both microbial community dynamics and metabolic outputs is essential for optimizing silage quality and enhancing its nutritional value under practical production scenarios. In particular, the integration of molecular techniques, such as 16S rRNA sequencing and metabolomic profiling, can provide valuable insights into the functional mechanisms of microbial additives in the ensiling process [
17].
In this study, we evaluated the effects of six commercial microbial additives on the fermentation characteristics, microbial communities, and metabolite profiles of king grass silage produced under field conditions. Our findings aim to inform the practical application of microbial additives in silage preparation, offering practical insights for enhancing feed quality, aerobic stability, and overall nutritional value in ruminant production systems under real-world conditions.
2. Materials and Methods
To select the representative microbial additives commonly used in the mountainous pastures of Southwest China, we surveyed 11 pastures across Sichuan and Guizhou provinces. Based on usage frequency and availability, six commercial additives were selected and designated as Treatments 1–6. Treatment 1 was a Qiangxing feed starter (Beihai Qiangxing Biotechnology Co., Ltd., Beihai, China). Treatments 2 and 3 were a Jingyu feed starter and Jingyu straw starter, respectively (Kaifeng Mubo Biotechnology Co., Ltd., Kaifeng, China). Treatments 4 and 5 were Fermentation Gold and a Nanhua feed starter (Henan Nanhua Qianmu Biotechnology Co., Ltd., Zhengzhou, China), while Treatment 6 was a Huaxu yeast powder (Xinxiang Huaxu Trading Co., Ltd., Xinxiang, China). As the microbial compositions of these commercial additives were not clearly disclosed by manufacturers, high-throughput sequencing was performed to determine the taxonomic profiles, which are presented in the Results
Section 3.
King grass was cultivated by Ruimu Feed Technology Co., Ltd. (Xichang, China) at an elevation of 2601 m above sea level. Approximately 60-day-old plants (1.2–1.5 m tall) were harvested at a stubble height of 10–12 cm and chopped into 10–20 mm lengths using a self-propelled forage harvester (H7, 4QZ-30, Wuzheng-Gaobei Agricultural Machinery Co., Ltd., Rizhao, China). The chopped forage was transported to the feed processing workshop by a tractor.
Each additive was applied to its respective treatment group (T1–T6) according to the manufacturer’s recommended dosage and thoroughly mixed using a TMR feed mixer (Qingdao Youhong Animal Husbandry Machinery Co., Ltd., Qingdao, China). The control group (CK) received no additive. The silage preparation was conducted using an industrial-scale wrapping process to simulate real-world production conditions, rather than small-scale laboratory bags. For each treatment, the mixed forage was compressed into cylindrical bales (115 × 100 cm, approximately 600 kg) using a silage baler (MW1230, Takakita Co., Ltd., Mie-ken, Japan) and wrapped with ≥6 layers of film to ensure airtightness and suppress butyric acid fermentation, thereby promoting stable lactic acid fermentation. Each bale was considered an experimental unit, and three bales were prepared per treatment, resulting in a total of 21 independent experimental units (7 treatments × 3 replicates). This replicate number was selected based on common practice in field-scale silage studies and to ensure feasibility given the large size (~600 kg) and resource-intensive nature of each bale, which involved independent preparation, fermentation, and downstream multi-omics analyses. All bales were stored under ambient conditions for 60 days.
After the fermentation period, samples were collected from each bale following standard multi-point sampling procedures for cylindrical silage bales. Upon unsealing, five subsamples were taken from different locations of each bale—including the top, bottom, center, and both sides—using a handheld silage corer to account for spatial variability and minimize the sampling bias. These subsamples were immediately combined and thoroughly homogenized to represent one biological replicate per bale. In total, 21 mixed samples (6 treatments + 1 control, each in triplicate) were collected. Each sample was placed in a sterile polyethylene bag, vacuum-sealed to prevent oxygen exposure, and immediately transported on ice to the laboratory for further analysis. All samples were processed within 12 h of collection to avoid secondary fermentation and ensure data integrity.
