Preparation of Synthesis Gas from CO2 for Fischer–Tropsch Synthesis—Comparison of Alternative Process Configurations



Abstract
1. Introduction
2. Technology Overview
2.1. Water Electrolysis
2.2. Synthesis Gas Preparation
2.3. Fischer–Tropsch Synthesis
3. Materials and Methods
3.1. Plant Configurations
- Allothermal from combustion;
- Autothermal from partial oxidation;
- Autothermal from electric resistance.
- once-through design (i.e., placement outside the FT recycle loop);
- recycle design (i.e., placement inside the FT recycle loop).
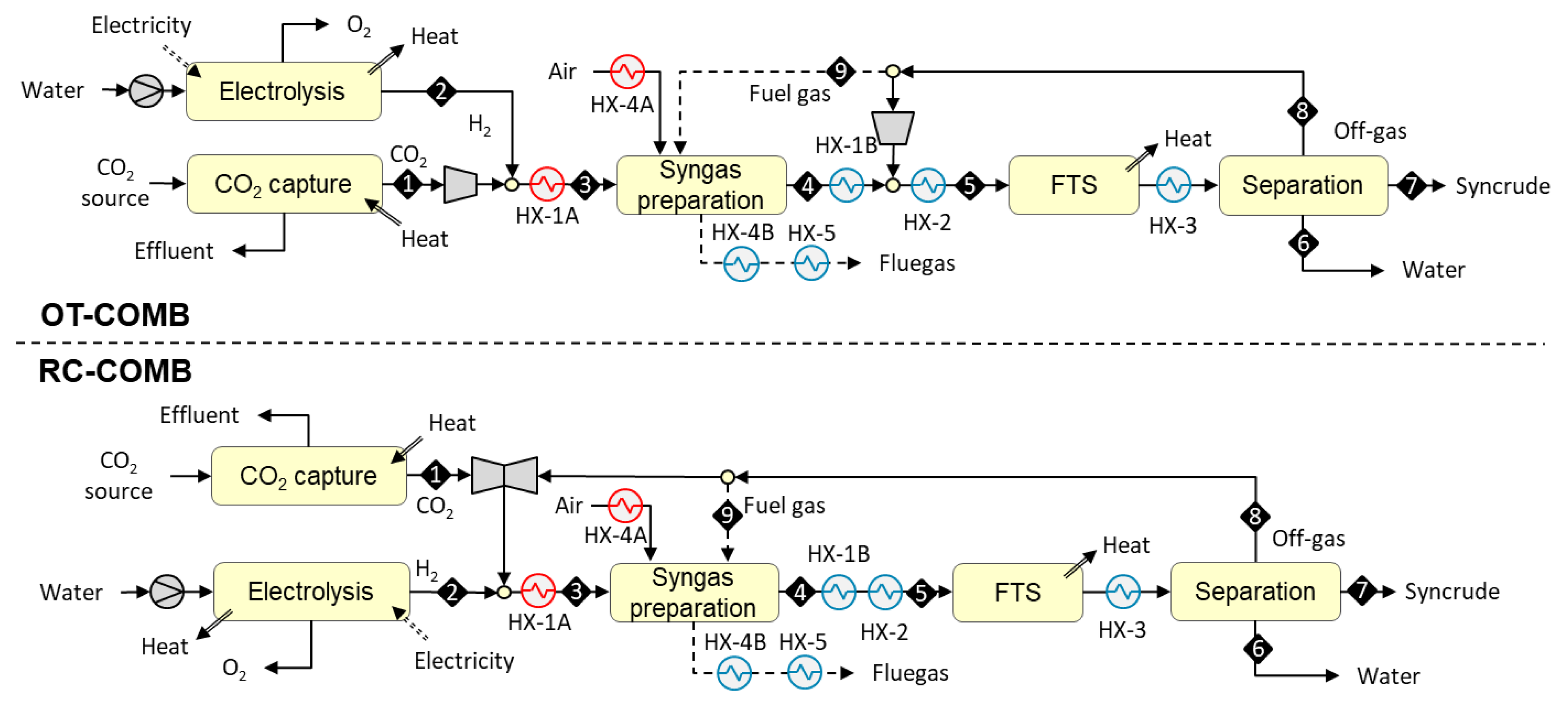
3.1.1. Designs Based on Allothermal Heating from Combustion (OT-COMB and RC-COMB)
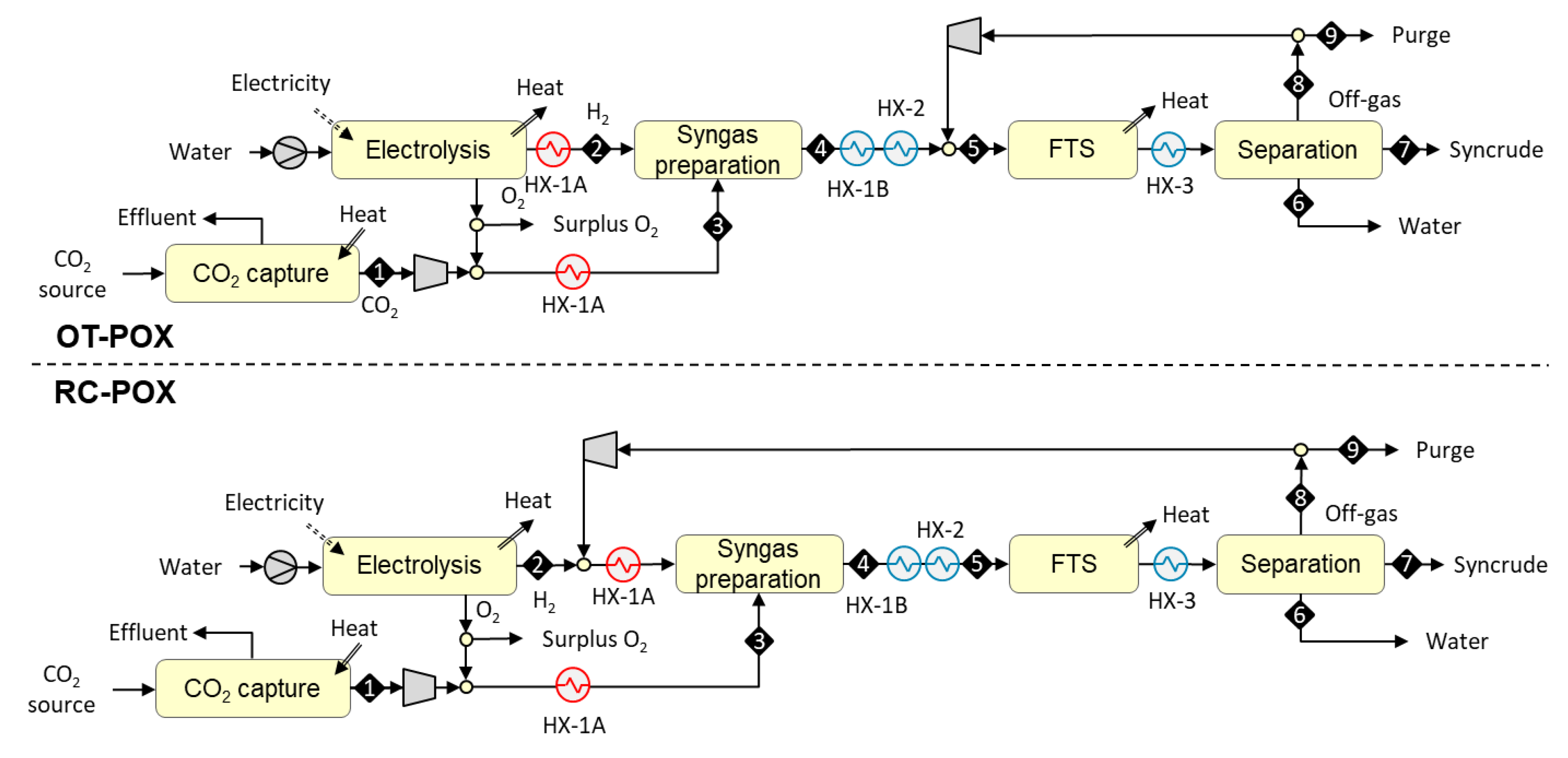
3.1.2. Designs Based on Autothermal Heating from Partial Oxidation (OT-POX and RC-POX)
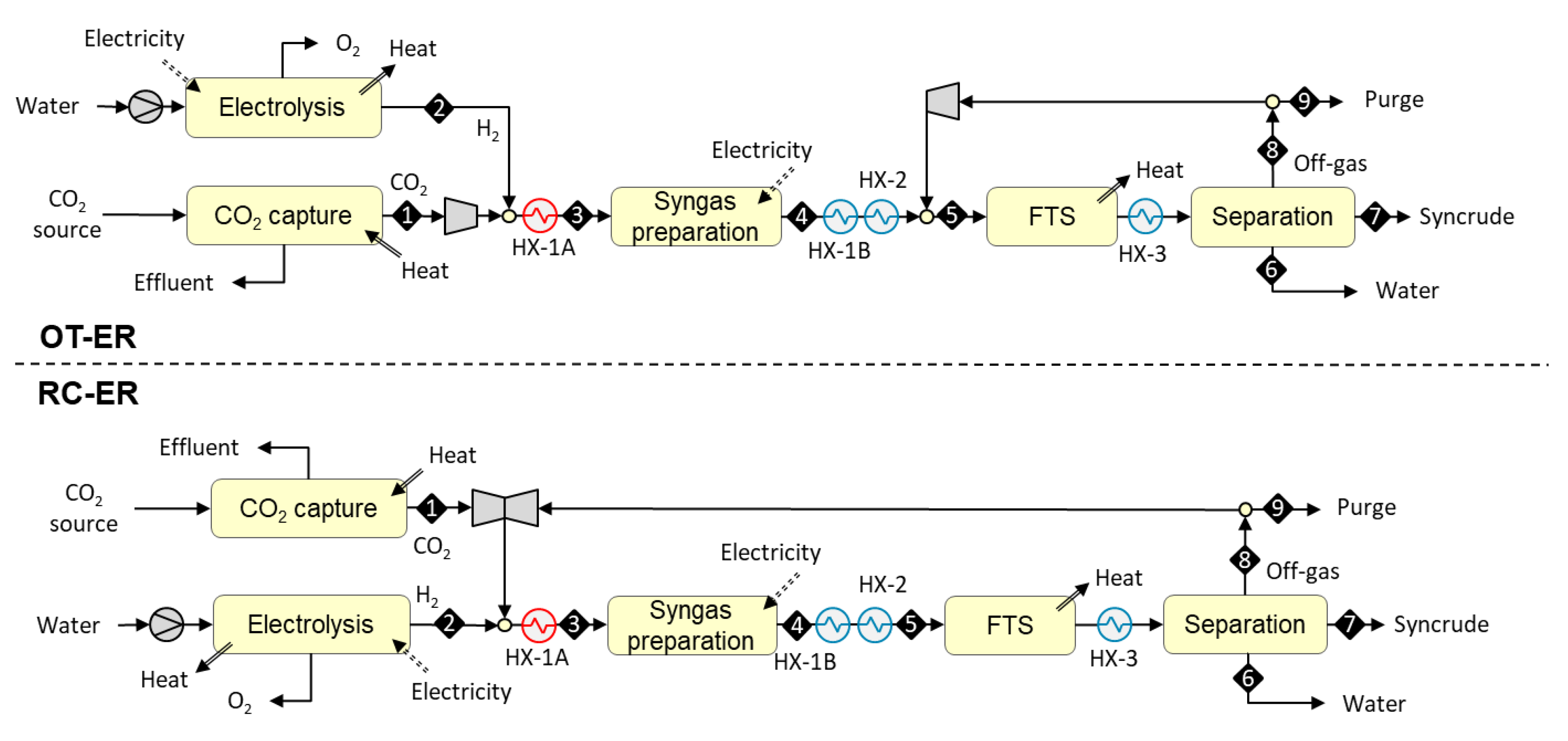
3.1.3. Designs Based on Autothermal Heating from Electric Resistance (OT-ER and RC-ER)
3.2. Evaluation Metrics and Design Parameters
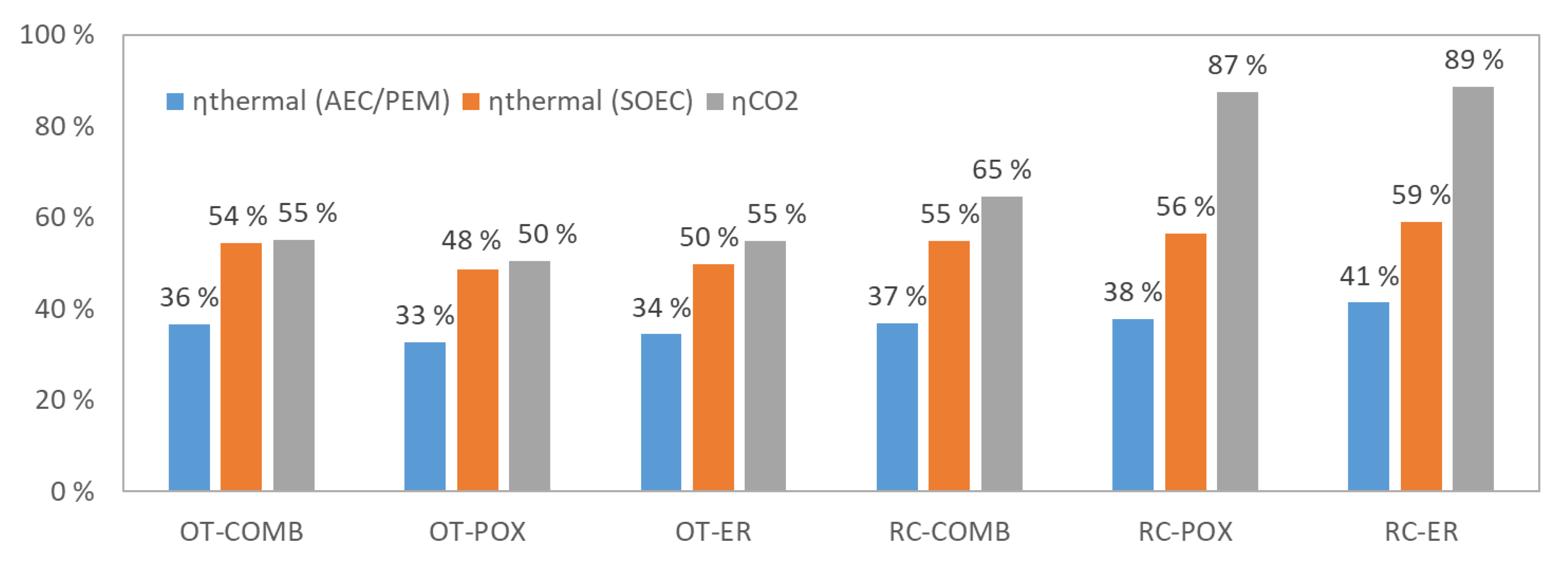
4. Results and Discussion
4.1. Mass and Energy Balances
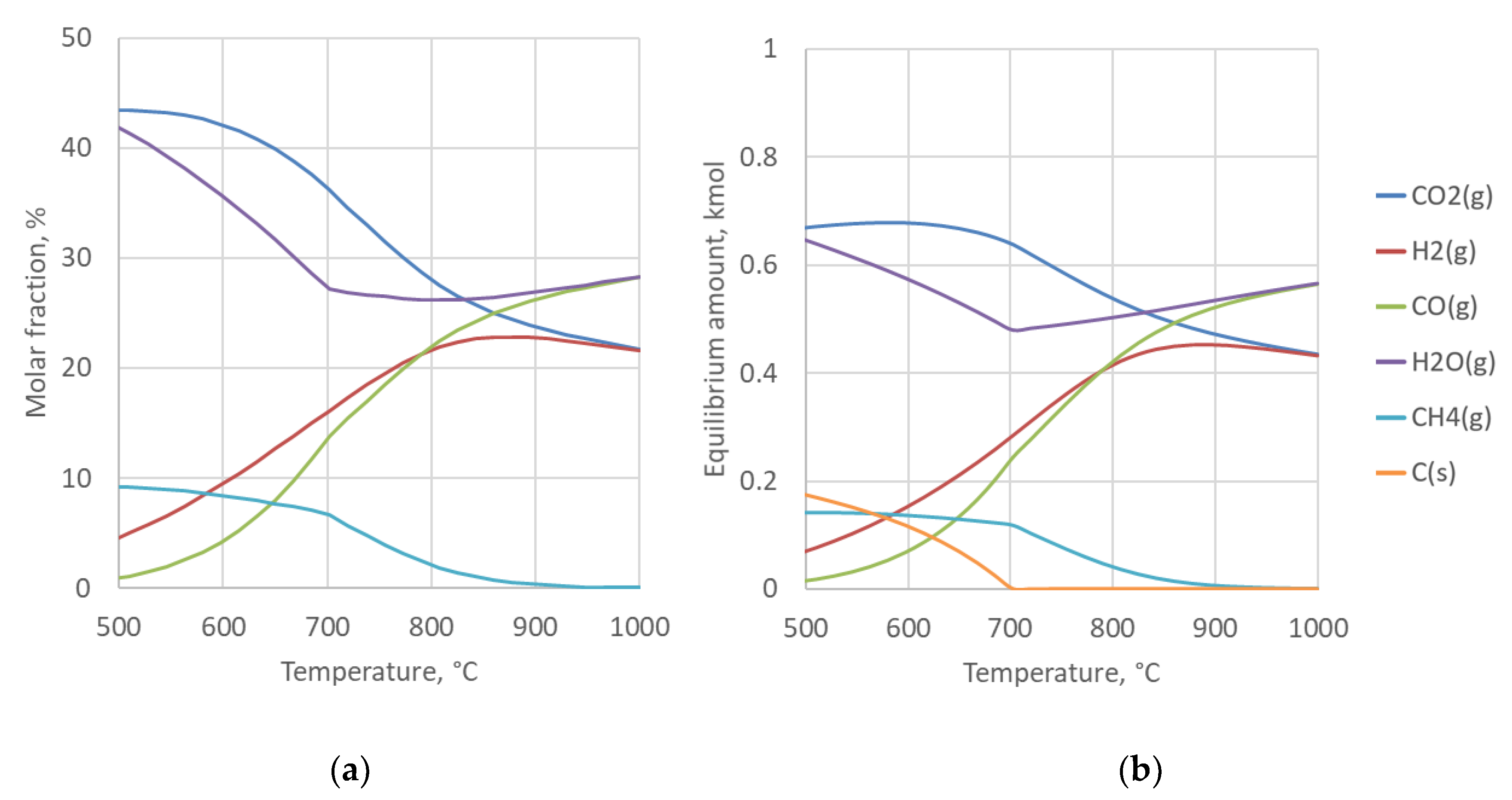
4.2. Sensitivity to Syngas Preparation Temperature
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stream | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature | C | 113 | 25 | 550 | 900 | 220 | 40 | 40 | 40 | 40 |
| Pressure | bar | 20 | 20 | 19.9 | 19.4 | 19.2 | 18.5 | 18.5 | 18.5 | 18.9 |
| Mole Flows | kmol/hr | 100.0 | 229.2 | 329.2 | 317.2 | 707.3 | 140.4 | 3.6 | 437.3 | 47.2 |
| Mole Fractions | ||||||||||
| CO | 0.206 | 0.119 | 0.048 | 0.048 | ||||||
| CO2 | 1.000 | 0.304 | 0.090 | 0.376 | 0.608 | 0.608 | ||||
| H2 | 1.000 | 0.696 | 0.441 | 0.246 | 0.088 | 0.088 | ||||
| N2 | ||||||||||
| CH4 | 0.019 | 0.138 | 0.234 | 0.234 | ||||||
| H2O | 0.000 | 0.244 | 0.109 | 1.000 | ||||||
| O2 | ||||||||||
| C2H6 | 0.004 | 0.008 | 0.008 | |||||||
