
Antisense-RNA-Mediated Gene Downregulation in Clostridium pasteurianum


Abstract
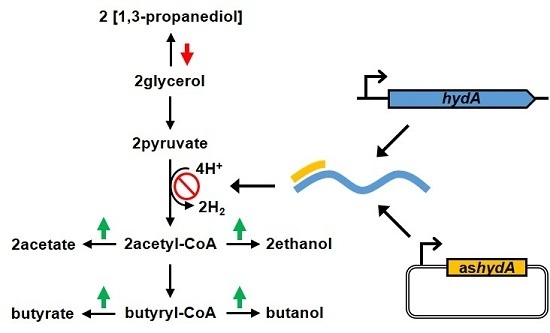
:
1. Introduction
2. Experimental Section
2.1. Bacterial Strains, Cultivation Conditions, and Electrotransformation

{kind=link}
{kind=link}
{kind=link}
| Strains or Plasmids | Relevant Characteristics | Source or Reference |
|---|---|---|
| Strains | ||
| Escherichia coli DH5α | F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG φ80dlacZ∆M15 ∆(lacZYA-argF)U169, hsdR17(rK−mK+), λ− | Lab stock |
| Escherichia coli ER1821 | F− endA1 glnV44 thi-1 relA1? e14−(mcrA−) rfbD1? spoT1? ∆(mcrC-mrr)114::IS10 | Lab stock; New England Biolabs |
| Clostridium pasteurianum ATCC 6013 | Wild-type | American type culture collection |
| Plasmids | ||
| pFnuDIIMKn | E. coli vector expressing the M.FnuDII methyltransferase for plasmid methylation prior to electrotransformation of C. pasteurianum (KnR; p15A ori) | [12] |
| pHT3 | Promoterless E. coli-C. pasteurianum β-galactosidase gene expression reporter vector (ApR; ColE1 ori; EmR; pIM13 ori) | [35] |
| pMTL85141 | E. coli-C. pasteurianum shuttle vector (ColE1 ori; TmR; pIM13 ori) | [36] |
| pSY6catP | E. coli-Clostridium group II intron expression vector (ApR; ColE1 ori; TmR; pIM13 ori) | [12] |
| pHT3catP | Thiamphenicol-resistant derivative of pHT3 | This study; [37] |
| pHT3catP-fdx | Derived by transcriptionally fusing the C. pasteurianum fdx promoter with the lacZ gene of pHT3catP | This study |
| pHT3catP-thl | Derived by transcriptionally fusing the C. pasteurianum thl promoter with the lacZ gene of pHT3catP | This study |
| pHTaslacZ110 | Vector expressing a 110 nt lacZ asRNA from the C. pasteurianum thl promoter | This study |
| pHTashydA | Vector expressing a 175 nt hydA asRNA from the C. pasteurianum thl promoter | This study |
| Oligonucleotide | Sequence (5ʹ–3ʹ) * | |
| Pfdx.NarI.S | CAGAACGGCGCCGAAGATATAAGAAAAAGACTCCCAAAGG | |
| Pfdx.XmaI.AS | ATCATACCCGGGCCATAACTTATTGTATCATGTTTTTAAAC | |
| Pthl.NarI.S | CTGTAGGCGCCGATATAGTCTATAAGCATTTAGATGGAGTTAG | |
| Pthl.XmaI.AS | AAATACCCGGGTCGATTGTTATTTAATTCACAATTTAATTATAACC | |
2.2. DNA Isolation, Manipulation, and Plasmid Construction
2.3. Strain Cultivation
2.4. Analytical Methods
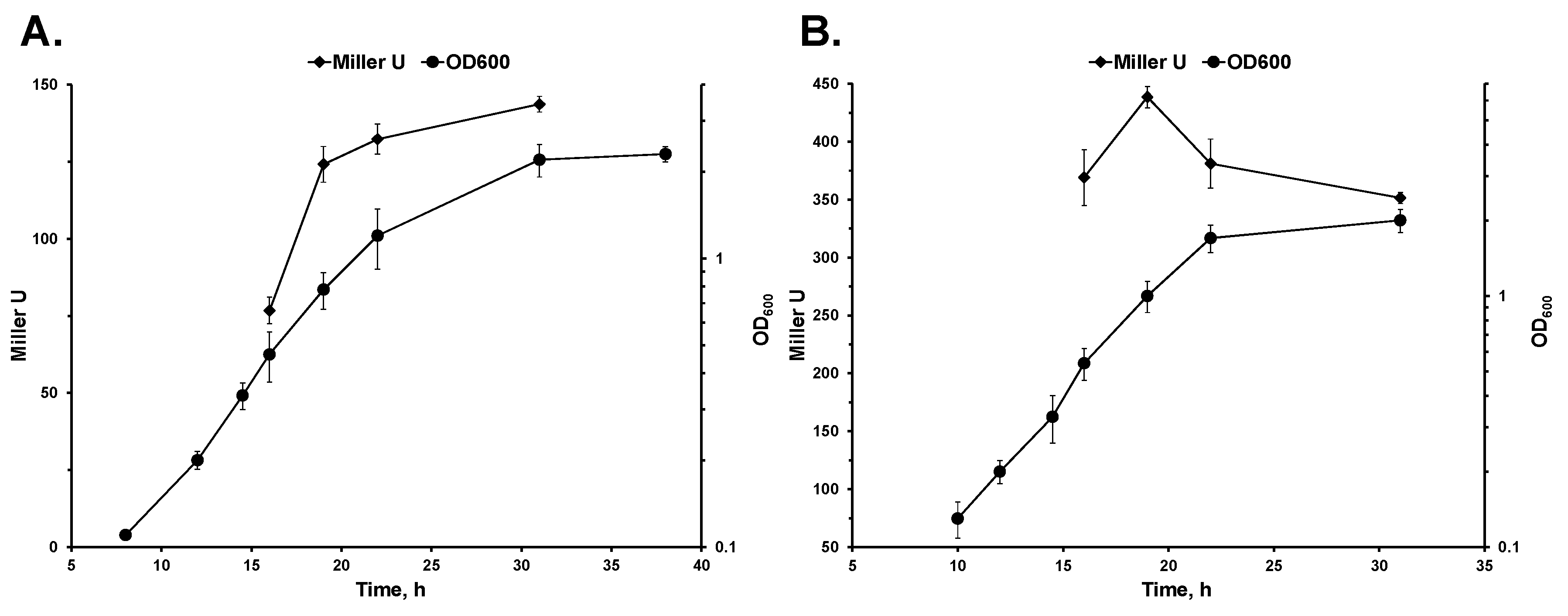
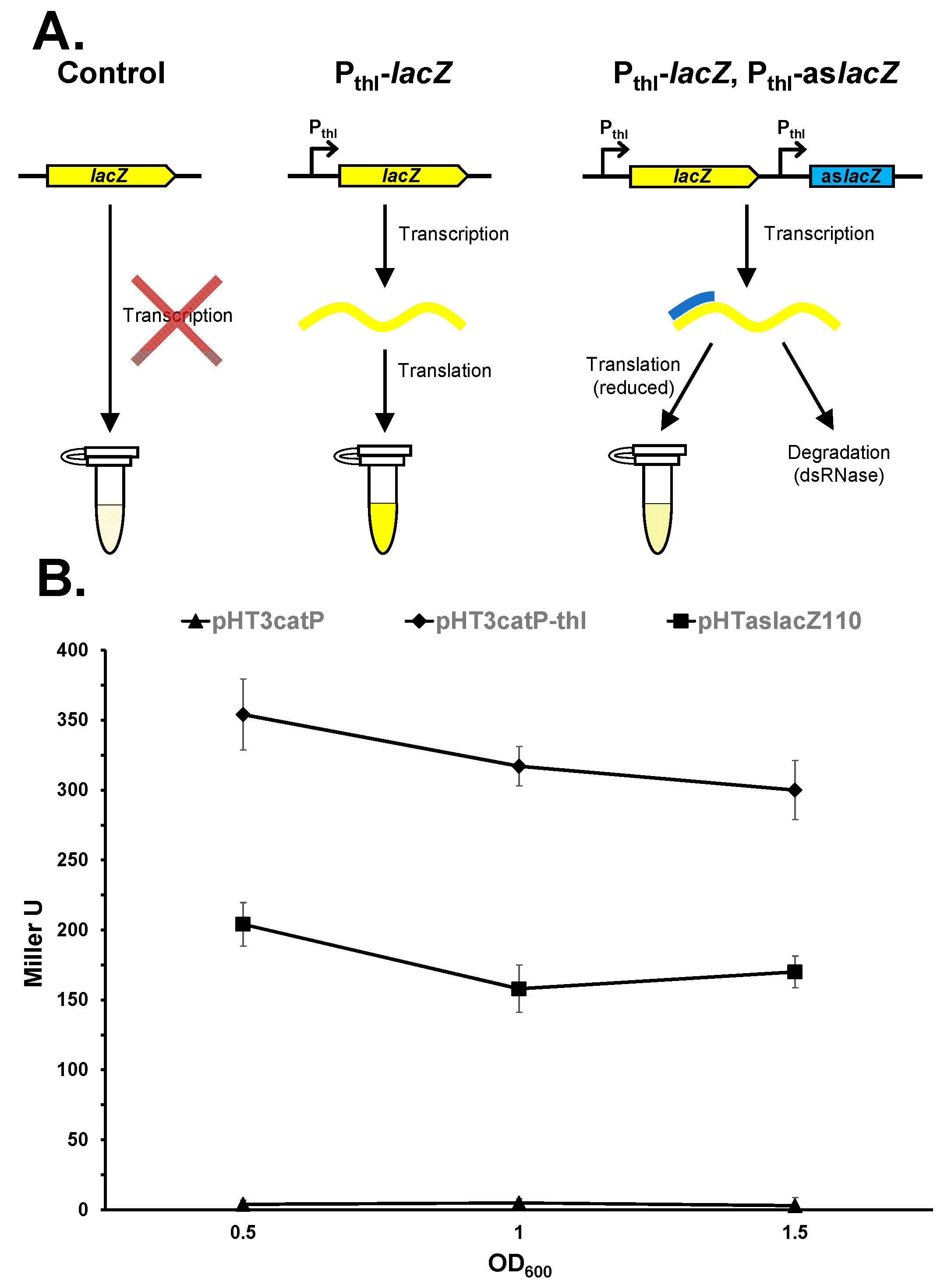