Fresh and ensiled king grass samples were analyzed for dry matter (DM), water-soluble carbohydrates (WSCs), crude protein (CP), ether extract (EE), crude fiber (CF), neutral detergent fiber (NDF), and acid detergent fiber (ADF). DM was determined using the AOAC method 934.01 (2016) by drying samples in an oven at 115 °C until a constant weight was achieved. CP and EE were determined according to AOAC official methods, with CP measured using the Kjeldahl method (AOAC 984.13, 2016) and EE analyzed using the AOAC method 920.39 (2005). NDF and ADF were measured using the method described by He et al. [
18]. The WSC content was estimated using the anthrone–sulfuric acid method, following the procedure outlined by Ke et al. [
19].
The fermentation quality was assessed using aqueous extracts of silage. Briefly, 50 g of wet silage was homogenized with 180 mL of sterile distilled water for 1 min in a blender and then incubated at 4 °C for 24 h. The mixture was filtered through four layers of sterile medical gauze. The pH of the filtrate was measured using a pH meter (FE28-Standard, Mettler Toledo, Zurich, Switzerland). Lactic acid and acetic acid concentrations were determined via high-performance liquid chromatography (HPLC) using an Agilent 1100 system (Agilent Technologies, Santa Clara, CA, USA), following the method of Zeng et al. [
20].
Microbial community analysis was conducted on both the six commercial additives and the ensiled king grass samples. For each additive, 20 g of dry powder was divided into two equal parts: one portion (10 g) was suspended in 200 mL of 5% brown sugar solution and shaken at 160 rpm for 2 h to activate microbial cells; the other 10 g was used directly for DNA extraction. For silage samples, 50 g of the silage sample was homogenized with 200 mL of sterile distilled water and incubated at 4 °C for 24 h. The mixture was filtered through two layers of sterile gauze, and the residue was rinsed several times with sterile water to collect surface-adhering microbes. The combined filtrates were centrifuged at 12,000× g for 5 min at 4 °C to pellet microbial cells.
Microbial genomic DNA was extracted using the E.Z.N.A.
® Soil DNA Kit (Omega Bio-Tek, Norcross, GA, USA) following the manufacturer’s instructions. Bacterial 16S rDNA was amplified using primers targeting the V3–V4 regions (338F: 5′-ACTCCTACGGGAGGCAGCAG-3′; 806R: 5′-GGACTACHVGGGTWTCTAAT-3′), while fungal internal transcribed spacer (ITS) sequences were amplified using ITS1-targeted primers (1737F: 5′-GGAAGTAAAAGTCGTAACAAGG-3′; 2043R: 5′-GCTGCGTTCTTCATCGATGC-3′) [
21,
22]. PCR amplification was performed using 10 ng of DNA as a template under the following conditions: initial denaturation at 95 °C for 3 min; 27 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s; and a final extension at 72 °C for 10 min. PCR products were purified, quantified, pooled in equimolar concentrations, and subjected to paired-end sequencing on an Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA) at Biomarker Technologies Corporation (Beijing, China).
Raw sequencing data were quality filtered using Trimmomatic [
23] and assembled with FLASH [
24]. Reads containing ≥3 ambiguous bases or with average Phred quality scores ≤ 20 were discarded. Chimeric sequences were identified and removed using UCHIME [
25]. High-quality reads were clustered into operational taxonomic units (OTUs) at 97% similarity using USEARCH [
26], and OTUs present in less than 5% of all samples were filtered out. Taxonomic annotation was performed using the SILVA 138 database for bacteria and UNITE 7.2 for fungi [
27]. Microbial diversity analyses were performed in R. The differential OTU abundance was assessed using the Wilcoxon rank-sum test based on OTUs with a median relative abundance >0.2% per group, and false discovery rate (FDR) correction was applied (FDR = 0.05). The alpha diversity was calculated using the core_diversity_analyses.py script from QIIME, while unconstrained principal coordinates analysis (PCoA) was performed using the capscale() function from the vegan package in R v4.4.0, based on the log2-transformed relative abundance (log2(RA)—1). Permutational multivariate analysis of variance (PERMANOVA) was conducted with the adonis() function (vegan package). All plots and figures were generated using the ggplot2 package.