| C3H8 | 0.004 | 0.007 | 0.007 | |||||||
| C4H10 | 0.004 | 0.007 | 0.007 | |||||||
| C5+ | 1.000 | |||||||||
| Mass Flows | kg/hr | 4401 | 462 | 4863 | 4863 | 17,725 | 2529 | 779 | 14,418 | 1556 |
| (LHV) Net heating value | MJ/kg | 0 | 119.96 | 11.40 | 11.75 | 9.07 | 0 | 43.89 | 8.06 | 8.06 |
| Duty | MW | 0.0 | 15.4 | 15.4 | 15.9 | 44.7 | 0.0 | 9.5 | 32.3 | 3.5 |
| Stream | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature | C | 105 | 25 | 152 | 900 | 220 | 40 | 40 | 40 | 40 |
| Pressure | bar | 20 | 20 | 19.9 | 19.4 | 19.2 | 18.5 | 18.5 | 18.5 | 18.5 |
| Mole Flows | kmol/hr | 100.0 | 234.1 | 116.3 | 327.8 | 689.9 | 156.7 | 3.3 | 414.5 | 52.4 |
| Mole Fractions | ||||||||||
| CO | 0.183 | 0.112 | 0.046 | 0.046 | ||||||
| CO2 | 1.000 | 0.860 | 0.112 | 0.421 | 0.701 | 0.701 | ||||
| H2 | 1.000 | 0.393 | 0.233 | 0.088 | 0.088 | |||||
| N2 | ||||||||||
| CH4 | 0.010 | 0.082 | 0.148 | 0.148 | ||||||
| H2O | 0.302 | 0.144 | 1.000 | |||||||
| O2 | 0.140 | |||||||||
| C2H6 | 0.003 | 0.006 | 0.006 | |||||||
| C3H8 | 0.003 | 0.006 | 0.006 | |||||||
| C4H10 | 0.003 | 0.005 | 0.005 | |||||||
| C5+ | 1.000 | |||||||||
| Mass Flows | kg/hr | 4401 | 472 | 4923 | 5395 | 18,226 | 2824 | 713 | 14,690 | 1858 |
| (LHV) Net heating value | MJ/kg | 0 | 119.96 | 0 | 9.39 | 6.52 | 0 | 43.89 | 5.31 | 5.31 |
| Duty | MW | 0.0 | 15.7 | 0.0 | 14.1 | 33.0 | 0.0 | 8.7 | 21.7 | 2.7 |
| Stream | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature | C | 105 | 25 | 550 | 900 | 220 | 40 | 40 | 40 | 40 |
| Pressure | bar | 20 | 20 | 19.9 | 19.4 | 19.2 | 18.5 | 18.5 | 18.5 | 18.5 |
| Mole Flows | kmol/hr | 100.0 | 229.5 | 329.5 | 317.6 | 739.2 | 140.6 | 3.6 | 468.5 | 46.9 |
| Mole Fractions | ||||||||||
| CO | 0.206 | 0.114 | 0.045 | 0.045 | ||||||
| CO2 | 1.000 | 0.303 | 0.090 | 0.387 | 0.611 | 0.611 | ||||
| H2 | 1.000 | 0.697 | 0.442 | 0.239 | 0.086 | 0.086 | ||||