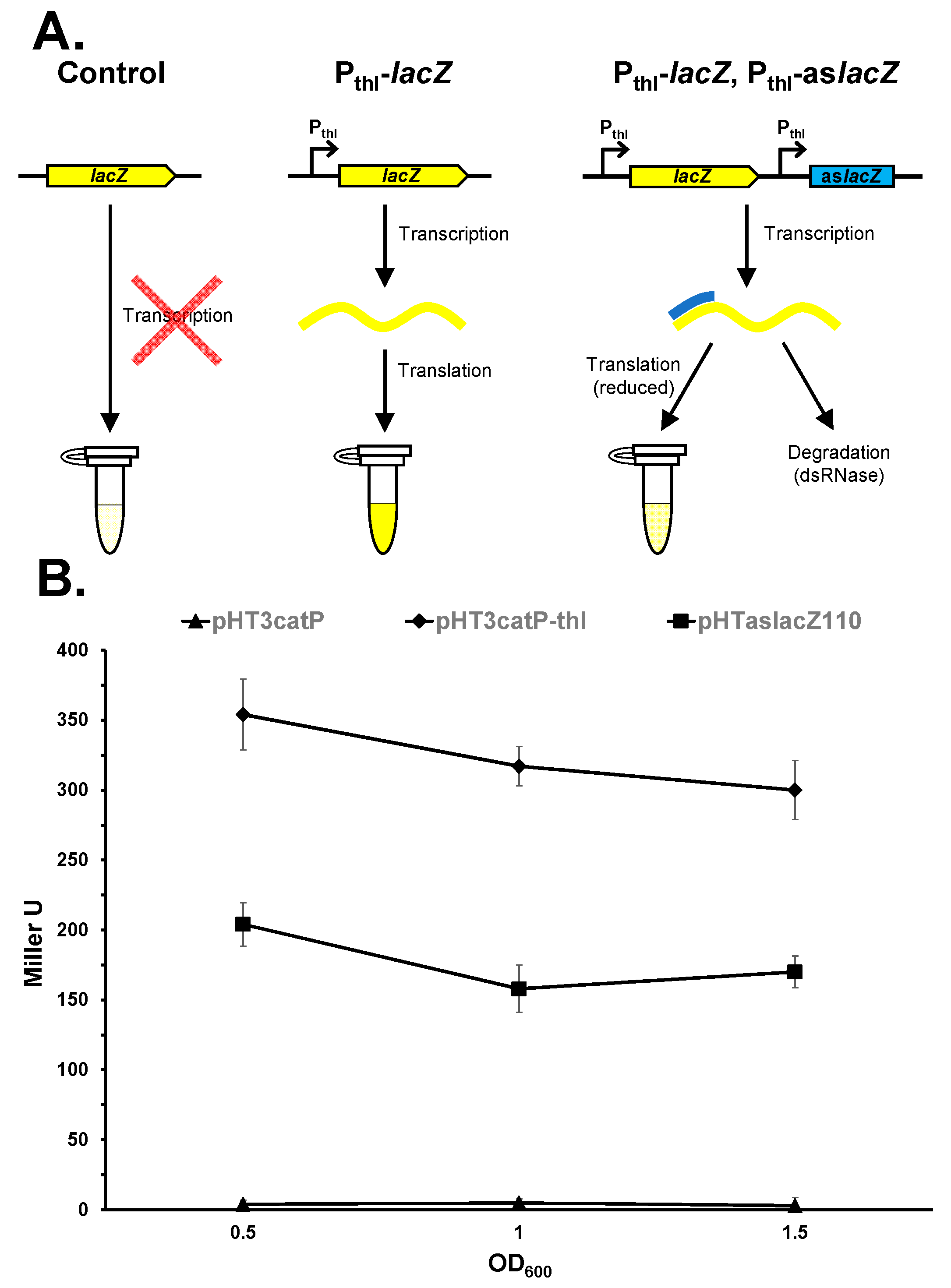
3. Results

| Strain | Culture time (h) | OD600 | Substrate and product titers (g L-1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Glycerol | Acetate | Butyrate | Ethanol | Butanol | 1,3-PDO | Total acids | Total alcohols | |||
| WT (pMTL85141) | 52.5 | 7.6 ± 0.4 | 11.4 ± 0.06 | 0.38 ± 0.0 | 0.15 ± 0.02 | 1.6 ± 0.04 | 8.9 ± 0.3 | 3.3 ± 0.3 | 0.53 ± 0.02 | 13.8 ± 0.05 |
| WT (pHTashydA) | 43.0 | 10.3 ± 0.07 | 12.3 ± 0.1 | 0.90 ± 0.4 | 0.21 ± 0.0 | 2.0 ± 0.1 | 10.0 ± 0.0 | 2.3 ± 0.03 | 1.1 ± 0.4 | 14.4 ± 0.07 |
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ahn, J.H.; Sang, B.I.; Urn, Y. Butanol production from thin stillage using Clostridium pasteurianum. Bioresour. Technol. 2011, 102, 4934–4937. [Google Scholar] [CrossRef] [PubMed]
- Ensen, T.O.; Kvist, T.; Mikkelsen, M.J.; Christensen, P.V.; Westermann, P. Fermentation of crude glycerol from biodiesel production by Clostridium pasteurianum. J. Ind. Microbiol. Biotechnol. 2012, 39, 709–717. [Google Scholar]
- Taconi, K.A.; Venkataramanan, K.P.; Johnson, D.T. Growth and solvent production by Clostridium pasteurianum ATCC (R) 6013 (Tm) utilizing biodiesel-derived crude glycerol as the sole carbon source. Environ. Prog. Sustain. Energy 2009, 28, 100–110. [Google Scholar] [CrossRef]
- Biebl, H. Fermentation of glycerol by Clostridium pasteurianum—Batch and continuous culture studies. J. Ind. Microbiol. Biotechnol. 2001, 27, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Park, J.H.; Jang, S.H.; Nielsen, L.K.; Kim, J.; Jung, K.S. Fermentative butanol production by Clostridia. Biotechnol. Bioeng. 2008, 101, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Pfromm, P.H.; Amanor-Boadu, V.; Nelson, R.; Vadlani, P.; Madl, R. Bio-butanol vs. bio-ethanol: A technical and economic assessment for corn and switchgrass fermented by yeast or Clostridium acetobutylicum. Biomass Bioenergy 2010, 34, 515–524. [Google Scholar] [CrossRef]
- Swana, J.; Yang, Y.; Behnam, M.; Thompson, R. An analysis of net energy production and feedstock availability for biobutanol and bioethanol. Bioresour. Technol. 2011, 102, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.T.; Taconi, K.A. The glycerin glut: Options for the value-added conversion of crude glycerol resulting from biodiesel production. Environ. Prog. 2007, 26, 338–348. [Google Scholar] [CrossRef]