For the metabolomic analysis, 50 mg of fresh silage from each homogenized composite sample was ground in liquid nitrogen and vortexed with 1000 μL of extraction solution containing 2 μL of the internal standard L-2-chlorophenylalanine (Aladdin, Shanghai, China). Ceramic beads were added, and samples were homogenized at 45 Hz for 10 min, followed by ultrasonication on ice for 10 min. The mixtures were then incubated at −20 °C for 1 h and centrifuged at 13,400× g at 4 °C for 15 min. A 500 μL aliquot of the supernatant was diluted with LC-MS-grade water to a final concentration of 60% methanol. The diluted solution was filtered through a 0.22 μm membrane, transferred to a new Eppendorf tube, and centrifuged again at 13,400× g at 4 °C for 15 min. Finally, 120 μL of the supernatant was transferred to a 2 mL LC vial for metabolomic analysis.
Samples were analyzed using a UPLC-MS/MS system (Waters UPLC I-Class PLUS coupled with a Xevo G2-XS QToF, Waters, Milford, MA, USA). Chromatographic separation was performed using gradient elution with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) under both positive and negative electrospray ionization (ESI) modes. Mass spectra were acquired in MSe mode using MassLynx V4.2 software (Waters), with alternating low (2 V) and high (10–40 V) collision energies at a scan rate of 0.2 s per cycle. The ESI source parameters were as follows: capillary voltage, +2000 V (positive)/−1500 V (negative); cone voltage, 30 V; ion source temperature, 150 °C; desolvation temperature, 500 °C; cone gas flow, 50 L/h; desolvation gas flow, 800 L/h.
Raw data were processed using Progenesis QI V2.3 (Nonlinear Dynamics, Newcastle, UK) for baseline correction, peak picking, retention time alignment, and feature extraction. The following parameters were used: precursor ion tolerance, 5 ppm; fragment ion tolerance, 10 ppm; product ion threshold, 5%. MS and MS/MS data were matched against the METLIN database and an in-house library (Biomarker Technologies, Beijing, China). Metabolite identification was based on the accurate mass (m/z), fragmentation pattern and isotope distribution, with the mass tolerance for precursor and fragment ions set to <100 ppm and <50 ppm, respectively. Only compounds with high MS/MS matching scores were retained for further analysis.
Data normalization was performed using Pareto scaling followed by log transformation. Differential metabolites were identified based on a fold change (FC) ≥ 2 or ≤0.5, p < 0.05 (t-test), and variable importance in projection (VIP) > 1 from the orthogonal partial least squares discriminant analysis (OPLS-DA) model. Metabolite annotation was conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and pathway analysis was performed using MetaboAnalyst combined with KEGG.
All statistical analyses were performed using R. Differences in chemical composition and fermentation quality parameters (e.g., pH, lactic acid, CP, NDF, ADF) among treatments were assessed using one-way analysis of variance (ANOVA) followed by a Least Significant Difference (LSD) test for pairwise comparisons. Data were tested for normality and homogeneity of variances prior to ANOVA. Results were considered statistically significant at p < 0.05.
For microbiome analysis, differential OTU abundances between treatments was assessed using the Wilcoxon rank-sum test, with false discovery rate (FDR) correction applied at a threshold of FDR = 0.05. Alpha diversity indices were calculated using the QIIME pipeline, and beta diversity was evaluated via principal coordinates analysis (PCoA) and PERMANOVA using the ‘adonis()’ function in the vegan R package.
For the metabolomic analysis, data were normalized using Pareto scaling and log transformation. Differential metabolites were identified based on the fold change (FC ≥ 2 or ≤0.5), p < 0.05 (Student’s t-test), and VIP > 1 from the orthogonal partial least squares discriminant analysis (OPLS-DA) model. Pathway enrichment analysis was conducted using MetaboAnalyst and KEGG annotations.
