BMIM-BF4 RTIL: Synthesis, Characterization and Performance Evaluation for Electrochemical CO2 Reduction to CO over Sn and MoSi2 Cathodes


Abstract
1. Introduction
2. Materials and Methods
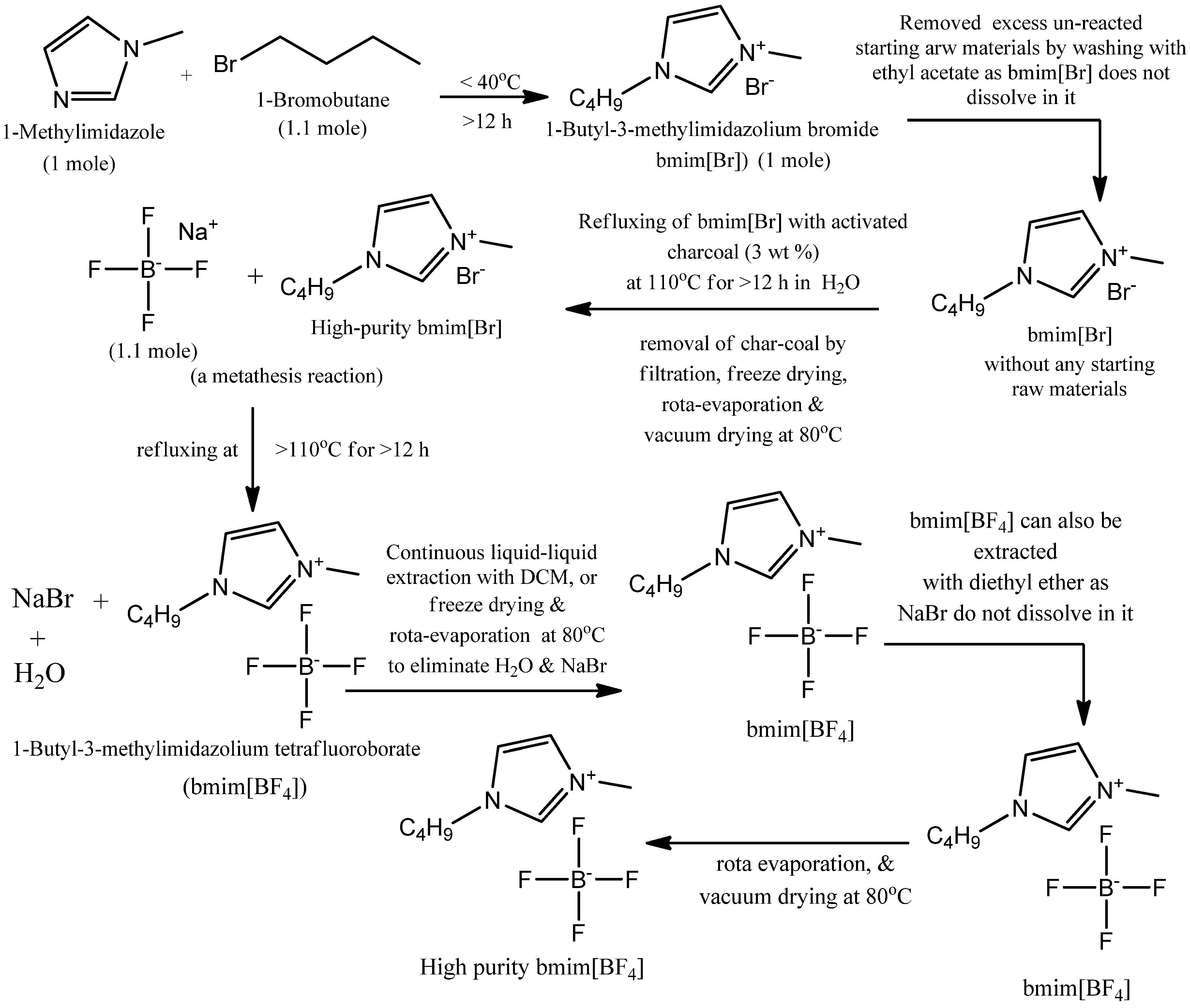
2.1. Synthesis of 1-Butyl-3-methylimidazolium Tetraflouroborate (bmim[BF4])
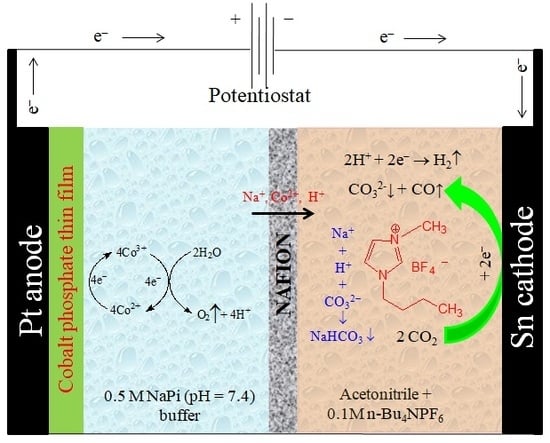
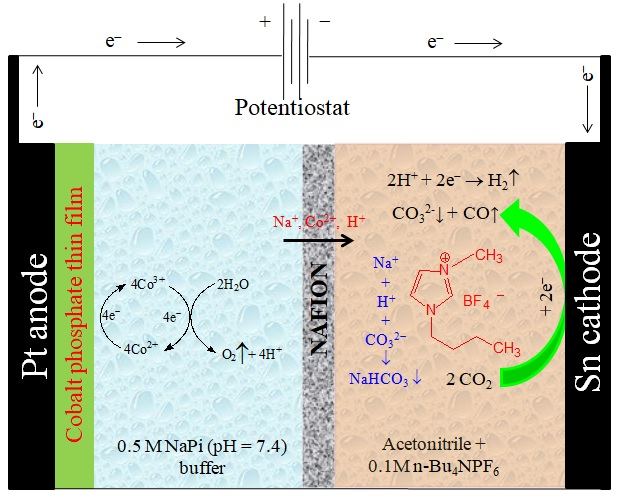
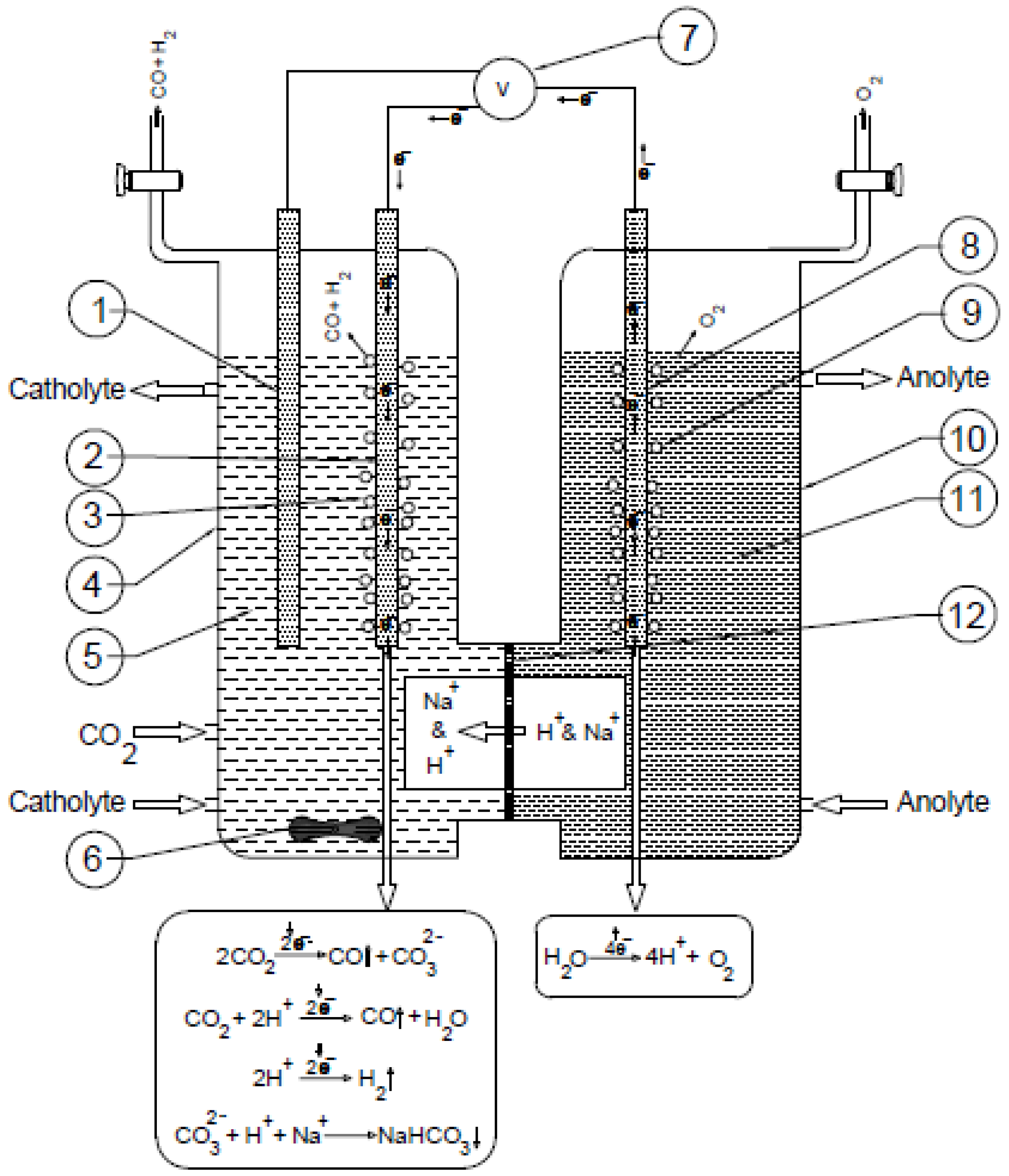
2.2. Electrochemical Experiments
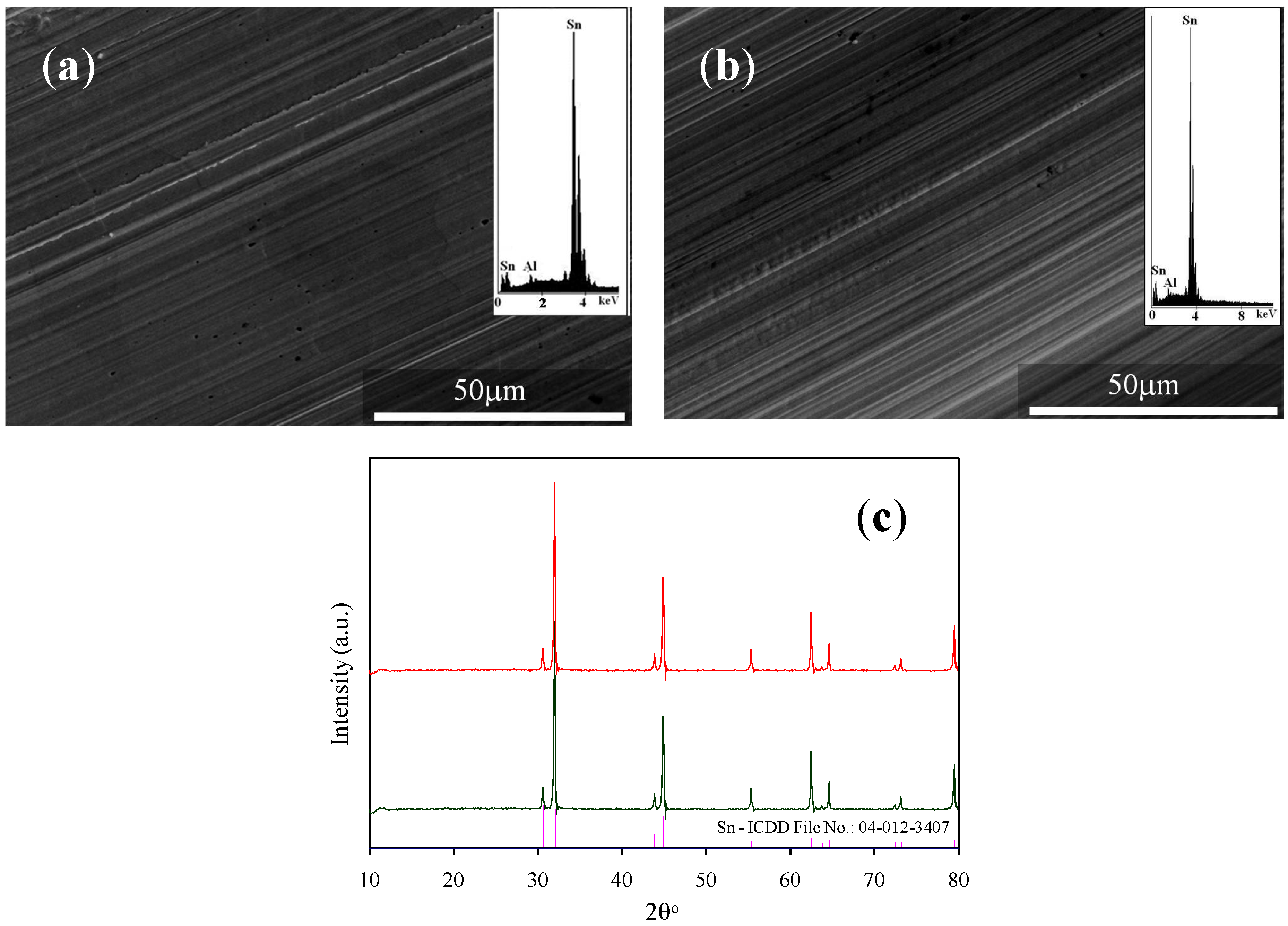
2.3. Material Characterization
3. Results
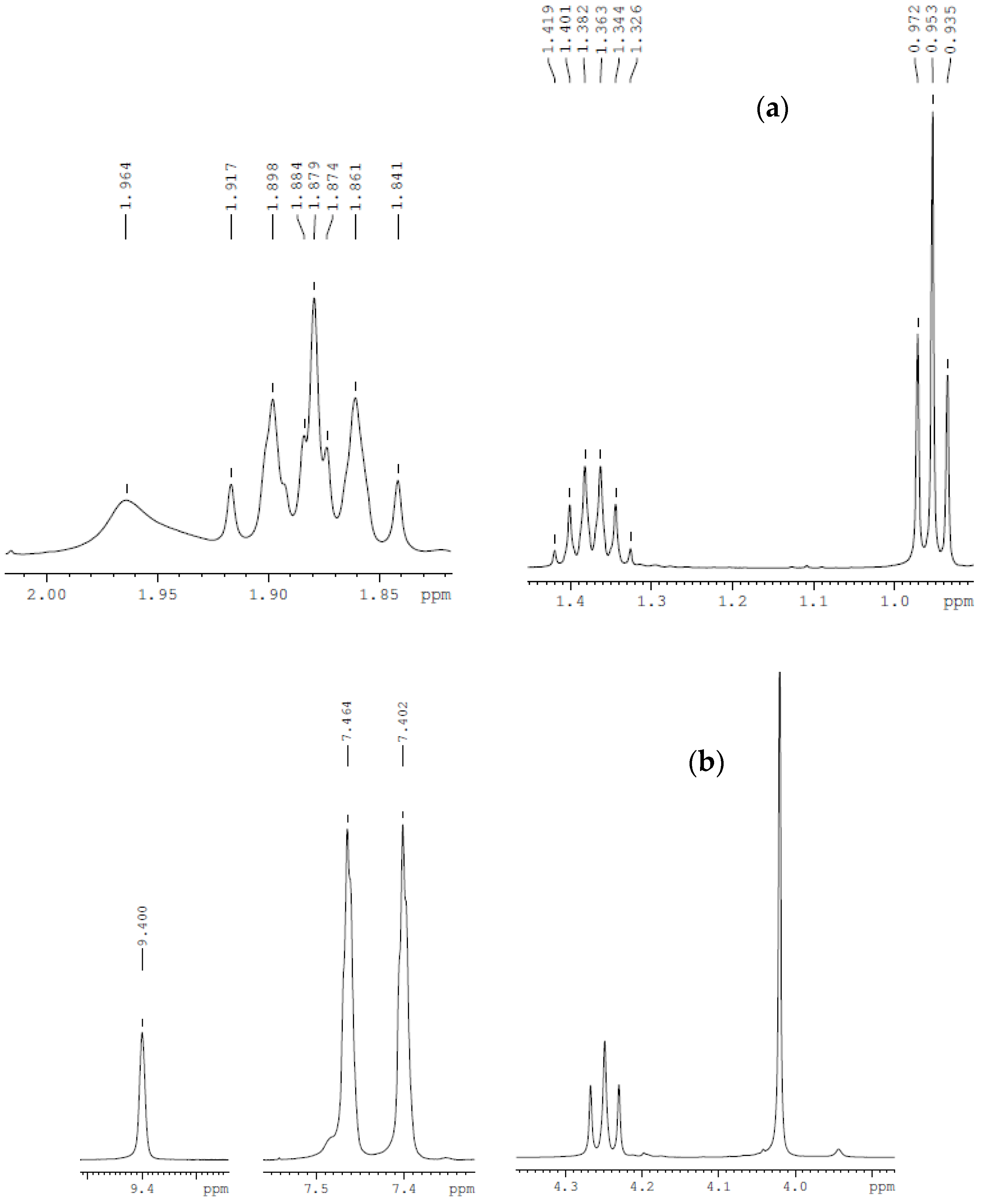
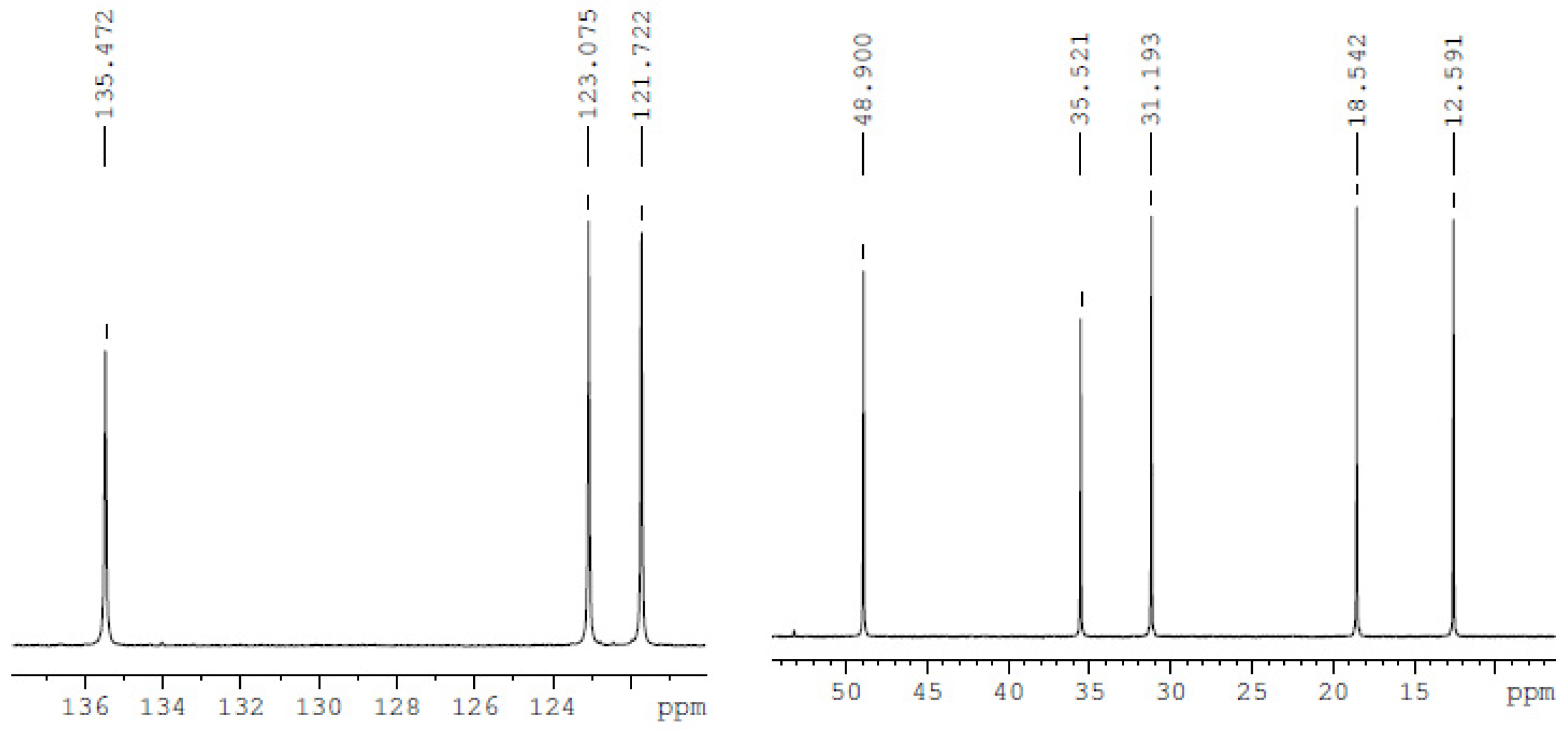
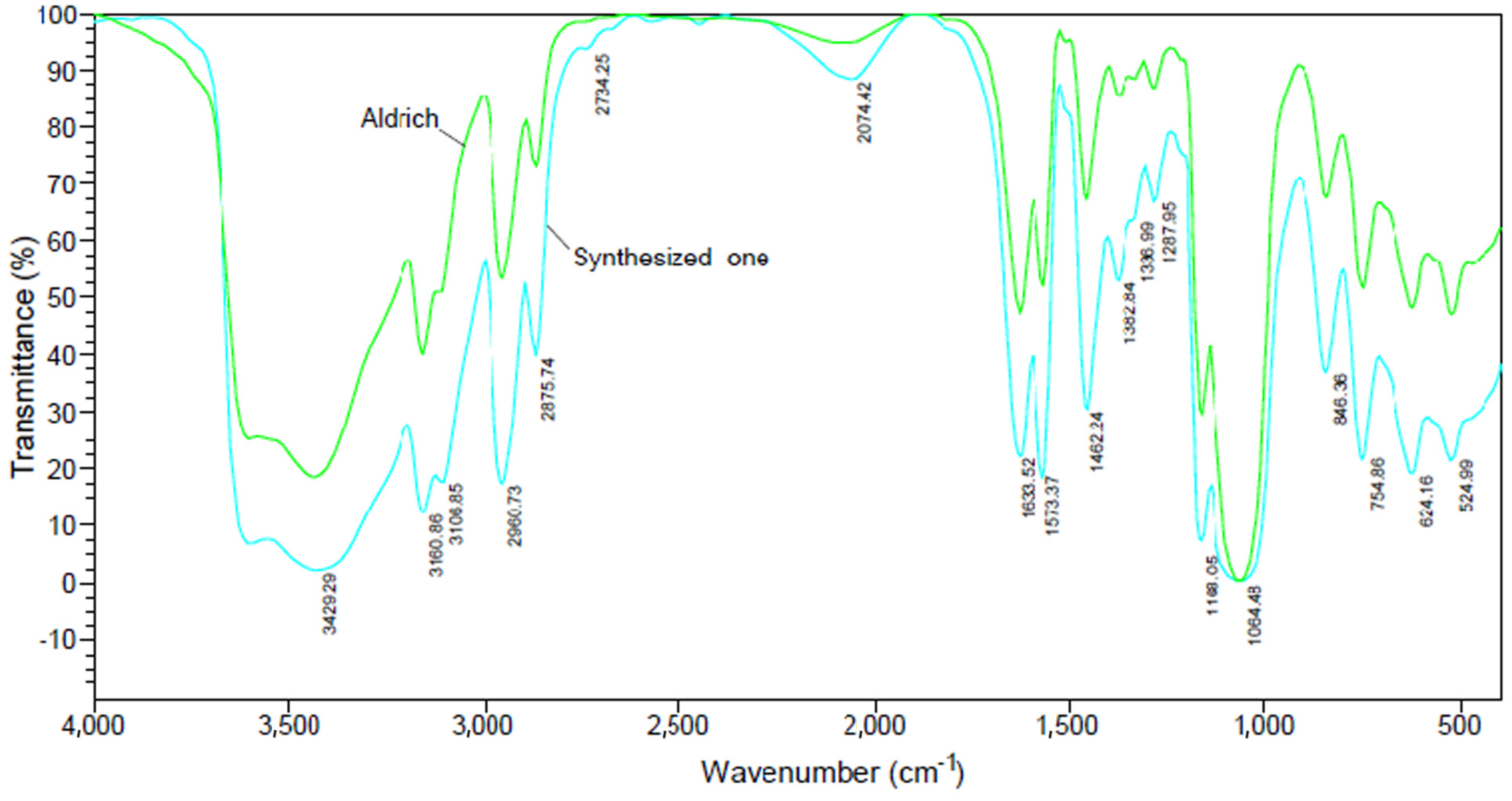
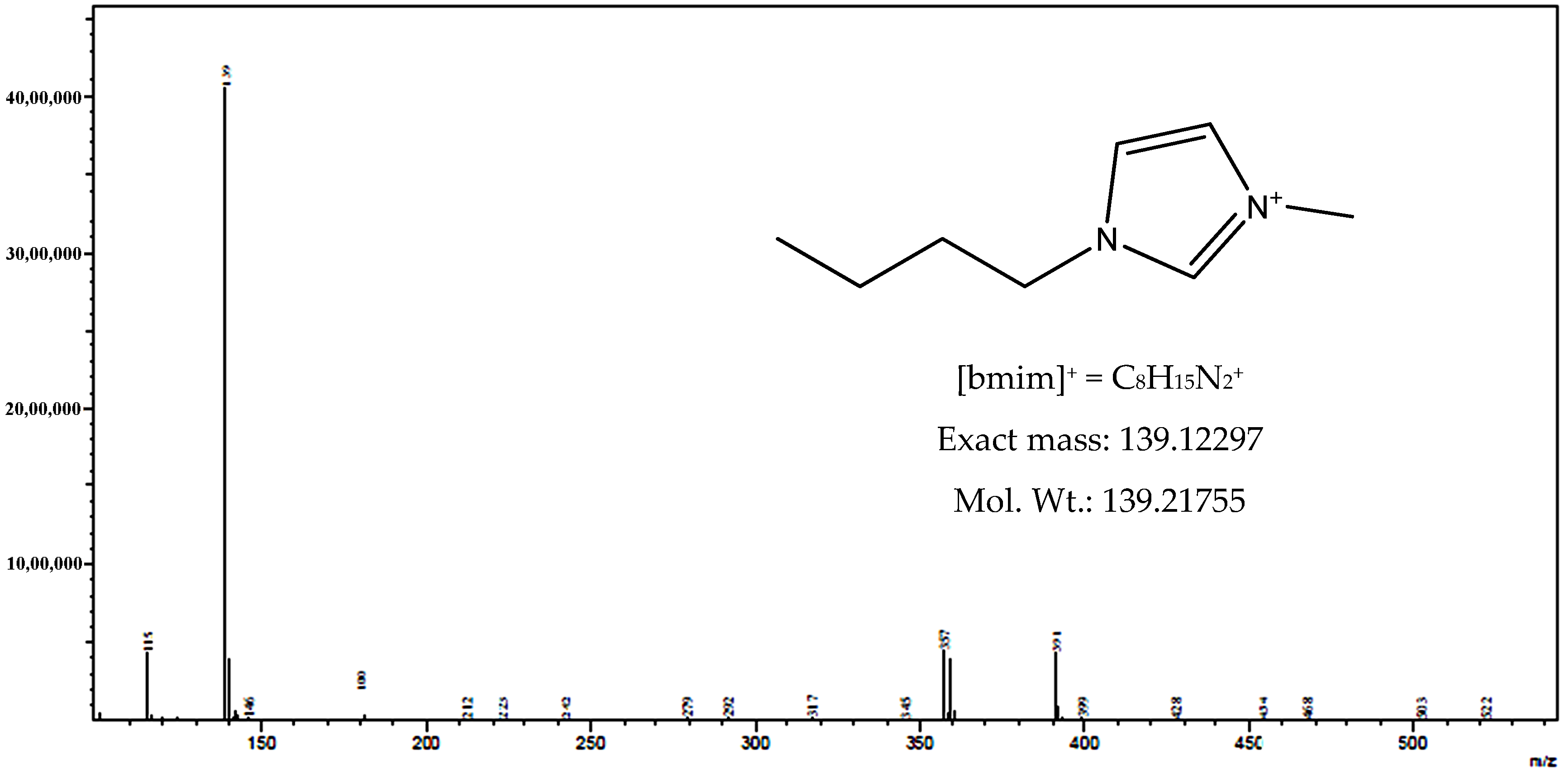
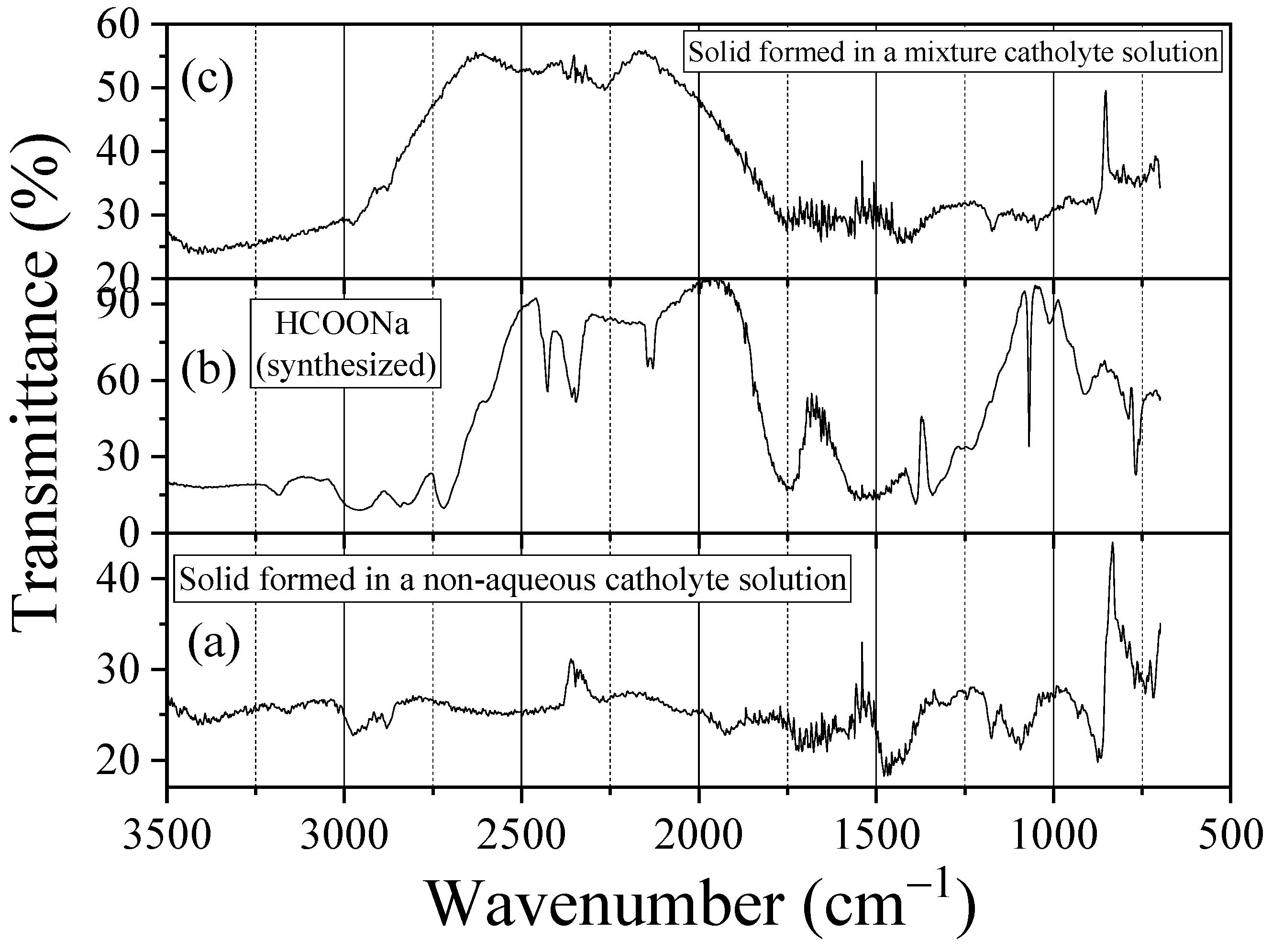
3.1. Characteristics of Bmim[Br] and Bmim[BF4]
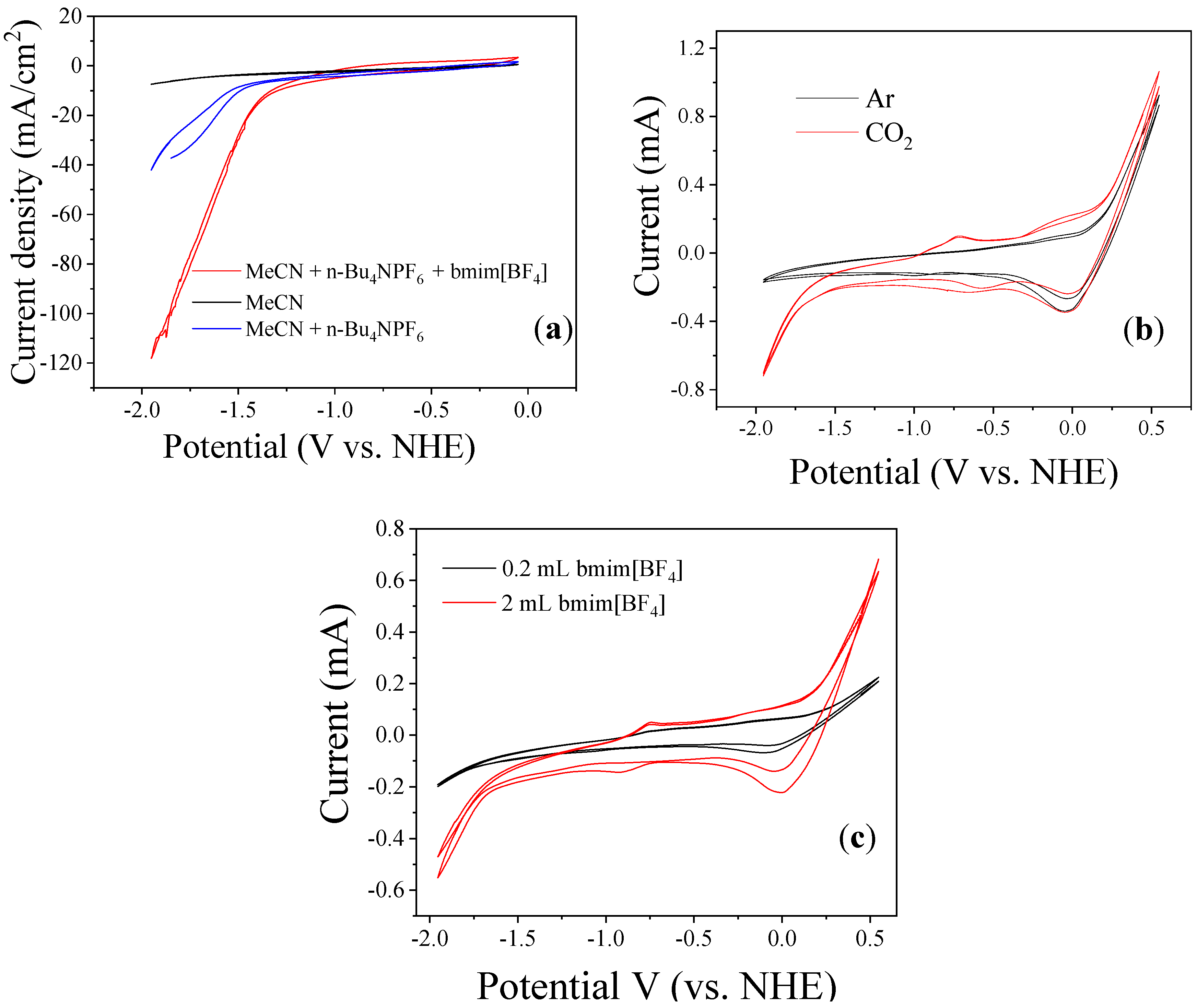
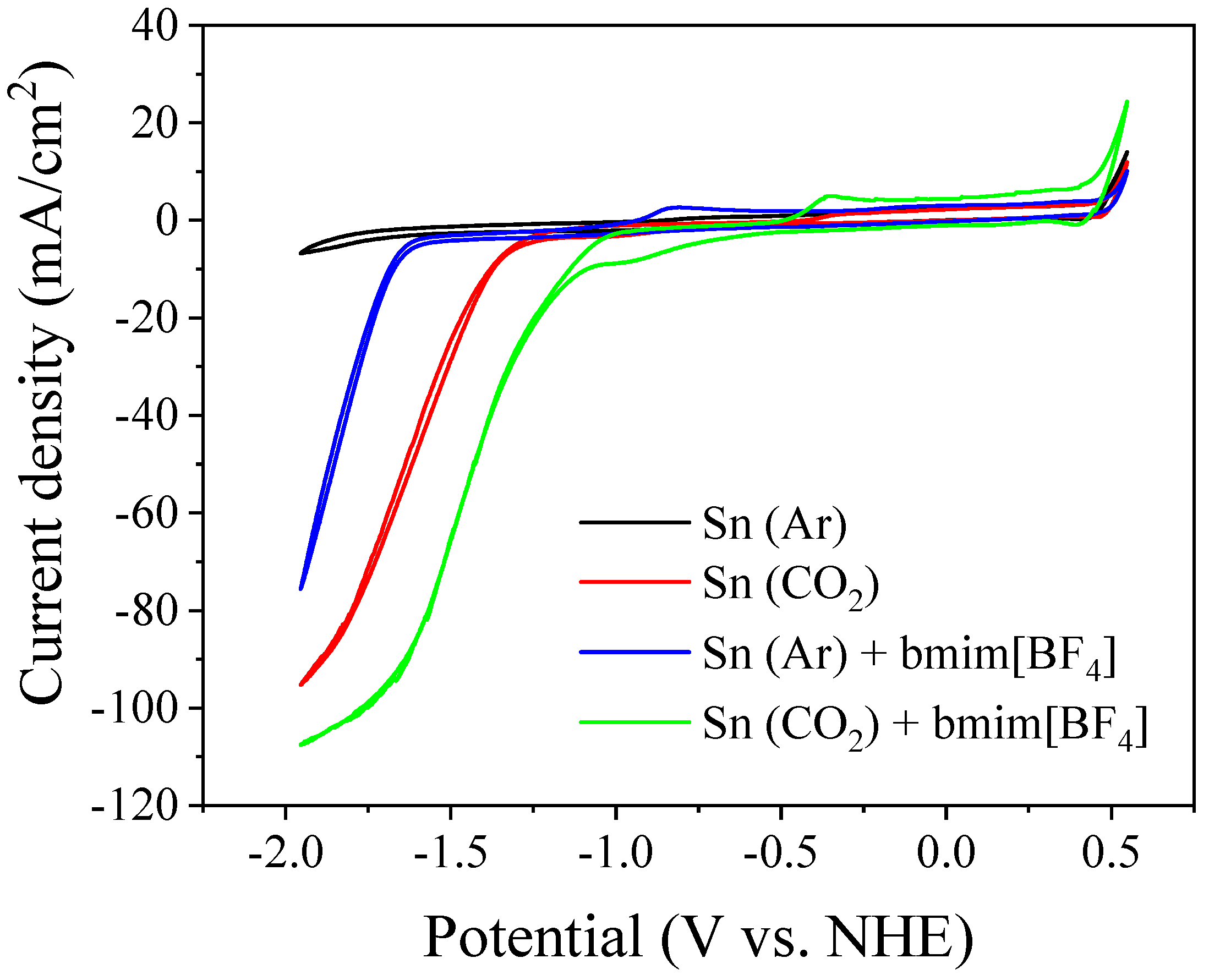
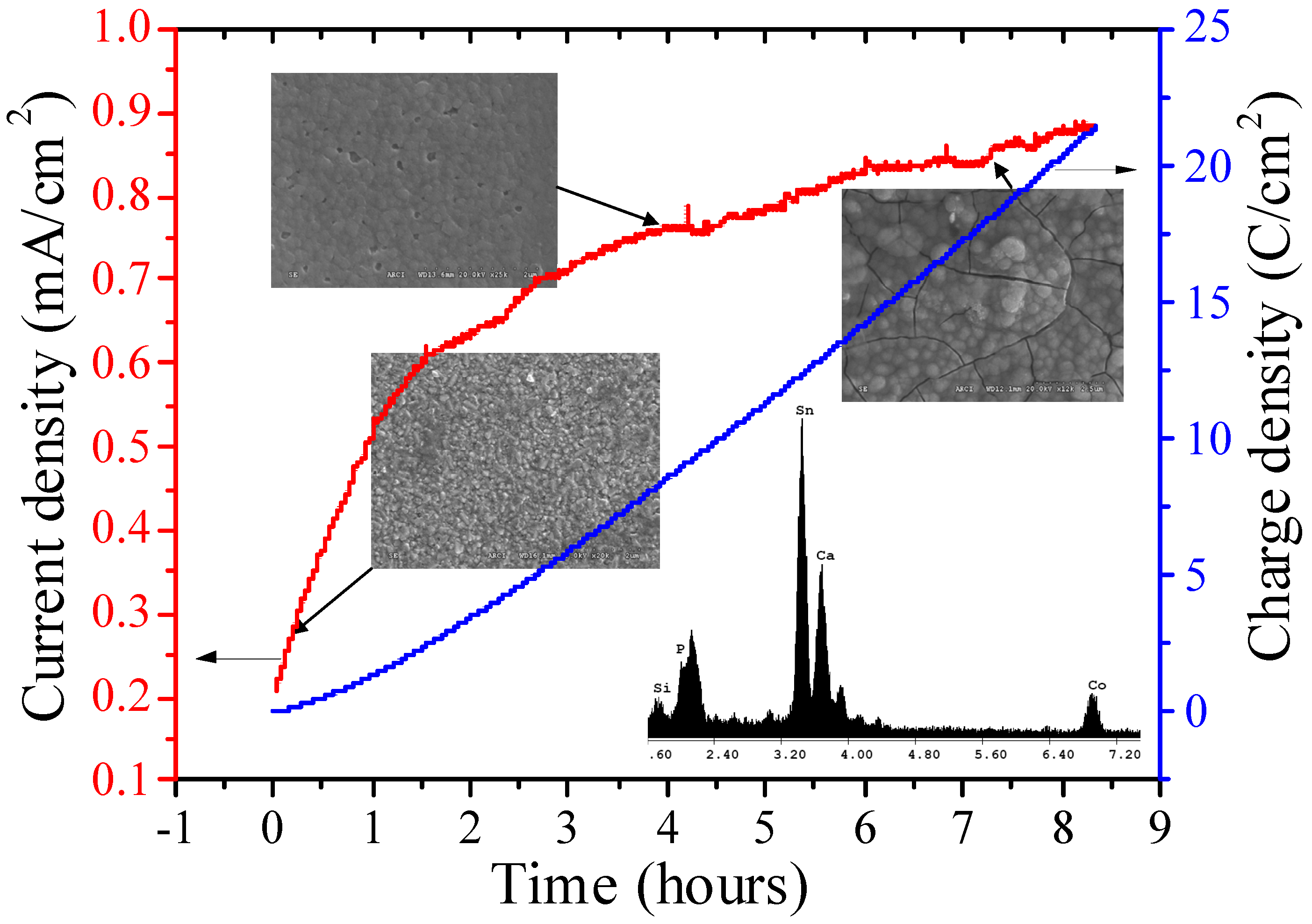
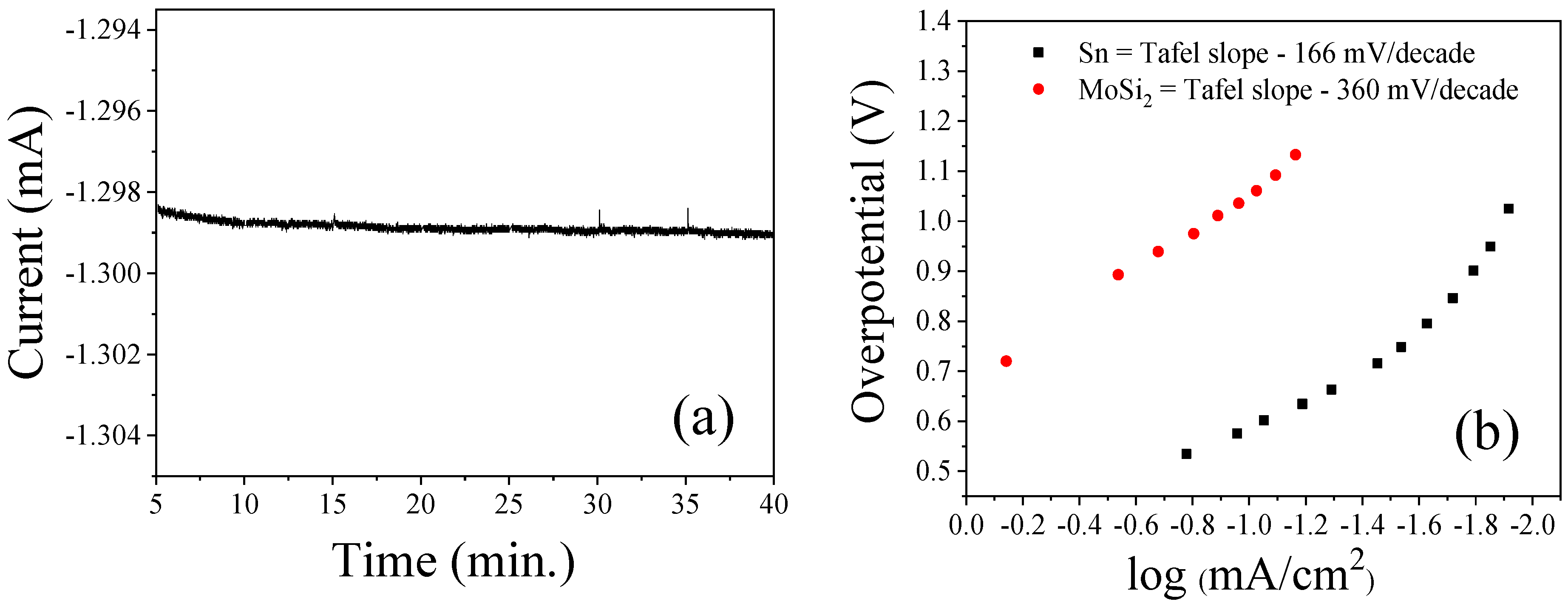
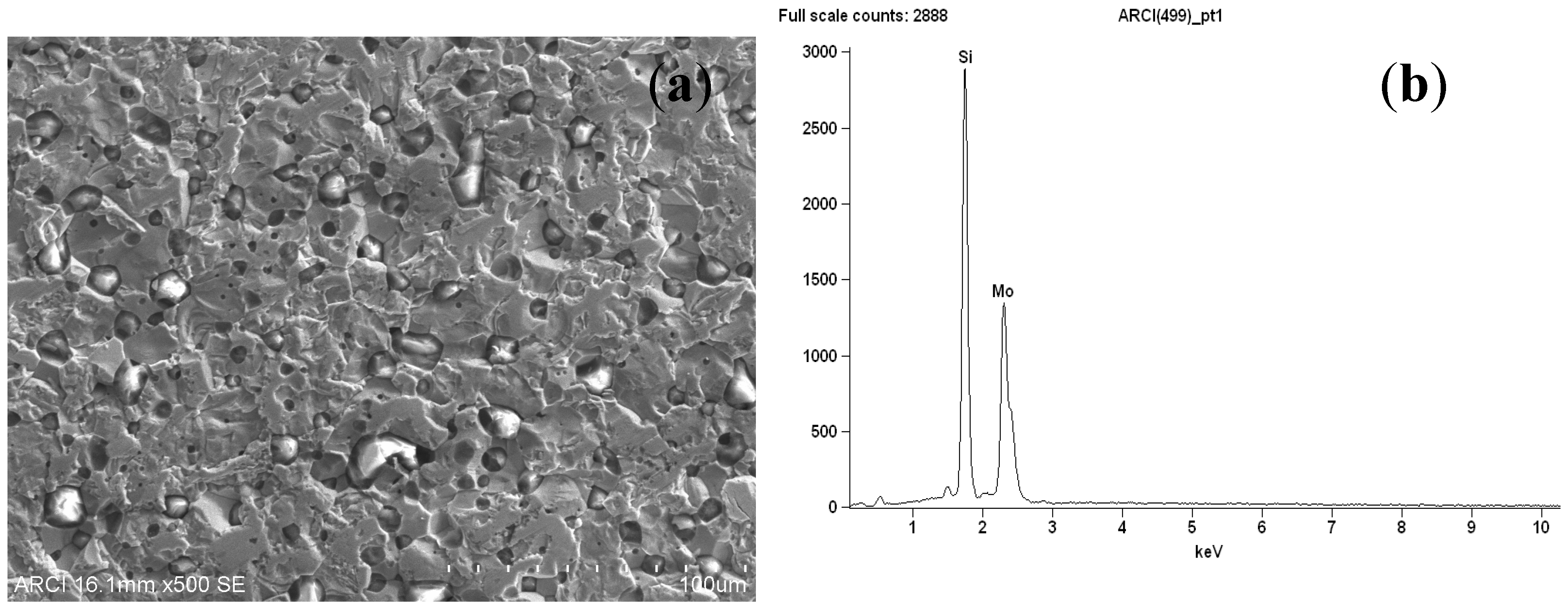