- Yazdani, S.S.; Gonzalez, R. Anaerobic fermentation of glycerol: A path to economic viability for the biofuels industry. Curr. Opin. Biotechnol. 2007, 18, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Van Haandel, A.C.; Catunda, P.F.C. Profitability increase of alcohol distilleries by the rational use of byproducts. Water Sci. Technol. 1994, 29, 117–124. [Google Scholar]
- Johnston, M.; Holloway, T. A global comparison of national biodiesel production potentials. Environ. Sci. Technol. 2007, 41, 7967–7973. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Development of an electrotransformation protocol for genetic manipulation of Clostridium pasteurianum. Biotechnol. Biofuels 2013, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Expansion of the genetic toolkit for metabolic engineering of Clostridium pasteurianum: Chromosomal gene disruption of the endogenous cpaai restriction enzyme. Biotechnol. Biofuels 2014, 7, 163. [Google Scholar] [CrossRef] [PubMed]
- Kolek, J.; Sedlář, K.; Provazník, I.; Patáková, P. Draft genome sequence of Clostridium pasteurianum NRRL B-598, a potential butanol or hydrogen producer. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Utturkar, S.; Brown, S.D.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Improved draft genome sequence of Clostridium pasteurianum strain ATCC 6013 (DSM 525) using a hybrid next-generation sequencing approach. Genome Announc. 2014, 2, e00790-14. [Google Scholar] [CrossRef] [PubMed]
- Rappert, S.; Song, L.; Sabra, W.; Wang, W.; Zeng, A.P. Draft genome sequence of type strain Clostridium pasteurianum DSM 525 (ATCC 6013), a promising producer of chemicals and fuels. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Poehlein, A.; Grosse-Honebrink, A.; Zhang, Y.; Minton, N.P.; Daniel, R. Complete genome sequence of the nitrogen-fixing and solvent-producing Clostridium pasteurianum DSM 525. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Rotta, C.; Poehlein, A.; Schwarz, K.; Mcclure, P.; Daniel, R.; Minton, N.P. Closed genome sequence of Clostridium pasteurianum ATCC 6013. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Liu, X.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Genomic-directed analysis of prophage excision, host defence systems, and central fermentative metabolism in Clostridium pasteurianum. unpublished work. 2015. [Google Scholar]