4. Discussion
The application of high-throughput sequencing technologies has substantially improved our understanding of microbial diversity in silage ecosystems and provided new insights into fermentation mechanisms and strategies for improving forage preservation. King grass, widely cultivated in the mountainous regions of southwest China, is a high-yield forage with great potential for silage. However, its high fiber content and low epiphytic lactic acid bacteria (LAB) populations often result in suboptimal fermentation when ensiled alone. Previous studies suggest that an LAB population exceeding 5.0 log
10 CFU/g fresh matter is critical for successful ensiling [
29], but the natural phyllosphere microbiota of king grass is typically insufficient to meet this threshold [
30,
31]. To address this limitation, microbial inoculants have been widely used to enhance the fermentation process by promoting lactic acid production and suppressing undesirable microorganisms. While single-strain LAB inoculants have been commonly applied, their performance under high-moisture conditions is often limited, with inconsistent effects on aerobic stability [
32,
33]. Therefore, exploring multi-strain or functionally diverse inoculants—particularly those adapted to local conditions—may provide more reliable improvements in the silage quality. In this context, our study evaluated the effects of six commercial microbial additives collected from pastures in the mountainous areas of Sichuan and Guizhou provinces, aiming to assess their influence on the fermentation performance, microbial community structure, and metabolomic characteristics of king grass silage under farm-scale production.
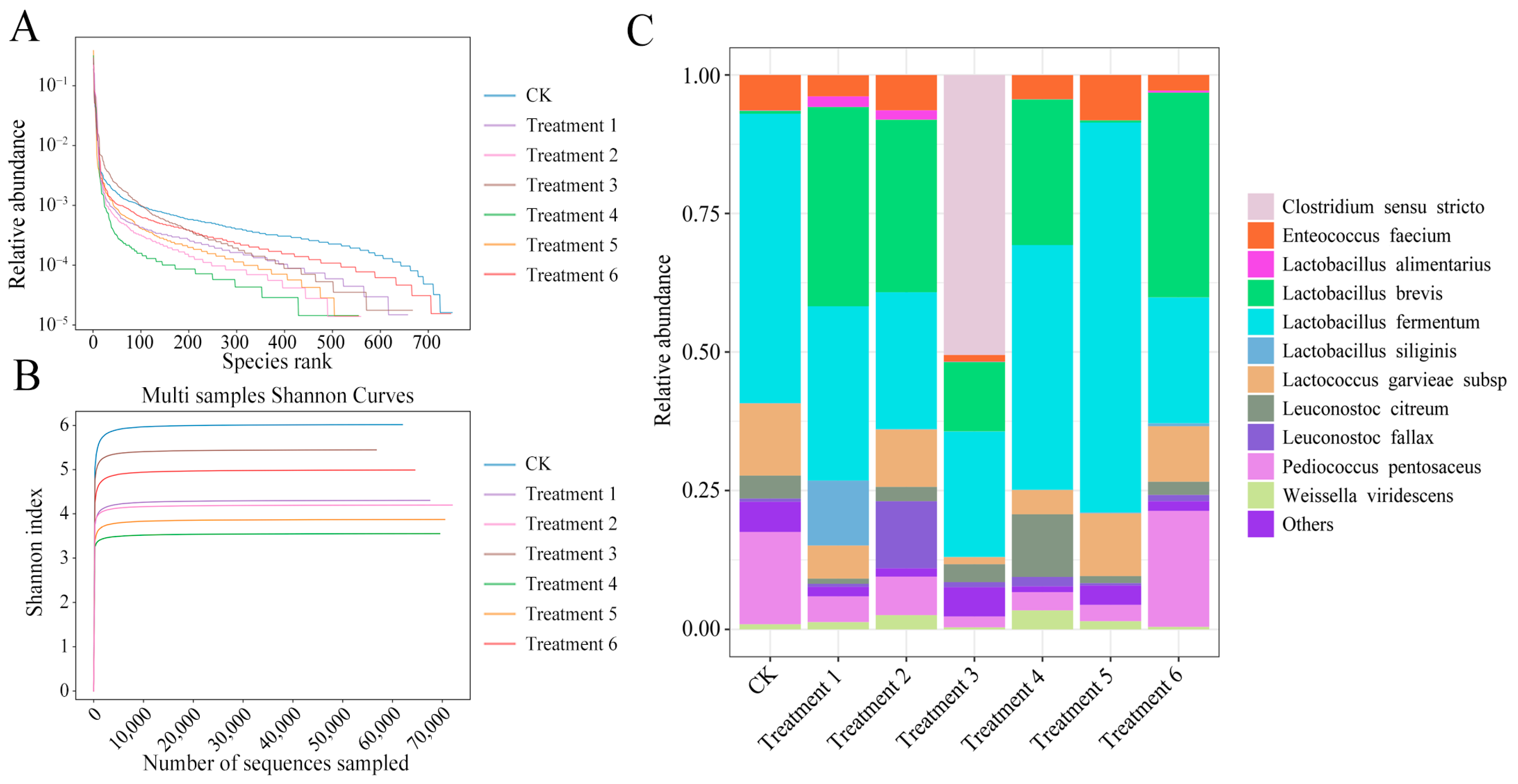
Microbial community analysis revealed that the six commercial additives, classified into three groups based on their dominant strains (
E. faecium-like and
Bacillus velezensis [Treatments 1, 2],
B. velezensis [Treatments 3, 6], and
Lactobacillus paraplantarum [Treatments 4, 5]), differentially shaped the bacterial composition of silage after 60 days of fermentation. Inoculation markedly increased the relative abundance of
L. brevis (Treatments 1, 2, 4, 6),
L. siliginis (Treatment 1),
L. fallax (Treatment 2),
L. citreum (Treatment 4), and
L. garvieae and
P. pentosaceus (Treatment 6), compared to the uninoculated control (CK). Notably, additives dominated by
B. velezensis (Treatments 1, 2, 3, and 6) promoted the proliferation of
L. brevis, suggesting a potential facilitative effect of