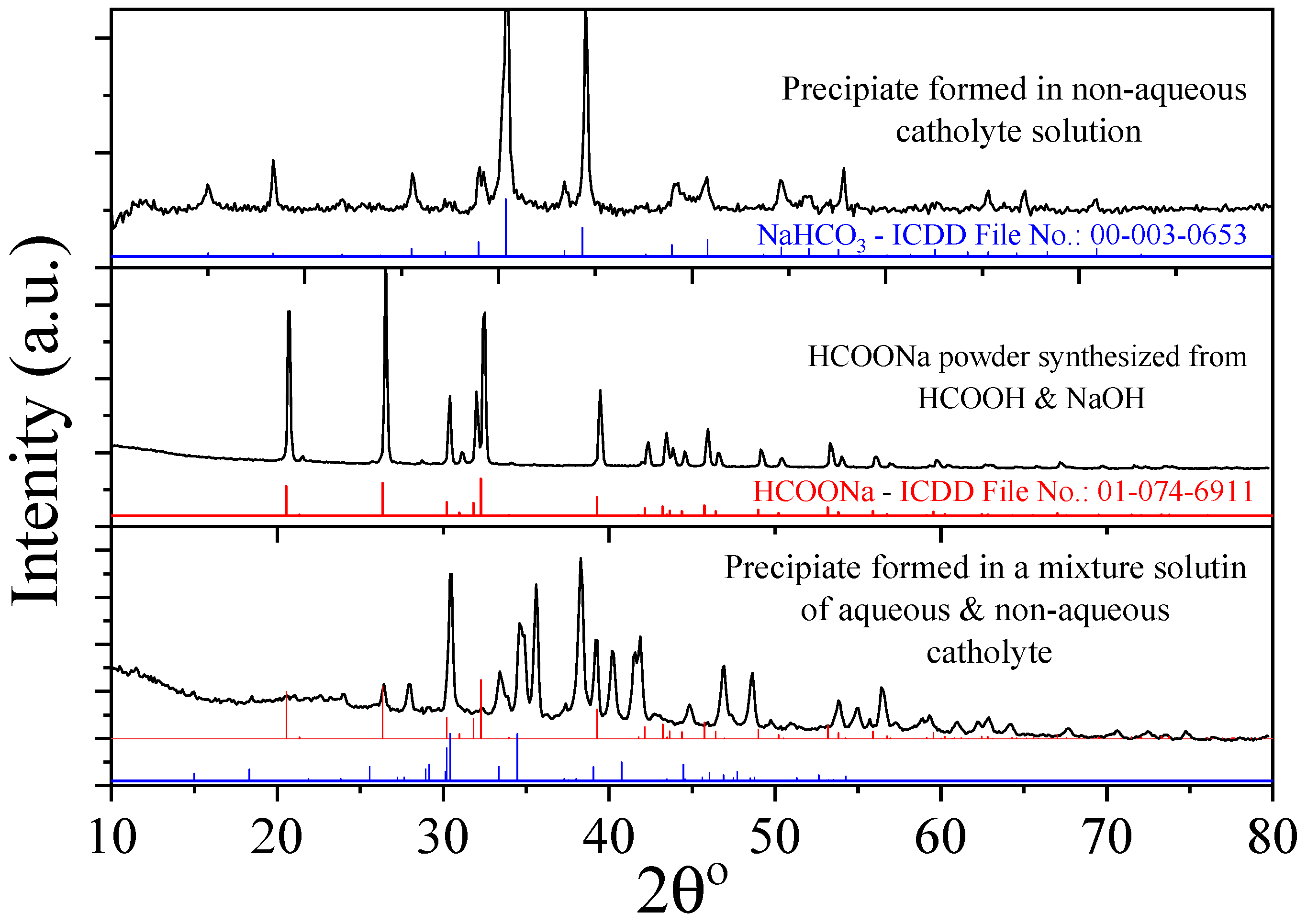
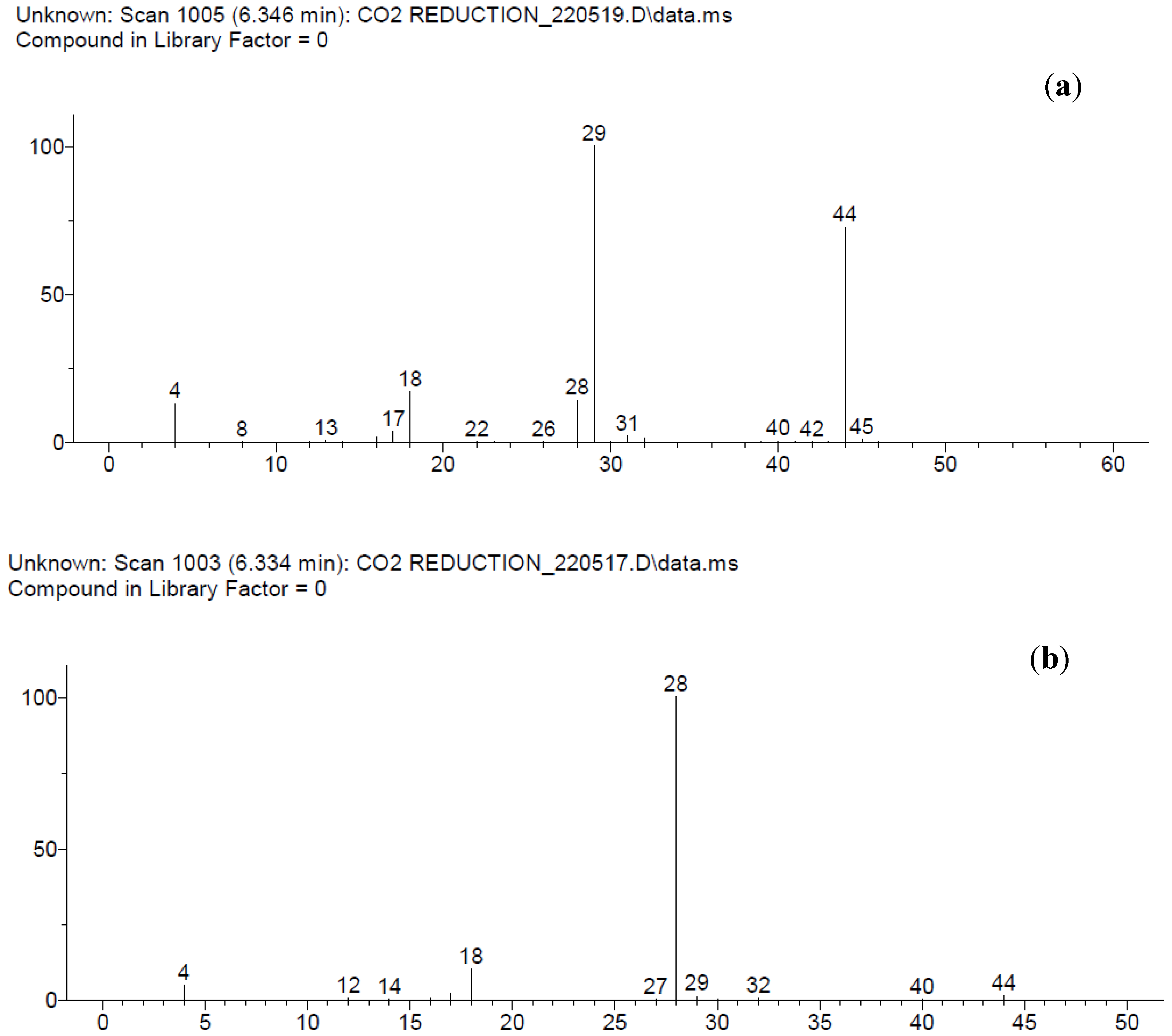
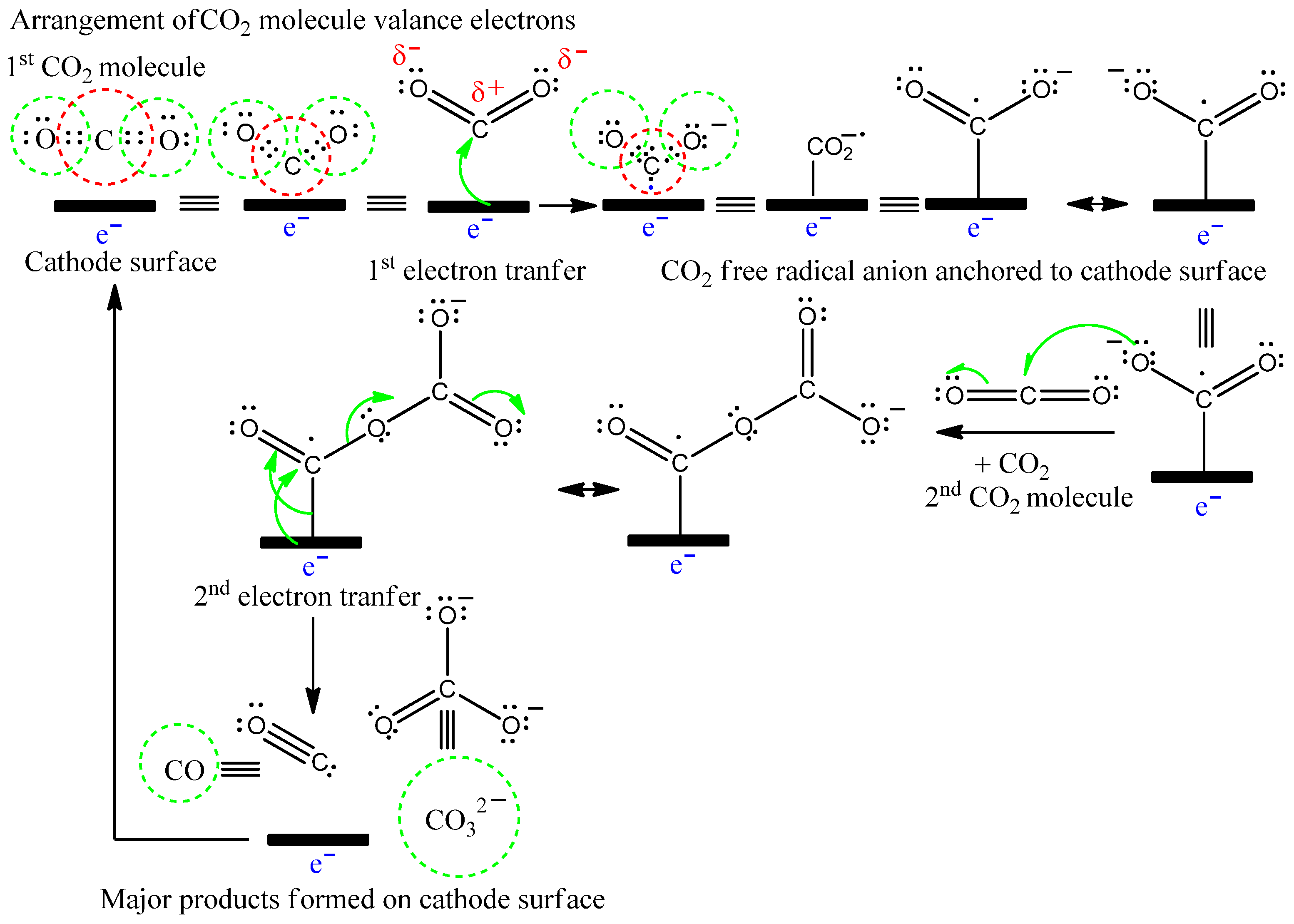
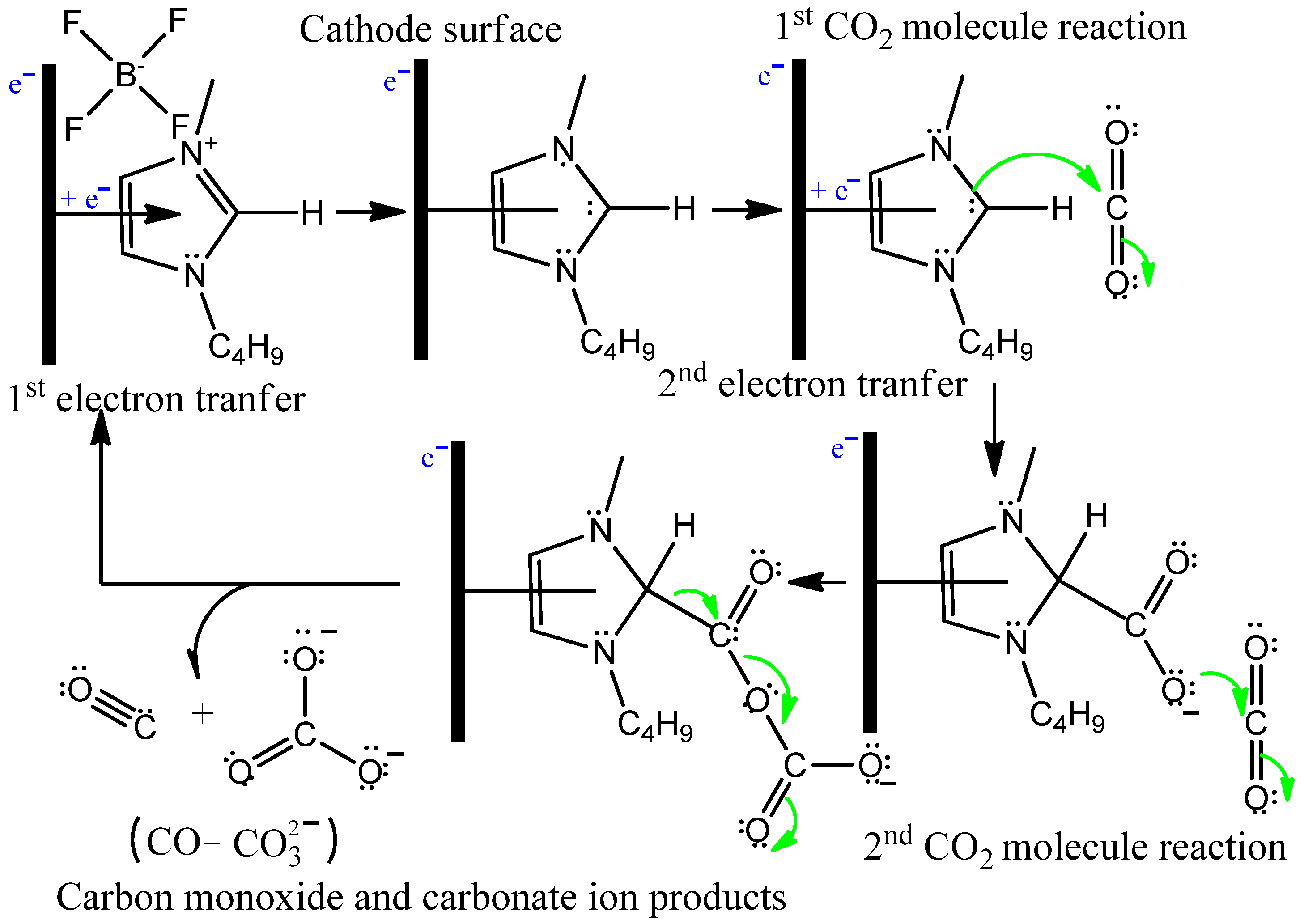
3.2. Bmim[BF4] Mediated ECR Reaction over MoSi2 and Sn Cathodes
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef]
- Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B.A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; et al. Robust Carbon Dioxide Reduction on Molybdenum Disulphide Edges. Nat. Commun. 2014, 4470, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.M.; Pupillo, R.C.; Keane, T.P.; DiMeglio, J.L.; Rosenthal, J. Efficient Conversion of CO2 to CO using Tin and Other Inexpensive and Easily Prepared Post-Transition Metal Catalysts. J. Am. Chem. Soc. 2015, 137, 5021–5027. [Google Scholar] [CrossRef]
- Rosen, B.A.; Khojin, A.S.; Thorson, M.R.; Zhu, W.; Whipple, D.T.; Kenis, P.J.A.; Masel, R.I. Ionic Liquid–Mediated Selective Conversion of CO2 to CO at Low Overpotentials. Science 2011, 334, 643–644. [Google Scholar] [CrossRef] [PubMed]