- Pyne, M.E.; Bruder, M.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Technical guide for genetic advancement of underdeveloped and intractable Clostridium. Biotechnol. Adv. 2014, 32, 623–641. [Google Scholar] [CrossRef] [PubMed]
- Papoutsakis, E.T. Engineering solventogenic Clostridia. Curr. Opin. Biotechnol. 2008, 19, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Lee, J.; Lee, S.Y. Genome engineering and gene expression control for bacterial strain development. Biotechnol. J. 2015, 10, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Cooksley, C.M.; Zhang, Y.; Wang, H.Z.; Redl, S.; Winzer, K.; Minton, N.P. Targeted mutagenesis of the Clostridium acetobutylicum acetone-butanol-ethanol fermentation pathway. Metab. Eng. 2012, 14, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.P.; Papoutsakis, E.T. Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl. Environ. Microbiol. 1999, 65, 936–945. [Google Scholar] [PubMed]
- Tummala, S.B.; Welker, N.E.; Papoutsakis, E.T. Design of antisense rna constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum. J. Bacteriol. 2003, 185, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Gheshlaghi, R.; Scharer, J.M.; Moo-Young, M.; Chou, C.P. Metabolic pathways of Clostridia for producing butanol. Biotechnol. Adv. 2009, 27, 764–781. [Google Scholar] [CrossRef] [PubMed]
- Dabrock, B.; Bahl, H.; Gottschalk, G. Parameters affecting solvent production by Clostridium pasteurianum. Appl. Environ. Microbiol. 1992, 58, 1233–1239. [Google Scholar] [PubMed]
- Simons, R.; Kleckner, N. Biological regulation by antisense RNA in prokaryotes. Annu. Rev. Genet. 1988, 22, 567–600. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.G.H.; Simons, R.W. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 1994, 48, 713–742. [Google Scholar] [CrossRef] [PubMed]
- Tummala, S.B.; Junne, S.G.; Papoutsakis, E.T. Antisense RNA downregulation of coenzyme a transferase combined with alcohol-aldehyde dehydrogenase overexpression leads to predominantly alcohologenic Clostridium acetobutylicum fermentations. J. Bacteriol. 2003, 185, 3644–3653. [Google Scholar] [CrossRef] [PubMed]