Bacillus species on LAB colonization during ensiling. While
E. faecium was dominant in Additives 1 and 2, it did not establish dominance in the final silage microbiota, likely due to limited competitiveness against other epiphytic or inoculated LAB, consistent with previous reports [
34]. Additives 4 and 5, although initially dominated by
L. paraplantarum, resulted in the dominance of
L. fermentum after ensiling, with bacterial profiles similar to the CK group. However, Treatment 4 retained higher abundances of
L. brevis and
L. citreum, possibly due to the presence of
B. coagulans in the inoculant. In contrast,
L. fallax, although introduced in Treatment 5, failed to colonize successfully, potentially due to competition from
L. paraplantarum. Treatment 6, containing both
B. velezensis and lactic acid-producing cocci (
P. pentosaceus,
L. garvieae), achieved the successful colonization of all three genera and yielded favorable fermentation outcomes. These results suggest that certain strains, such as cocci LAB and
Bacillus, may not dominate the final community but can modulate the microbial environment to favor desirable LAB succession.
The composition of the bacterial community was closely associated with the silage fermentation quality and nutritional parameters. Treatments 2, 4, and 6 significantly increased crude protein (CP) and lactic acid (LA) contents, while reducing neutral detergent fiber (NDF) and acid detergent fiber (ADF), indicating improved fermentation efficiency and nutrient preservation. In addition to the nutrient profiles, variations in the dry matter (DM) content also reflected the effectiveness of fermentation and the properties of the additives. Most treatments showed an increase in the DM content compared to fresh king grass, likely due to the addition of dry carriers in the commercial inoculants and partial water loss during fermentation. However, Treatment 3 was the only group with a lower DM content than the fresh material. This may be attributed to its poor fermentation performance, as indicated by high pH and low lactic acid contents, suggesting incomplete acidification and possible moisture retention. Moreover, the formulation of the additive used in Treatment 3 may have contributed to this outcome, potentially due to a higher intrinsic moisture content or hygroscopic components. These observations underscore that not all commercial additives equally promote dehydration or DM preservation during ensiling.
