

Microbiological and Physicochemical Dynamics in Traditional and Industrial Fermentation Processes of Koumiss


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Koumiss Fermentation and Sampling
2.2. Microbial Isolation, Counts, and Identification
2.3. Culture-Independent Analyses
2.3.1. Total DNA Extraction
2.3.2. Amplification and Sequencing Parameters
2.3.3. Bioinformatics
2.4. Physicochemical Analyses
2.5. Statistical Analyses
3. Results
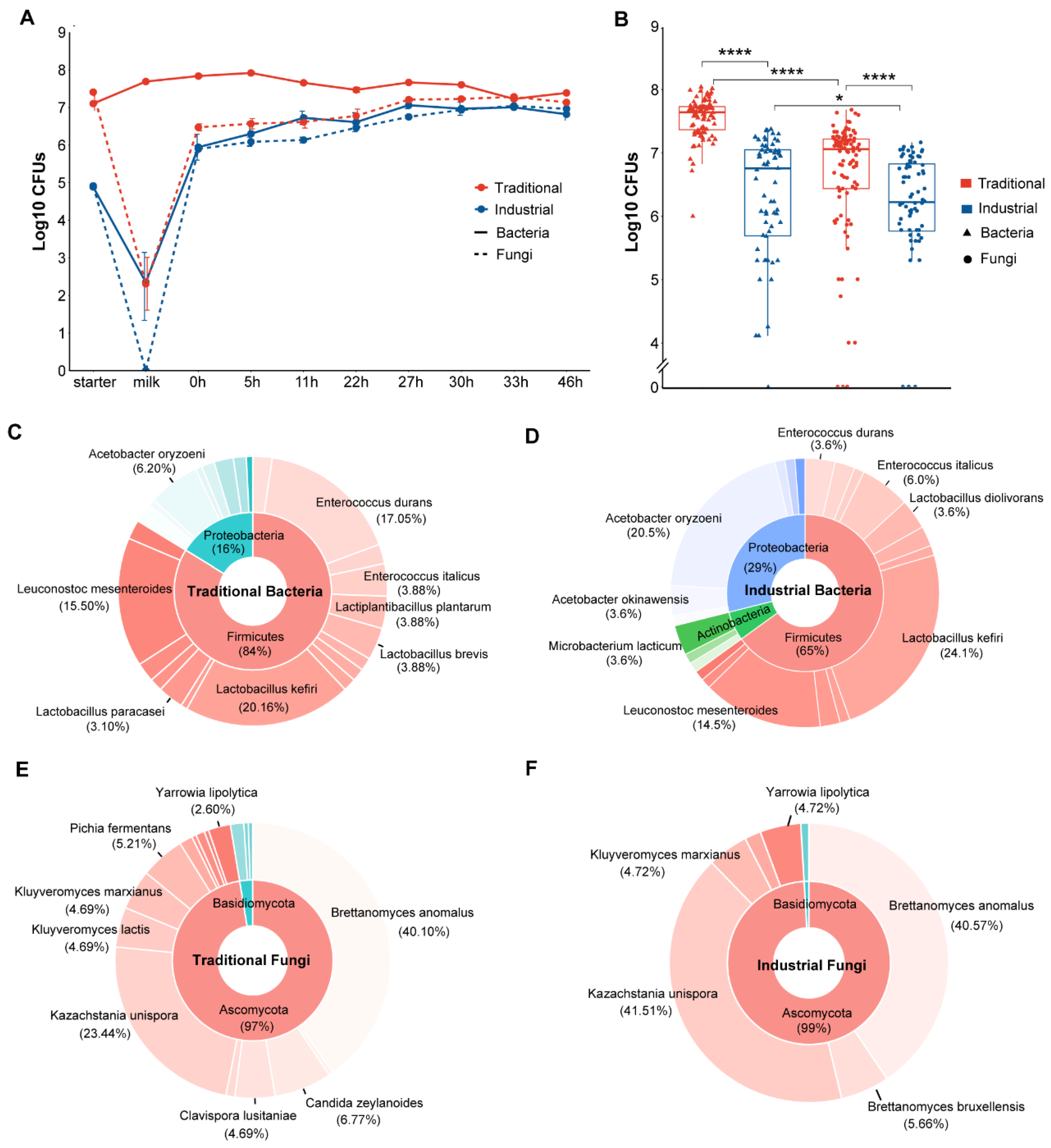
3.1. Changes and Composition of Culturable Microbes during Koumiss Fermentation
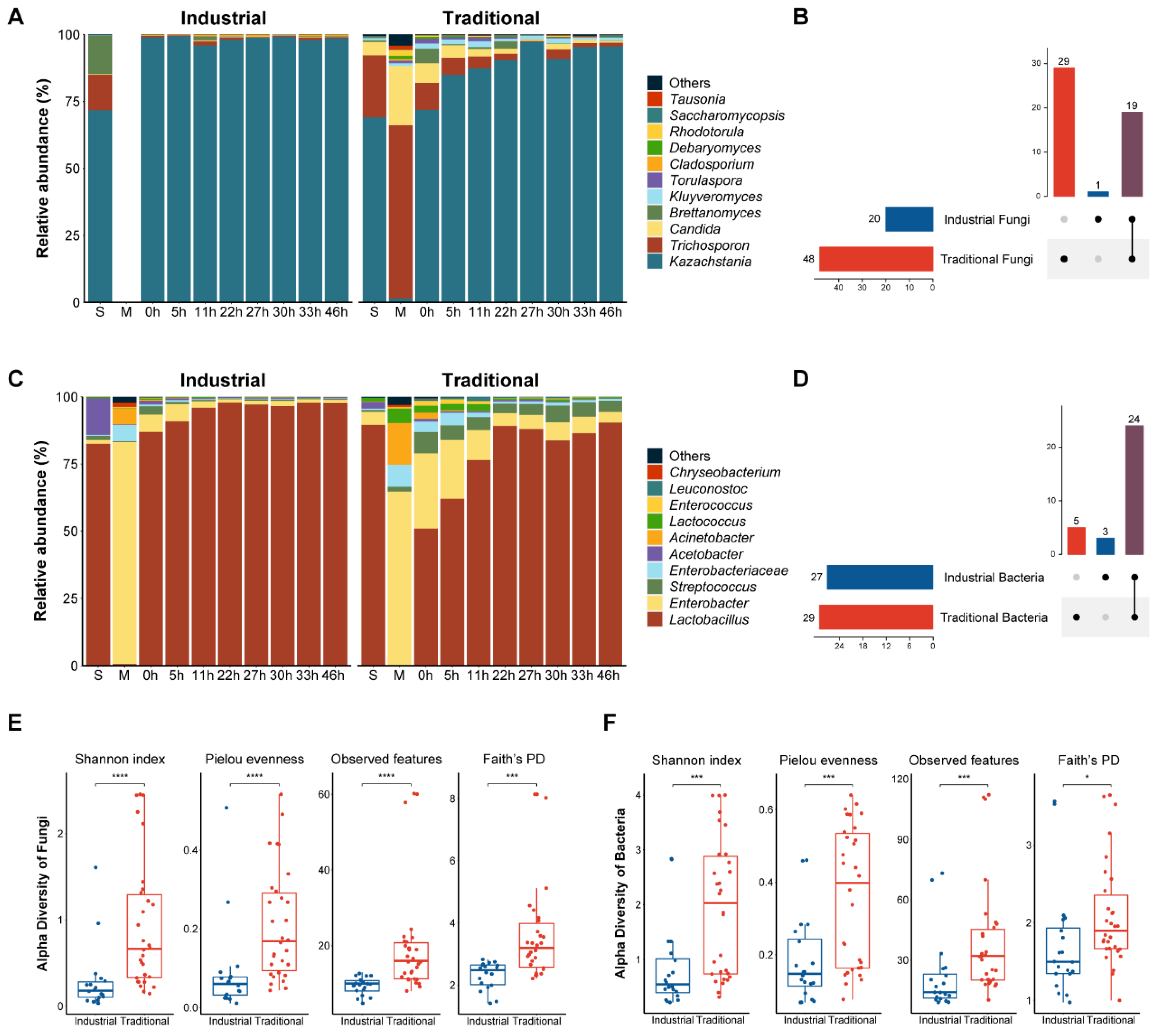
3.2. Microbial Community Dynamics during Koumiss Fermentation Revealed by Culture-Independent Analyses
3.2.1. Fungal Dynamics
3.2.2. Bacterial Dynamics
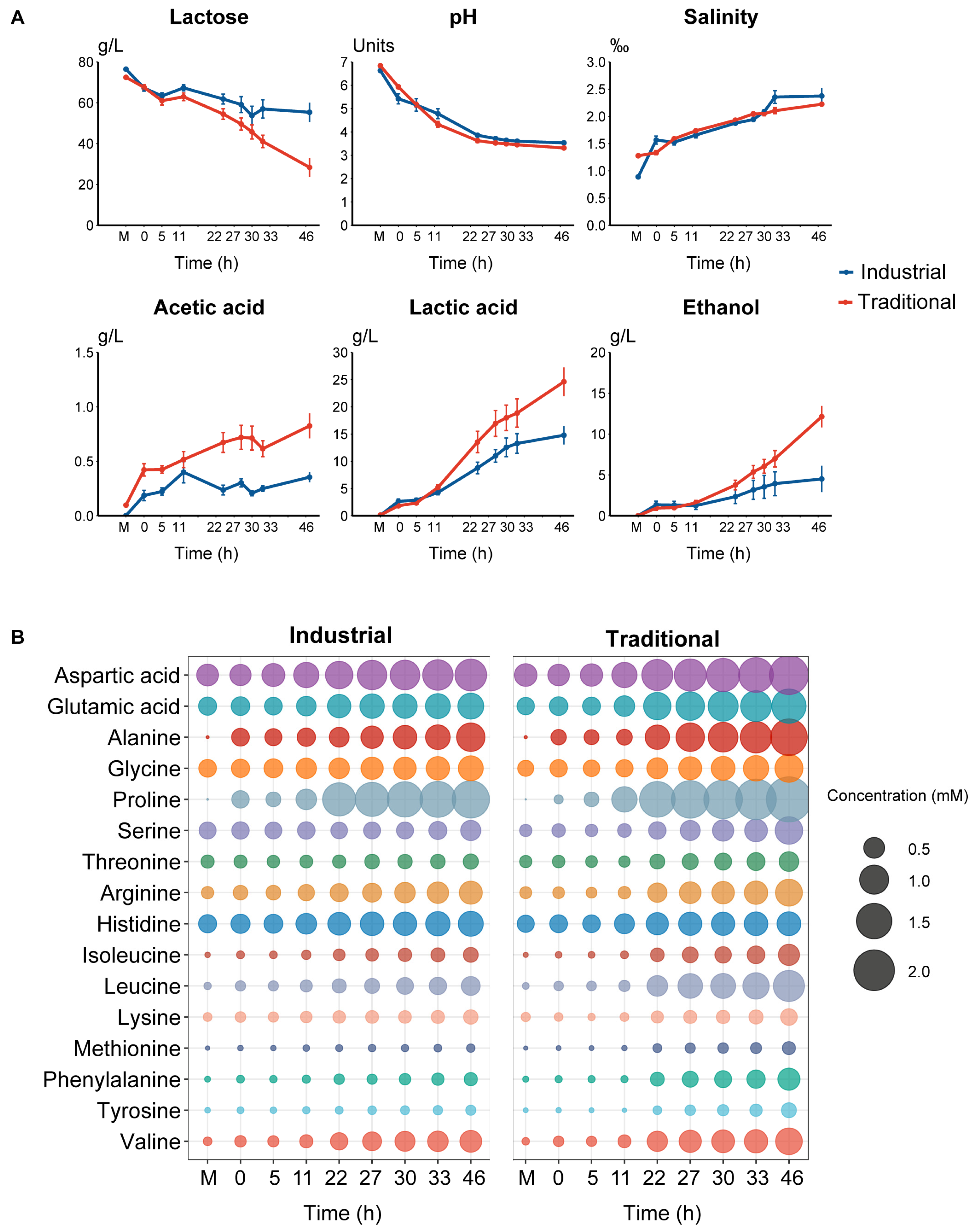
3.3. Physicochemical Dynamics during Fermentation
3.4. Correlation between Microbes and Physicochemical Profile during Fermentation
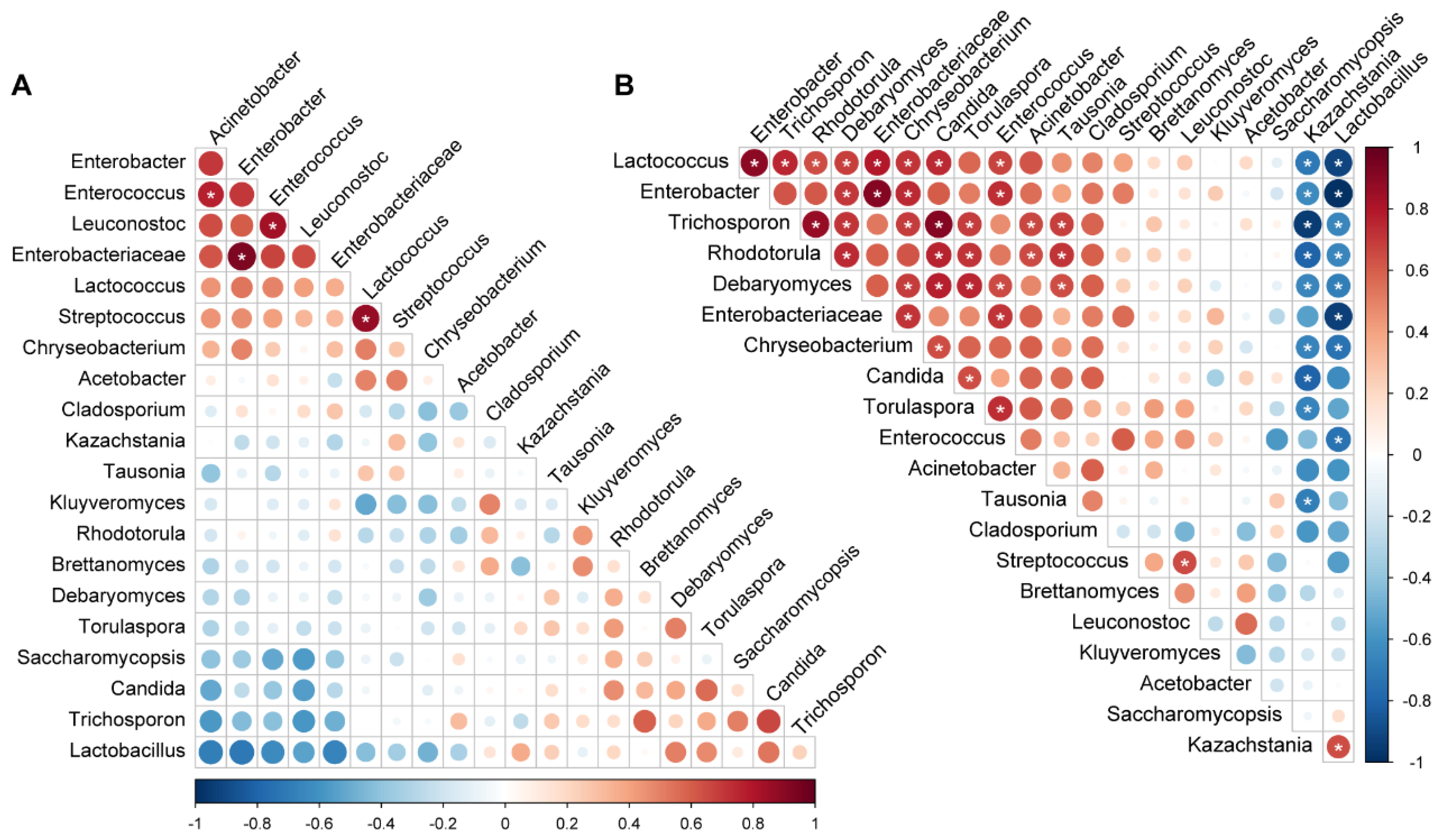
3.5. Microbial Co-Occurrence during Fermentation
3.6. Source Tracking of Koumiss Microbial Community
3.7. Microbial Genera Distinguishing Fermentation Modes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uniacke-Lowe, T. Koumiss. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., McNamara, J.P., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; Volume 3, pp. 446–452. [Google Scholar]