| N2 | ||||||||||
| CH4 | 0.019 | 0.143 | 0.236 | 0.236 | ||||||
| H2O | 0.243 | 0.105 | 1.000 | |||||||
| O2 | ||||||||||
| C2H6 | 0.004 | 0.008 | 0.008 | |||||||
| C3H8 | 0.004 | 0.007 | 0.007 | |||||||
| C4H10 | 0.004 | 0.007 | 0.007 | |||||||
| C5+ | 1.000 | |||||||||
| Mass Flows | kg/hr | 4401 | 463 | 4864 | 4864 | 18,802 | 2533 | 782 | 15,487 | 1549 |
| (LHV) Net heating value | MJ/kg | 0 | 119.96 | 11.41 | 11.76 | 9.01 | 0 | 43.89 | 8.05 | 8.05 |
| Duty | MW | 0.0 | 15.4 | 15.4 | 15.9 | 47.1 | 0.0 | 9.5 | 34.6 | 3.5 |
| Stream | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature | C | 113 | 25 | 550 | 900 | 220 | 40 | 40 | 40 | 40 |
| Pressure | bar | 20 | 20 | 19.9 | 19.4 | 19.2 | 18.5 | 18.5 | 18.5 | 18.5 |
| Mole Flows | kmol/hr | 100.0 | 270.4 | 441.8 | 440.2 | 440.2 | 160.6 | 4.2 | 127.4 | 55.9 |
| Mole Fractions | ||||||||||
| CO | 0.031 | 0.224 | 0.224 | 0.194 | 0.194 | |||||
| CO2 | 1.000 | 0.268 | 0.074 | 0.074 | 0.256 | 0.256 | ||||
| H2 | 1.000 | 0.674 | 0.474 | 0.474 | 0.386 | 0.386 | ||||
| N2 | ||||||||||
| CH4 | 0.025 | 0.032 | 0.032 | 0.156 | 0.156 | |||||
| H2O | 0.197 | 0.197 | 1.000 | |||||||
| O2 | ||||||||||
| C2H6 | 0.001 | 0.003 | 0.003 | |||||||
| C3H8 | 0.000 | 0.003 | 0.003 | |||||||
| C4H10 | 0.000 | 0.003 | 0.003 | |||||||
| C5+ | 1.000 | |||||||||
| Mass Flows | kg/hr | 4401 | 545 | 6400 | 6400 | 6400 | 2892 | 915 | 2593 | 1139 |
| (LHV) Net heating value | MJ/kg | 0 | 119.96 | 13.47 | 13.98 | 13.98 | 0 | 43.89 | 14.32 | 14.32 |
| Duty | MW | 0.0 | 18.2 | 23.9 | 24.9 | 24.9 | 0.0 | 11.2 | 10.3 | 4.5 |
| Stream | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature | C | 105 | 25 | 188 | 900 | 220 | 40 | 40 | 40 | 40 |
| Pressure | bar | 20 | 20 | 19.9 | 19.4 | 19.2 | 18.5 | 18.5 | 18.5 | 18.5 |
| Mole Flows | kmol/hr | 100.0 | 352.5 | 131.7 | 628.3 | 628.3 | 247.1 | 5.6 | 176.1 | 19.9 |