Redundancy analysis (RDA) further confirmed that
L. brevis abundance was positively correlated with higher DM, CP, and LA contents, particularly in Treatments 1, 2, 4, and 6. Silage pH, a key indicator of fermentation success, was markedly lower in Treatments 2 and 4 (final pH = 3.85), reaching levels sufficient to inhibit spoilage microorganisms such as Clostridium, molds, and yeasts [
7]. The strong acidification capacity of these inoculants was associated with a higher LA content and the dominance of LAB genera. Notably, Treatment 2 demonstrated the most efficient lactic acid synthesis, further confirming its superior fermentation performance. Taken together, these findings indicate that microbial inoculants—particularly those containing combinations of
Bacillus,
Pediococcus, and
Lactobacillus species—can reshape the fermentation trajectory of king grass silage, resulting in enhanced preservation, improved nutrient profiles, and better microbial stability. While redundancy analysis (RDA) revealed significant associations between specific bacterial taxa and fermentation parameters, it is important to acknowledge that these correlations may, in part, reflect underlying treatment effects. Since both the microbial community composition and fermentation outcomes were influenced by the applied additives, their covariation could result from shared treatment-driven changes rather than direct causal relationships. Nevertheless, we employed multivariate statistical tools, including RDA and Mantel tests, to reduce such confounding and to capture meaningful ecological associations. Moreover, the observed relationships—such as the positive correlation of
Lactobacillus brevis with lactic acid and crude protein, or the link between
Clostridium sensu stricto and a higher pH—are consistent with the known metabolic functions of these taxa in silage fermentation. These results suggest that, despite potential treatment effects, the microbial indicators identified are ecologically relevant and may play functional roles in the fermentation dynamics. Future studies using factorial experimental designs or simplified microbial models could help disentangle the treatment-based and microbe-specific effects more precisely.