- Afzaal, M.; Saeed, F.; Anjum, F.; Waris, N.; Husaain, M.; Ikram, A.; Ateeq, H.; Muhammad Anjum, F.; Suleria, H. Nutritional and ethnomedicinal scenario of koumiss: A concurrent review. Food Sci. Nutr. 2021, 9, 6421–6428. [Google Scholar] [CrossRef] [PubMed]
- Lomer, M.C.; Parkes, G.; Sanderson, J. Review article: Lactose intolerance in clinical practice—Myths and realities. Aliment. Pharmacol. Ther. 2008, 27, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Li, C.; Liu, Y.; Li, W.; Chen, Y.; Siqinbateer; Bao, Y.; Saqila, W.; Zhang, H.; Menghe, B. Koumiss consumption modulates gut microbiota, increases plasma high density cholesterol, decreases immunoglobulin G and albumin. J. Funct. Foods 2019, 52, 469–478. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Wang, H.; Fan, H.; Mi, Z.; Kwok, L.-Y.; Zhang, H.; Menghe, B.; Sun, Z.; Chen, Y. Koumiss consumption induced changes in the fecal metabolomes of chronic atrophic gastritis patients. J. Funct. Foods 2019, 62, 103522. [Google Scholar]
- Guo, C.-F.; Zhang, S.; Yuan, Y.-H.; Yue, T.-L.; Li, J.-Y. Comparison of lactobacilli isolated from Chinese suan-tsai and koumiss for their probiotic and functional properties. J. Funct. Foods 2015, 12, 294–302. [Google Scholar]
- Zhao, H.-Z.; Song, Q.-J.; Guo, H.; Liu, C.-Y.; Yang, C.; Li, X.; Wang, Y.-X.; Ma, Z.-P.; Wang, F.-X.; Wen, Y.-J. Characterization of a Potential Probiotic Strain in Koumiss. Fermentation 2023, 9, 87. [Google Scholar] [CrossRef]
- Montanari, G.; Zambonelli, C.; Grazia, L.; Kamesheva, G.K.; Shigaeva, M.K. Saccharomyces unisporus as the principal alcoholic fermentation microorganism of traditional koumiss. J. Dairy Res. 1996, 63, 327–331. [Google Scholar] [CrossRef]
- Mu, Z.; Yang, X.; Yuan, H. Detection and identification of wild yeast in Koumiss. Food Microbiol. 2012, 31, 301–308. [Google Scholar] [CrossRef]
- Biçer, Y.; Telli, A.E.; Sönmez, G.; Turkal, G.; Telli, N.; Uçar, G. Comparison of commercial and traditional kefir microbiota using metagenomic analysis. Int. J. Dairy Technol. 2021, 74, 528–534. [Google Scholar] [CrossRef]
- Guo, L.; Ya, M.; Guo, Y.S.; Xu, W.L.; Li, C.D.; Sun, J.P.; Zhu, J.J.; Qian, J.P. Study of bacterial and fungal community structures in traditional koumiss from Inner Mongolia. J. Dairy Sci. 2019, 102, 1972–1984. [Google Scholar] [CrossRef]