| Mole Fractions | ||||||||||
| CO | 0.212 | 0.212 | 0.189 | 0.189 | ||||||
| CO2 | 1.000 | 0.759 | 0.089 | 0.089 | 0.317 | 0.317 | ||||
| H2 | 1.000 | 0.445 | 0.445 | 0.367 | 0.367 | |||||
| N2 | ||||||||||
| CH4 | 0.021 | 0.021 | 0.119 | 0.119 | ||||||
| H2O | 0.234 | 0.234 | 1.000 | |||||||
| O2 | 0.241 | |||||||||
| C2H6 | 0.003 | 0.003 | ||||||||
| C3H8 | 0.003 | 0.003 | ||||||||
| C4H10 | 0.003 | 0.003 | ||||||||
| C5+ | 1.000 | |||||||||
| Mass Flows | kg/hr | 4401 | 711 | 5415 | 9603 | 9603 | 4451 | 1233 | 3920 | 443 |
| (LHV) Net heating value | MJ/kg | 0 | 119.96 | 0 | 12.03 | 12.03 | 0 | 43.89 | 11.48 | 11.48 |
| Duty | MW | 0.0 | 23.7 | 0.0 | 32.1 | 32.1 | 0.0 | 15.0 | 12.5 | 1.4 |
| Stream | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Temperature | C | 105 | 25 | 550 | 900 | 220 | 40 | 40 | 40 | 40 |
| Pressure | bar | 20 | 20 | 19.9 | 19.4 | 19.2 | 18.5 | 18.5 | 18.5 | 18.5 |
| Mole Flows | kmol/hr | 100.0 | 297.0 | 537.7 | 555.4 | 555.4 | 189.1 | 5.7 | 157.4 | 16.7 |
| Mole Fractions | ||||||||||
| CO | 0.056 | 0.244 | 0.244 | 0.215 | 0.215 | |||||
| CO2 | 1.000 | 0.244 | 0.062 | 0.062 | 0.220 | 0.220 | ||||
| H2 | 1.000 | 0.641 | 0.489 | 0.489 | 0.338 | 0.338 | ||||
| N2 | ||||||||||
| CH4 | 0.057 | 0.047 | 0.047 | 0.217 | 0.217 | |||||
| H2O | 0.158 | 0.158 | 1.000 | |||||||
| O2 | ||||||||||
| C2H6 | 0.001 | 0.004 | 0.004 | |||||||
| C3H8 | 0.001 | 0.003 | 0.003 | |||||||
| C4H10 | 0.001 | 0.003 | 0.003 | |||||||
| C5+ | 1.000 | |||||||||
| Mass Flows | kg/hr | 4401 | 599 | 7860 | 7860 | 7860 | 3406 | 1255 | 3198 | 338 |
| (LHV) Net heating value | MJ/kg | 0 | 119.96 | 15.17 | 15.89 | 15.89 | 0 | 43.89 | 16.59 | 16.59 |
| Duty | MW | 0.0 | 19.9 | 33.1 | 34.7 | 34.7 | 0.0 | 15.3 | 14.7 | 1.6 |
References