- DiMeglio, J.L.; Rosenthal, J. Selective Conversion of CO2 to CO with High Efficiency using an Inexpensive Bismuth-Based Electrocatalyst. J. Am. Chem. Soc. 2013, 135, 8798–8801. [Google Scholar] [CrossRef]
- Schreier, M.; Curvat, L.; Giordano, F.; Steier, L.; Abate, A.; Zakeeruddin, S.M.; Luo, J.; Mayer, M.T.; Gratzel, M. Efficient Photosynthesis of Carbon Monoxide from CO2 using Perovskite Photovoltaics. Nat. Commun. 2015, 7326, 1–6. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic Reduction of Carbon Dioxide in Aqueous Suspensions of Semiconductor Powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Ganesh, I. Electrochemical Conversion of Carbon Dioxide into Renewable Fuel Chemicals—The Role of Nanomaterials and the Commercialization. Renew. Sustain. Energy Rev. 2016, 59, 1269–1297. [Google Scholar] [CrossRef]
- Ganesh, I. Solar Fuels vis-a´-vis Electricity Generation from Sunlight: The Current State-of-the-Art (a Review). Renew. Sustain. Energy Rev. 2015, 44, 904–932. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of Carbon Dioxide into Methanol—A Potential Liquid Fuel: Fundamental Challenges and Opportunities (a Review). Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef]
- Matter, J.M.; Stute, M.; Snæbjörnsdottir, S.Ó.; Oelkers, E.H.; Gislason, S.R.; Aradottir, E.S.; Sigfusson, B.; Gunnarsson, I.; Sigurdardottir, H.; Gunnlaugsson, E.; et al. Rapid Carbon Mineralization for Permanent Disposal of Anthropogenic Carbon Dioxide Emissions. Science 2016, 352, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Zhang, D. Long-term viability of carbon sequestration in deep-sea sediments. Sci. Adv. 2018, 4, eaao6588. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.; Ridgwell, A. Ocean Acidification. Nat. Educ. Knowl. 2012, 3, 21. [Google Scholar]