- Xia, Y.; Yu, J.; Miao, W.; Shuang, Q. A UPLC-Q-TOF-MS-based metabolomics approach for the evaluation of fermented mare’s milk to koumiss. Food Chem. 2020, 320, 126619. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Chen, X.; Sun, Z.; Li, Y.; Chen, D.; Fang, S.; Chen, J. Exploring core microbiota responsible for the production of volatile flavor compounds during the traditional fermentation of Koumiss. LWT-Food Sci. Technol. 2021, 135, 110049. [Google Scholar]
- Xia, Y.; Yu, J.; Liu, H.; Feng, C.; Shuang, Q. Novel insight into physicochemical and flavor formation in koumiss based on microbial metabolic network. Food Res. Int. 2021, 149, 110659. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Gesudu, Q.; Zhang, J.; Sun, Z.; Halatu, H.; Menghe, B.; Liu, W. Bacterial composition and function during fermentation of Mongolia koumiss. Food Sci. Nutr. 2021, 9, 4146–4155. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Ma, H.; Hou, Q.; Li, W.; Xu, H.; Liu, W.; Sun, Z.; Haobisi, H.; Menghe, B. Profiling of koumiss microbiota and organic acids and their effects on koumiss taste. BMC Microbiol. 2020, 20, 85. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, Y.; Siqin, B.; Xilin, T.; Zhang, N.; Li, M. Changes in Microbial Diversity and Nutritional Components of Mare Milk Before and After Traditional Fermentation. Front. Sustain. Food Syst. 2022, 6, 913763. [Google Scholar]
- Kurtzman, C.; Robnett, C. Identification of clinically important ascomycetous yeasts based on nucleotide divergence in the 5’end of the large-subunit (26S) ribosomal DNA gene. J. Clin. Microbiol. 1997, 35, 1216–1223. [Google Scholar]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar]
- DeLong, E.F. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. USA 1992, 89, 5685–5689. [Google Scholar] [CrossRef]
- Srinivasan, S.; Hoffman, N.G.; Morgan, M.T.; Matsen, F.A.; Fiedler, T.L.; Hall, R.W.; Ross, F.J.; McCoy, C.O.; Bumgarner, R.; Marrazzo, J.M. Bacterial communities in women with bacterial vaginosis: High resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS ONE 2012, 7, e37818. [Google Scholar] [CrossRef]
- Ihrmark, K.; Bodeker, I.T.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandstrom-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A. Global diversity and geography of soil fungi. Science 2014, 346, 1256688. [Google Scholar] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar]
- R Core Team. 2023. R: Alanguage and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 8 December 2023).