- Sims, R.; Schaeffer, R.; Creutzig, F.; Cruz-Núñez, X.; D’Agosto, M.; Dimitriu, D.; Meza, M.J.F.; Fulton, L.; Kobayashi, S.; Lah, O.; et al. 2014: Transport. In Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Edenhofer, O., Pichs-Madruga, R., Sokona, Y., Farahani, E., Kadner, S., Seyboth, K., Adler, A., Baum, I., Brunner, S., Eickemeier, P., et al., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2014. [Google Scholar]
- Hannula, I.; Reiner, D.M. The role of carbon-neutral synthetic fuels and battery electric vehicles in a sustainable transport system. Joule 2019, 3, 2390–2402. [Google Scholar] [CrossRef]
- Tracking Clean Energy Progress—Topics—IEA. Available online: www.iea.org/tcep (accessed on 16 June 2020).
- Mohr, A.; Raman, S. Lessons from first generation biofuels and implications for the sustainability appraisal of second generation biofuels. Energy Policy 2013, 63, 114–122. [Google Scholar] [CrossRef] [PubMed]
- International Energy Agency. Delivering Sustainable Bioenergy, Technology Roadmap, OECD, Paris; IEA: Paris, France, 2017. [Google Scholar]
- Steinberg, M. Synthetic carbonaceous fuels and feedstocks from oxides of carbon and nuclear power. Fuel 1978, 57, 460–468. [Google Scholar] [CrossRef]
- Zeman, F.S.; Keith, D.W. Carbon neutral hydrocarbons. Philos. Trans. R. Soc. A 2008, 366, 3901–3918. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, I.; Garcia-Gutiérrez, P.; Elder, R.H.; Cuéllar-Franca, R.M.; Azapagic, A.; Allen, R.W.K. Carbon dioxide utilisation for production of transport fuels: Process and economic analysis. Energy Environ. Sci. 2015, 8, 1775–1789. [Google Scholar] [CrossRef]
- Abanades, J.C.; Rubin, E.S.; Mazzotti, M.; Herzog, H.J. On the climate change mitigation potential of CO2 conversion to fuels. Energy Environ. Sci. 2017, 10, 2491–2499. [Google Scholar] [CrossRef]