- Perret, S.; Maamar, H.; Belaich, J.P.; Tardif, C. Use of antisense rna to modify the composition of cellulosomes produced by Clostridium cellulolyticum. Mol. Microbiol. 2004, 51, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.I.; Kosaka, T.; Hirakawa, H.; Matsuura, K.; Yoshino, S.; Furukawa, K. Metabolic engineering for solvent productivity by downregulation of the hydrogenase gene cluster Hupcba in Clostridium saccharoperbutylacetonicum Strain N1-4. Appl. Microbiol. Biotechnol. 2008, 78, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, H.; Young, M.; Kashket, E.R. Butanol tolerance of Clostridium Beijerinckii NCIMB 8052 associated with down-regulation of GLDA by antisense RNA. J. Mol. Microbiol. Biotechnol. 2000, 2, 87–93. [Google Scholar] [PubMed]
- Sillers, R.; A-Hinai, M.A.; Papoutsakis, E.T. Aldehyde-alcohol dehydrogenase and/or thiolase overexpression coupled with coa transferase downregulation lead to higher alcohol titers and selectivity in Clostridium acetobutylicum fermentations. Biotechnol. Bioeng. 2009, 102, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Tummala, S.B.; Welker, N.E.; Papoutsakis, E.T. Development and characterization of a gene expression reporter system for Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 1999, 65, 3793–3799. [Google Scholar] [PubMed]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Minton, N.P. A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods 2009, 78, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bruder, M.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Elimination of carbon catabolite repression in Clostridium acetobutylicum—A journey toward simultaneous use of xylose and glucose. Appl. Microbiol. Biotechnol. 2015, 99, 7579–7588. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Miller, J.H. Experiments in Molecular Genetics; Gold Spring Harbor Laboratory: Gold Spring Harbor, NY, USA, 1972. [Google Scholar]
- Moon, C.; Lee, C.H.; Sang, B.I.; Um, Y. Optimization of medium compositions favoring butanol and 1,3-propanediol production from glycerol by Clostridium pasteurianum. Bioresour. Technol. 2011, 102, 10561–10568. [Google Scholar] [CrossRef] [PubMed]
- Graves, M.; Rabinowitz, J. In vivo and in vitro transcription of the Clostridium pasteurianum ferredoxin gene. evidence for “extended” promoter elements in gram-positive organisms. J. Biol. Chem. 1986, 261, 11409–11415. [Google Scholar] [PubMed]