- Olszewski, T.D. A unified mathematical framework for the measurement of richness and evenness within and among multiple communities. Oikos 2004, 104, 377–387. [Google Scholar]
- Kers, J.G.; Saccenti, E. The Power of Microbiome Studies: Some Considerations on Which Alpha and Beta Metrics to Use and How to Report Results. Front. Microbiol. 2021, 12, 796025. [Google Scholar]
- Heinrikson, R.L.; Meredith, S.C. Amino acid analysis by reverse-phase high-performance liquid chromatography: Precolumn derivatization with phenylisothiocyanate. Anal. Biochem. 1984, 136, 65–74. [Google Scholar] [CrossRef]
- Schiffman, S.S.; Sennewald, K.; Gagnon, J. Comparison of taste qualities and thresholds of D-and L-amino acids. Physiol. Behav. 1981, 27, 51–59. [Google Scholar]
- Penland, M.; Deutsch, S.M.; Falentin, H.; Pawtowski, A.; Poirier, E.; Visenti, G.; Le Meur, C.; Maillard, M.B.; Thierry, A.; Mounier, J.; et al. Deciphering Microbial Community Dynamics and Biochemical Changes During Nyons Black Olive Natural Fermentations. Front. Microbiol. 2020, 11, 586614. [Google Scholar]
- de Melo Pereira, G.V.; de Carvalho Neto, D.P.; Maske, B.L.; De Dea Lindner, J.; Vale, A.S.; Favero, G.R.; Viesser, J.; de Carvalho, J.C.; Goes-Neto, A.; Soccol, C.R. An updated review on bacterial community composition of traditional fermented milk products: What next-generation sequencing has revealed so far? Crit. Rev. Food Sci. Nutr. 2022, 62, 1870–1889. [Google Scholar] [CrossRef] [PubMed]
- Quigley, L.; O’Sullivan, O.; Stanton, C.; Beresford, T.P.; Ross, R.P.; Fitzgerald, G.F.; Cotter, P.D. The complex microbiota of raw milk. FEMS Microbiol. Rev. 2013, 37, 664–698. [Google Scholar] [PubMed]
- Xie, Z.-B.; Zhang, K.-Z.; Kang, Z.-H.; Yang, J.-G. Saccharomycopsis fibuligera in liquor production: A review. Eur. Food Res. Technol. 2021, 247, 1569–1577. [Google Scholar] [CrossRef]
- Ogundeji, A.O.; Li, Y.; Liu, X.; Meng, L.; Sang, P.; Mu, Y.; Wu, H.; Ma, Z.; Hou, J.; Li, S. Eggplant by grafting enhanced with biochar recruits specific microbes for disease suppression of Verticillium wilt. Appl. Soil Ecol. 2021, 163, 103912. [Google Scholar] [CrossRef]
- Xia, Y.; Oyunsuren, E.; Yang, Y.; Shuang, Q. Comparative metabolomics and microbial communities associated network analysis of black and white horse-sourced koumiss. Food Chem. 2021, 370, 130996. [Google Scholar]
- de Vos, W.M.; Vaughan, E.E. Genetics of lactose utilization in lactic acid bacteria. FEMS Microbiol. Rev. 1994, 15, 217–237. [Google Scholar] [CrossRef]
- Korcari, D.; Ricci, G.; Capusoni, C.; Fortina, M.G. Physiological performance of Kazachstania unispora in sourdough environments. World J. Microbiol. Biotechnol. 2021, 37, 88. [Google Scholar]
- Smith, M.T.; Yamazaki, M.; Poot, G.J.Y. Dekkera, Brettanomyces and Eeniella: Electrophoretic comparison of enzymes and DNA–DNA homology. Yeast 1990, 6, 299–310. [Google Scholar]
- Hayaloglu, A.A. Microbiology of Cheese. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., McNamara, J.P., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; Volume 3, pp. 225–237. [Google Scholar]