- Blanco, H.; Faaij, A. A review of the role of storage in energy systems with a focus on Power to Gas and long-term storage. Renew. Sustain. Energ. Rev. 2018, 81, 1049–1086. [Google Scholar] [CrossRef]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What should we make with CO2 and how can we make it? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Quaranta, E. State of the art and perspectives in catalytic processes for CO2 conversion into chemicals and fuels: The distinctive contribution of chemical catalysis and biotechnology. J. Catal. 2016, 343, 2–45. [Google Scholar] [CrossRef]
- Bruhn, T.; Naims, H.; Olfe-Krautlein, B. Separating the debate on CO2 utilisation from carbon capture and storage. Environ. Sci. Policy 2016, 60, 38–43. [Google Scholar] [CrossRef]
- Brynolf, S.; Taljegard, M.; Grahn, M.; Hansson, J. Electrofuels for the transport sector: A review of production costs. Renew. Sustain. Energy Rev. 2018, 81, 1887–1905. [Google Scholar] [CrossRef]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Wulf, C.; Linßen, J.; Zapp, P. Review of Power-to-Gas Projects in Europe. Energy Procedia 2018, 155, 367–378. [Google Scholar] [CrossRef]
- CRI—Carbon Recycling International. Available online: www.carbonrecycling.is (accessed on 16 June 2020).
- Otten, R. The First Industrial PtG Plant—Audi E-Gas as Driver for the Energy Turnaround. CEDEC Gas Day Verona Italy. 2014. Available online: http://bit.ly/2zTg5kW (accessed on 16 June 2020).
- Soletair—Sustainable Technologies. Available online: https://soletair.fi (accessed on 16 June 2020).
- Vázquez, F.V.; Koponen, J.; Ruuskanen, V.; Bajamundi, C.; Kosonen, A.; Simell, P.; Ahola, J.; Frilund, C.; Elfving, J.; Reinikainen, M.; et al. Power-to-X technology using renewable electricity and carbon dioxide from ambient air: SOLETAIR proof-of-concept and improved process concept. J. CO2 Util. 2018, 28, 235–246. [Google Scholar] [CrossRef]
- Sunfire—Energy Everywhere. Available online: https://www.sunfire.de/en/ (accessed on 16 June 2020).