In addition to RDA, multivariate analyses such as PCoA and UPGMA also provided complementary insights into bacterial community patterns. Notably, although Treatment 4 was positioned farthest from the cluster center in the PCoA plot, it was grouped together with Treatments 1 and 6 in the UPGMA dendrogram. This apparent discrepancy reflects methodological differences between ordination-based and clustering-based approaches. PCoA visualizes sample separation based on a limited number of principal coordinates that explain part of the variance, whereas UPGMA considers the entire beta-diversity matrix to represent the overall community similarity. Therefore, Treatment 4 may differ from other groups along dominant axes but still share broader microbial community characteristics with Treatments 1 and 6. This highlights the importance of using multiple complementary analytical methods to gain a more comprehensive understanding of the silage microbiome structure.
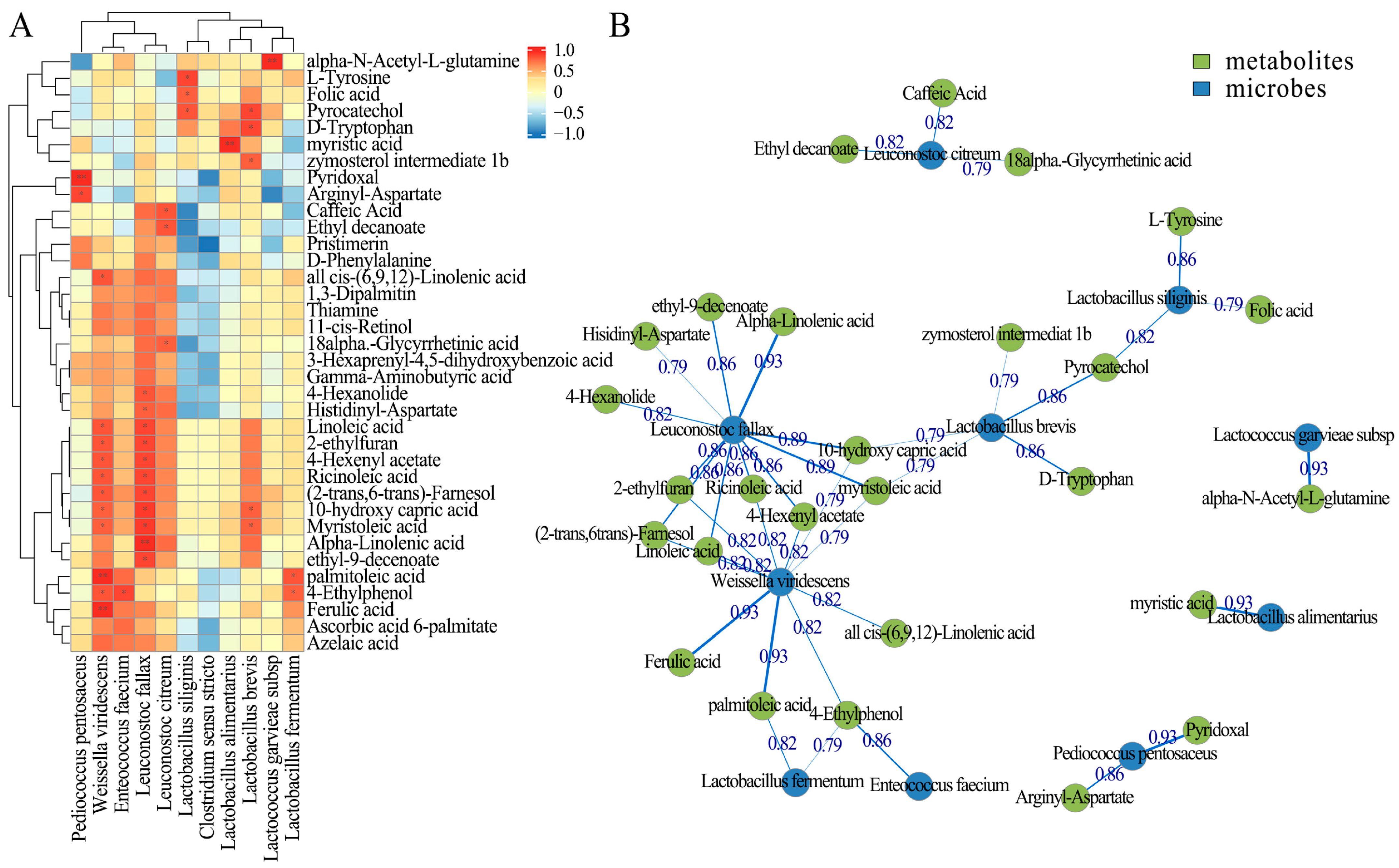
Beyond changes in the microbial diversity, our findings revealed potential synergistic interactions among dominant LAB species that may underlie the enhanced fermentation quality and metabolite production in treated silages. Correlation analyses between bacterial species and functional metabolites showed that several LAB taxa—especially
Lactobacillus brevis,
L. siliginis,
Leuconostoc fallax, and
Weissella viridescens—were positively associated with key biofunctional metabolites including pyrocatechol, linolenic acid, farnesol, ricinoleic acid, and 4-hexenyl acetate. These compounds possess antimicrobial, antioxidant, and flavor-enhancing properties, suggesting that specific bacterial consortia may co-contribute to the biofunctional enrichment of silage. For example,
L. brevis has been reported to degrade phenolic acids, leading to the production of compounds like pyrocatechol, which exhibit antioxidant activity [
35]. In contrast,
Clostridium sensu stricto, an undesirable genus for silage fermentation, showed negative correlations with several beneficial metabolites such as azelaic acid, pristimerin, and γ-aminobutyric acid (GABA), reinforcing its suppressive effect on silage quality. The presence of
C. sensu stricto has been associated with poor fermentation quality in silages, characterized by increased butyric acid and ammoniacal nitrogen concentrations [
36]. The network analysis further supported the presence of a cooperative microbial framework.