- Centi, G.; Perathoner, S. CO2-Based Energy Vectors for the Storage of Solar Energy. Greenh. Gas. Sci. Technol. 2011, 1, 21–35. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and Prospects in the Chemical Recycling of Carbon Dioxide to Fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Ganesh, I. The Electrochemical Conversion of Carbon Dioxide to Carbon Monoxide Over Nanomaterial Based Cathodic Systems: Measures to Take to Apply This Laboratory Process Industrially. In Applications of Nanomaterials: Advances and Key Technologies in the Micro and NanoTechnologies Series, Elsevier; Oluwafemi, S., Thomas, N., Kalarikkal, S.M., Eds.; Woodhead Publishing: Sawston, UK, 2018; Volume III, pp. 1–53. [Google Scholar]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical Recycling of Carbon Dioxide to Methanol and Dimethyl Ether: From Greenhouse Gas to Renewable, Environmentally Carbon Neutral Fuels and Synthetic Hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Kember, M.R.; Knight, P.D.; Reung, P.T.R.; Williams, C.K. Highly Active Dizinc Catalyst for the Copolymerization of Carbon Dioxide and Cyclohexene Oxide at One Atmosphere Pressure. Angew. Chem. Int. Ed. 2009, 48, 931–933. [Google Scholar] [CrossRef]