- Bertuccioli, L.; Chan, A.; Hart, D.; Lehner, F.; Madden, B.; Standen, E. Study on Development of Water Electrolysis in the EU; FCH JU: Bruxelles, Belgium, 2014. [Google Scholar]
- Buttler, A.; Spliethoff, H. Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: A review. Renew. Sustain. Energy Rev. 2018, 82, 2440–2454. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Kaiser, P.; Unde, R.; Kern, C.; Jess, A. Production of Liquid Hydrocarbons with CO2 as Carbon Source based on Reverse Water—Gas Shift and Fischer—Tropsch Synthesis. Chem. Ing. Tech. 2013, 85, 489–499. [Google Scholar] [CrossRef]
- Hansen, J.B.; Christiansen, N.; Nielsen, J.U. Production of Sustainable Fuels by Means of Solid Oxide Electrolysis. ECS Trans. 2011, 35, 2941–2948. [Google Scholar] [CrossRef]
- Simell, P.; Hannula, I.; Tuomi, S.; Nieminen, M.; Kurkela, E.; Hiltunen, I.; Kaisalo, N.; Kihlman, J. Clean syngas from biomass—Process development and concept assessment. Biomass Convers. Biorefinery 2014, 4, 357–370. [Google Scholar] [CrossRef]
- Sie, S.; Krishna, R. Fundamentals and selection of advanced Fischer-Tropsch reactors. Appl. Catal. A Gen. 1999, 186, 55–70. [Google Scholar] [CrossRef]
- Sie, S.; Senden, M.; Wechem, H.V. Conversion of natural gas to transportation fuels via the shell middle distillate synthesis process (SMDS). Catal. Today 1991, 8, 371–394. [Google Scholar] [CrossRef]
- Anderson, R. Catalysts for the Fischer-Tropsch Synthesis; Van Nostrand-Reinhold: New York, NY, USA, 1956; Volume 4. [Google Scholar]
- De Klerk, A. Fischer-Tropsch fuels refinery design. Energy Environ. Sci. 2011, 4, 1177–1205. [Google Scholar] [CrossRef]
- Eilers, J.; Posthuma, S.; Sie, S. The shell middle distillate synthesis process (smds). Catal. Lett. 1990, 7, 253–269. [Google Scholar] [CrossRef]
- de Klerk, A. Fischer-Tropsch Refining; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- de Klerk, A.; Li, Y.W.; Zennaro, R. Fischer-Tropsch Technology. In Greener Fischer-Tropsch Processes for Fuels and Feedstocks; Maitlis, P.M., de Klerk, A., Eds.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Rytter, E.; Tsakoumis, N.E.; Holmen, A. On the selectivity to higher hydrocarbons in Co-based Fischer-Tropsch synthesis. Catal. Today 2016, 261, 3–16. [Google Scholar] [CrossRef]
- LeViness, S.; Deshmukh, S.R.; Richard, L.A.; Robota, H.J. Velocys Fischer–Tropsch Synthesis Technology—New Advances on State-of-the-Art. Top. Catal. 2014, 57, 518–525. [Google Scholar] [CrossRef]
- Okoye-Chine, C.G.; Moyo, M.; Liu, X.; Hildebrandt, D. A critical review of the impact of water on cobalt-based catalysts in Fischer-Tropsch synthesis. Fuel Process. Technol. 2019, 192, 105–129. [Google Scholar] [CrossRef]
- Wismann, S.T.; Engbæk, J.S.; Vendelbo, S.B.; Bendixen, F.B.; Eriksen, W.L.; Aasberg-Petersen, K.; Frandsen, C.; Chorkendorff, I.; Mortensen, P.M. Electrified methane reforming: A compact approach to greener industrial hydrogen production. Science 2019, 364, 756–759. [Google Scholar] [CrossRef]
- Hannula, I. Co-production of synthetic fuels and district heat from biomass residues, carbon dioxide and electricity: Performance and cost analysis. Biomass Bioenergy 2015, 74, 26–46. [Google Scholar] [CrossRef]
- Hannula, I. Hydrogen enhancement potential of synthetic biofuels manufacture in the European context: A techno-economic assessment. Energy 2016, 104, 199–212. [Google Scholar] [CrossRef]
- Hombach, L.E.; Doré, L.; Heidgen, K.; Maas, H.; Wallington, T.J.; Walther, G. Economic and environmental assessment of current (2015) and future (2030) use of E-fuels in light-duty vehicles in Germany. J. Clean. Prod. 2019, 207, 153–162. [Google Scholar] [CrossRef]






| Allothermal from Combustion | Autothermal from Partial Oxidation | Autothermal from Electric Resistance | |
|---|---|---|---|
| Once-through | OT-COMB | OT-POX | OT-ER |
| Recycle | RC-COMB | RC-POX | RC-ER |
| Item | Design Parameters |
|---|---|
| Electrolyser | H2 and O2 purity 100%, Both delivered at 20 bar and 25 °C. Nominal system efficiency from electricity to hydrogen is 60% (LHV) for AEC/PEM and 90% (LHV) for SOEC. |