L. fallax and
W. viridescens emerged as central network nodes, sharing metabolite associations with other LAB including
L. brevis,
L. siliginis,
L. fermentum, and
E. faecium. For instance,
L. brevis co-occurred with
L. siliginis through shared positive correlations with pyrocatechol and with
L. fallax via associations with 10-hydroxy capric acid and myristoleic acid. These patterns suggest functional complementarity, where different LAB species contribute distinct but interrelated metabolic outputs during fermentation. Such interactions may be explained by metabolic cross-feeding or environmental modification. For example, early colonizers such as
P. pentosaceus or
L. garvieae may acidify the environment, facilitating the growth and metabolic activation of later-colonizing
Lactobacillus and
Leuconostoc species. Meanwhile,
Bacillus velezensis, although not dominant in the final microbiota, likely played a facilitative role by providing enzymes or inhibitory compounds that suppressed spoilage microbes and supported LAB proliferation. These results underscore the importance of selecting complementary strains in silage inoculants rather than relying on single-strain products. Designing multispecies inoculants that intentionally combine early acidifiers, bioactive-compound-producers, and robust fermenters may improve both fermentation efficiency and the biofunctional quality of the final product.
Inoculation with microbial additives significantly reshaped the silage metabolome, with 1523 metabolites detected and 56–84 differentially abundant compounds identified per treatment relative to the control. These metabolites were enriched in various functional categories, including antimicrobial agents (e.g., pyrocatechol, caffeic acid, ferulic acid), cholesterol-lowering compounds (e.g., γ-aminobutyric acid, linoleic acid, pyridoxal), and aromatic flavor precursors (e.g., ethyl decanoate, 2-ethylfuran, 4-ethylphenol). Notably, many of these compounds were positively correlated with dominant LAB species, suggesting microbially mediated enrichment during ensiling. KEGG pathway enrichment analysis revealed that these up-accumulated metabolites were primarily involved in amino acid metabolism, nucleotide metabolism, cofactor and vitamin metabolism, and energy-related pathways. Among them, the arginine and proline metabolism pathways were significantly activated across all treatment groups. These pathways are closely linked to bacterial energy production via amino acid decarboxylation, arginine deamination, and malate decarboxylation—processes previously reported to promote lactic acid accumulation under anaerobic conditions [
37]. In our study, arginine levels declined while intermediate products such as β-alanine, histamine, and γ-aminobutyric acid (GABA) significantly increased in inoculated silages, indicating enhanced amino acid turnover and energy metabolism. These findings align with reports before, which suggested that amino acid degradation supports silage acidification and stabilization by providing ATP and reducing equivalents [
38]. Several amino acids were found in greater abundance in treated silages and were positively associated with specific LAB strains. For example, tyrosine and tryptophan were enriched and correlated with
L. siliginis, while arginyl-aspartate and phenylalanine were associated with
P. pentosaceus. These associations imply that certain LAB may actively contribute to amino acid biosynthesis or preservation, potentially enhancing the protein quality of silage and explaining the observed increase in crude protein. Additionally, vitamin biosynthesis pathways were upregulated. Key compounds including thiamine, folic acid, pyridoxal, and 11-cis-retinol were significantly increased in treated silages. Their accumulation was positively correlated with LAB such as
L. siliginis,
P. pentosaceus, and
L. fermentum, suggesting that microbial fermentation not only helps preserve but may also actively enrich vitamins in the final product. Since many vitamins are prone to degradation during ensiling, the use of functional LAB may represent a strategy to maintain or even enhance their concentrations, supporting the nutritional value and health benefits of silage in ruminant diets.
Future research should further explore the ecological mechanisms behind LAB synergy, such as metabolic cross-feeding, quorum sensing, or niche complementarity, and identify key keystone species within silage microbiomes. Advanced tools like genome-resolved metagenomics, metabolite flux analysis, and co-culture experiments could help to elucidate causal links between microbial interactions and metabolite production. Moreover, the development of designer inoculants combining early acidifiers, bioactive compound producers, and resilient fermenters tailored to specific forage types or regional environments may represent a promising strategy for improving both the nutritional quality and functional safety of silages. Finally, field-scale validation under diverse climatic conditions and feeding trials are needed to evaluate the practical benefits of these multispecies formulations on animal performance and health.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}