- Sujith, S.; Min, J.K.; Seong, J.E.; Na, S.J.; Lee, B.Y. A Highly Active and Recyclable Catalytic System for CO2/Propylene Oxide Copolymerization. Angew. Chem. Int. Ed. Engl. 2008, 47, 7306–7309. [Google Scholar]
- Darensbourg, D.J. Chemistry of Carbon Dioxide Relevant to Its Utilization: A Personal Perspective. Inorg. Chem. 2010, 49, 10765–10780. [Google Scholar] [CrossRef]
- Arakawa, H.; Aresta, M.; Armor, J.N.; Barteau, M.A.; Beckman, E.J.; Bell, A.T.; Bercaw, J.E.; Creutz, C.; Dinjus, E.; Dixon, D.A.; et al. Catalysis Research of Relevance to Carbon Management: Progress, Challenges, and Opportunities. Chem. Rev. 2001, 101, 953–996. [Google Scholar] [CrossRef]
- Ganesh, I.; Kumar, P.P.; Annapoorna, I.; Sumliner, J.M.; Ramakrishna, M.; Hebalkar, N.Y.; Padmanabham, G.; Sundararajan, G. Preparation and Characterization of Cu-Doped TiO2 Materials for Electrochemical, Photoelectrochemical, and Photocatalytic Applications. Appl. Surf. Sci. 2014, 293, 229–247. [Google Scholar] [CrossRef]