- Blasche, S.; Kim, Y.; Mars, R.A.; Machado, D.; Maansson, M.; Kafkia, E.; Milanese, A.; Zeller, G.; Teusink, B.; Nielsen, J. Metabolic cooperation and spatiotemporal niche partitioning in a kefir microbial community. Nat. Microbiol. 2021, 6, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Wurihan; Bao, L.; Hasigaowa; Bao, X.; Dai, Y.; Jia, S. Bacterial community succession and metabolite changes during the fermentation of koumiss, a traditional Mongolian fermented beverage. Int. Dairy J. 2019, 98, 1–8. [Google Scholar] [CrossRef]
- Gram, L.; Ravn, L.; Rasch, M.; Bruhn, J.B.; Christensen, A.B.; Givskov, M. Food spoilage—Interactions between food spoilage bacteria. Int. J. Food Microbiol. 2002, 78, 79–97. [Google Scholar] [PubMed]
- Yang, Y.; Fan, Y.; Li, T.; Yang, Y.; Zeng, F.; Wang, H.; Suo, H.; Song, J.; Zhang, Y. Microbial composition and correlation between microbiota and quality-related physiochemical characteristics in chongqing radish paocai. Food Chem. 2022, 369, 130897. [Google Scholar] [CrossRef]
- Liang, C.; Liu, L.-X.; Liu, J.; Aihaiti, A.; Tang, X.-J.; Liu, Y.-G. New Insights on Low-Temperature Fermentation for Food. Fermentation 2023, 9, 477. [Google Scholar] [CrossRef]
- Du, B.; Meng, L.; Wu, H.; Yang, H.; Liu, H.; Zheng, N.; Zhang, Y.; Zhao, S.; Wang, J. Source Tracker Modeling Based on 16S rDNA Sequencing and Analysis of Microbial Contamination Sources for Pasteurized Milk. Front. Nutr. 2022, 9, 845150. [Google Scholar] [CrossRef]
- Mounier, J.; Coton, M. Kluyveromyces spp. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., McNamara, J.P., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; Volume 4, pp. 569–574. [Google Scholar]
- Johnson, E.A.; Echavarri-Erasun, C. Yeast Biotechnology. In The Yeasts, 5th ed.; Kurtzman, C.P., Fell, J.W., Boekhout, T., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2011; pp. 21–44. [Google Scholar]
- Salimei, E.; Park, Y.W. Mare milk. In Handbook of Milk of Non-Bovine Mammals, 2nd ed.; Park, Y.W., Haenlein, G.F.W., Wendorff, W.L., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 369–408. [Google Scholar]
- Li, L.; Renye, J.A., Jr.; Feng, L.; Zeng, Q.; Tang, Y.; Huang, L.; Ren, D.; Yang, P. Characterization of the indigenous microflora in raw and pasteurized buffalo milk during storage at refrigeration temperature by high-throughput sequencing. J. Dairy Sci. 2016, 99, 7016–7024. [Google Scholar] [CrossRef]
- Singh, B.; Anand, S. Enterobacteriaceae. In Encyclopedia of Dairy Sciences, 3rd ed.; McSweeney, P.L.H., McNamara, J.P., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; Volume 4, pp. 482–489. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Song, L.; Han, D.; Han, P.; Bai, F. Microbiological and Physicochemical Dynamics in Traditional and Industrial Fermentation Processes of Koumiss. Fermentation 2024, 10, 66. https://doi.org/10.3390/fermentation10010066
Zhao X, Song L, Han D, Han P, Bai F. Microbiological and Physicochemical Dynamics in Traditional and Industrial Fermentation Processes of Koumiss. Fermentation. 2024; 10(1):66. https://doi.org/10.3390/fermentation10010066
Chicago/Turabian StyleZhao, Xin, Liang Song, Dayong Han, Peijie Han, and Fengyan Bai. 2024. "Microbiological and Physicochemical Dynamics in Traditional and Industrial Fermentation Processes of Koumiss" Fermentation 10, no. 1: 66. https://doi.org/10.3390/fermentation10010066
APA StyleZhao, X., Song, L., Han, D., Han, P., & Bai, F. (2024). Microbiological and Physicochemical Dynamics in Traditional and Industrial Fermentation Processes of Koumiss. Fermentation, 10(1), 66. https://doi.org/10.3390/fermentation10010066