- Meng, Y.; Li, J.L. Cloning, expression and characterization of a thiolase gene from Clostridium pasteurianum. Biotechnol. Lett. 2006, 28, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Guedon, E.; Desvaux, M.; Petitdemange, H. Improvement of cellulolytic properties of Clostridium cellulolyticum by metabolic engineering. Appl. Environ. Microbiol. 2002, 68, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Carter, G.P.; Minton, N.P. The clostron: A universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 2007, 70, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J. Preparation and properties of clostridial ferredoxins. Methods Enzymol. 1971, 24, 431–446. [Google Scholar]
- Heyndrickx, M.; Devos, P.; Deley, J. Fermentation characteristics of Clostridium pasteurianum LMG 3285 grown on glucose and mannitol. J. Appl. Bacteriol. 1991, 70, 52–58. [Google Scholar] [CrossRef]
- Higashide, W.; Li, Y.C.; Yang, Y.F.; Liao, J.C. Metabolic engineering of Clostridium cellulolyticum for production of isobutanol from cellulose. Appl. Environ. Microbiol. 2011, 77, 2727–2733. [Google Scholar] [CrossRef] [PubMed]
- Mermelstein, L.D.; Welker, N.E.; Bennett, G.N.; Papoutsakis, E.T. Expression of cloned homologous fermentative genes in Clostridium acetobutylicum ATCC 824. Bio-Technology 1992, 10, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Bellows, P.; Datta, R.; Zeikus, J.G. Control of carbon and electron flow in Clostridium acetobutylicum fermentations—Utilization of carbon monoxide to inhibit hydrogen production and to enhance butanol yields. Appl. Environ. Microbiol. 1984, 48, 764–770. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyne, M.E.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Antisense-RNA-Mediated Gene Downregulation in Clostridium pasteurianum. Fermentation 2015, 1, 113-126. https://doi.org/10.3390/fermentation1010113
Pyne ME, Moo-Young M, Chung DA, Chou CP. Antisense-RNA-Mediated Gene Downregulation in Clostridium pasteurianum. Fermentation. 2015; 1(1):113-126. https://doi.org/10.3390/fermentation1010113
Chicago/Turabian StylePyne, Michael E., Murray Moo-Young, Duane A. Chung, and C. Perry Chou. 2015. "Antisense-RNA-Mediated Gene Downregulation in Clostridium pasteurianum" Fermentation 1, no. 1: 113-126. https://doi.org/10.3390/fermentation1010113