| Syngas preparation reactor | Reactors modelled with RGibbs using Redlich-Kwong-Soave equation of state with Boston-Mathias modification (RKS-BM). Both phase and chemical equilibrium calculated. All components considered as products. Toutlet = 900 °C, ∆p = −0.5 bar. Target H2/CO ratio at the FT reactor inlet is 2.0 for all examined configurations. |
| Fischer-Tropsch synthesis | Treaction = 220 °C, Pfeed = 19–20 bar, ∆p =−0.5 bar, Boiling-water reactor using cobalt catalysts modelled with RStoic using Redlich-Kwong-Soave equation of state with Boston-Mathias modification (RKS-BM). Carbon monoxide reacts with hydrogen to form n-paraffins at 0.92 α value, with methane selectivity set to 9%. The per-pass CO conversion is set to 75%. Input H2O, CO2, N2 as well as methane, ethane and longer hydrocarbons are considered as inert. For RC-POX and RC-ER configurations, the amount of recycle is chosen to achieve 98% conversion of hydrogen (2% of hydrogen lost in purge). |
| Heat exchangers | ∆p = −0.1 bar, ∆T min = 15 °C (gas-liq), 30 °C (gas-gas). Heat loss = 1% of heat transferred. |
| Compressors | Stage pressure ratio < 2, ηpolytropic = 0.81, ηdriver = 0.90, ηmechanical = 0.98. Tintercooler = 35 °C, ∆p/pintercooler = 1%. |
| Pumps | ηhydraulic = 0.75, ηdriver = 0.90, ηmechanical = 0.98. |
| CONFIGURATION | OT-COMB | OT-POX | OT-ER | RC-COMB | RC-POX | RC-ER | |
|---|---|---|---|---|---|---|---|
| Feedstocks and intermediates | |||||||
| Captured and concentrated CO2 | kg/h | 1000 | 1000 | 1000 | 1000 | 1000 | 1000 |
| Water for electrolysis | kg/h | 938 | 959 | 936 | 1092 | 1446 | 1216 |
| Electrolytic oxygen | kg/h | 833 | 852 | 831 | 969 | 1285 | 1080 |
| Electrolytic hydrogen | kg/h | 105 | 107 | 105 | 122 | 162 | 136 |
| MW | 3.5 | 3.6 | 3.5 | 4.1 | 5.4 | 4.5 | |
| Products | |||||||
| C5–C12 hydrocarbons | kg/h | 46 | 42 | 46 | 54 | 73 | 74 |
| MW | 0.6 | 0.5 | 0.6 | 0.7 | 0.9 | 0.9 | |
| C13+ hydrocarbons | kg/h | 131 | 120 | 130 | 154 | 208 | 211 |
| MW | 1.6 | 1.5 | 1.6 | 1.9 | 2.5 | 2.6 | |
| Total syncrude (C5+) | kg/h | 177 | 162 | 176 | 208 | 281 | 285 |
| MW | 2.2 | 2.0 | 2.1 | 2.5 | 3.4 | 3.5 | |
| Purge gas | kg/h | 422 | 356 | 97 | 77 | ||
| MW | 0.6 | 0.8 | 0.3 | 0.4 | |||
| Saturated steam | kg/h | 933 | 663 | 762 | 1097 | 1464 | 1500 |
| Condensed water | kg/h | 575 | 642 | 573 | 655 | 1015 | 774 |
| Surplus oxygen | kg/h | 833 | 733 | 831 | 969 | 1053 | 1080 |
| Electricity use | |||||||
| Electrolyser | |||||||
| Low temperature (AEC/PEM) | kW | 5830 | 5961 | 5814 | 6784 | 8989 | 7554 |
| High temperature (SOEC) | kW | 3887 | 3974 | 3876 | 4523 | 5993 | 5036 |
| Syngas preparation | kW | 334 | 774 | ||||
| Main compressor | kW | 74 | 73 | 73 | 77 | 73 | 73 |
| Recycle compressor | kW | 5 | 28 | 26 | 1 | 13 | 11 |
| Combustion air blower | kW | 10 | 12 | ||||
| Water pumps | kW | 1 | 1 | 1 | 2 | 2 | 2 |
| Sum total (AEC/PEM) | kW | 5920 | 6064 | 6249 | 6876 | 9077 | 8415 |
| Sum total (SOEC) | kW | 3976 | 4077 | 4311 | 4615 | 6081 | 5897 |
| Heat recovery | |||||||
| Electrolyser heat (AEC/PEM) | kW | 1846 | 1887 | 1841 | 2148 | 2 846 | 2 392 |
| Syngas cooling | kW | −21 | 274 | 136 | 194 | 418 | 264 |
| Syncrude cooling | kW | 636 | 684 | 610 | 561 | 877 | 671 |
| Flue gas cooling | kW | 404 | 388 | ||||
| FT reaction exotherm | kW | 660 | 469 | 539 | 777 | 1 036 | 1 062 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannula, I.; Kaisalo, N.; Simell, P. Preparation of Synthesis Gas from CO2 for Fischer–Tropsch Synthesis—Comparison of Alternative Process Configurations. C 2020, 6, 55. https://doi.org/10.3390/c6030055
Hannula I, Kaisalo N, Simell P. Preparation of Synthesis Gas from CO2 for Fischer–Tropsch Synthesis—Comparison of Alternative Process Configurations. C. 2020; 6(3):55. https://doi.org/10.3390/c6030055
Chicago/Turabian StyleHannula, Ilkka, Noora Kaisalo, and Pekka Simell. 2020. "Preparation of Synthesis Gas from CO2 for Fischer–Tropsch Synthesis—Comparison of Alternative Process Configurations" C 6, no. 3: 55. https://doi.org/10.3390/c6030055
APA StyleHannula, I., Kaisalo, N., & Simell, P. (2020). Preparation of Synthesis Gas from CO2 for Fischer–Tropsch Synthesis—Comparison of Alternative Process Configurations. C, 6(3), 55. https://doi.org/10.3390/c6030055