- Ramos, J.M.; DiMeglio, J.L.; Rosenthal, J. Efficient Reduction of CO2 to CO With High Current Density Using In Situ or Ex Situ Prepared Bi-Based Materials. J. Am. Chem. Soc. 2014, 136, 8361–8367. [Google Scholar] [CrossRef] [PubMed]
- Khojin, A.S.; Jhong, H.-R.M.; Rosen, B.A.; Zhu, W.; Ma, S.; Kenis, P.J.A.; Masel, R.I. Nanoparticle Silver Catalysts That Show Enhanced Activity for Carbon Dioxide Electrolysis. J. Phys. Chem. C 2013, 117, 1627–1632. [Google Scholar] [CrossRef]
- Rosen, B.A.; Zhu, W.; Kaul, G.; Khojin, A.S.; Masel, R.I. Water Enhancement of CO2 Conversion on Silver in 1-Ethyl-3-Methylimidazolium Tetrafluoroborate. J. Electrochem. Soc. 2013, 160, H138–H141. [Google Scholar] [CrossRef]
- Rosen, B.A.; Haan, J.L.; Mukherjee, P.; Braunschweig, B.; Zhu, W.; Khojin, A.S.; Dlott, D.D.; Masel, R.I. In Situ Spectroscopic Examination of a Low Overpotential Pathway for Carbon Dioxide Conversion to Carbon Monoxide. J. Phys. Chem. C 2012, 116, 15307–15312. [Google Scholar] [CrossRef]
- Ganesh, I. BMIM-BF4 Mediated Electrochemical CO2 Reduction to CO is a Reverse Reaction of CO Oxidation in Air—An Experimental Evidence. J. Phyc. Chem. C 2019, 123, 30198–30212. [Google Scholar] [CrossRef]
- Frese, K.W.J.; Leach, S.C.; Summers, D.P. Electrochemical Reduction of Aqueous Carbon Dioxide to Methanol. U.S. Patent 4,609,441, 2 September 1986. [Google Scholar]
- Oh, Y.; Hu, X. Ionic liquids enhance the electrochemical CO2 reduction catalyzed by MoO2. Chem. Commun. 2015, 51, 13698–13701. [Google Scholar] [CrossRef]
- Handoko, A.D.; Chen, H.; Lum, Y.; Zhang, Q.; Anasori, B.; She, Z.W. Two-dimensional titanium and molybdenum carbide MXenes as electrocatalysts for CO2 reduction. Iscience 2020, 23, 101181. [Google Scholar] [CrossRef]
- Deevi, S.C. Self-propagating high-temperature synthesis of molybdenum disilicide. J. Mater. Sci. 1991, 26, 3343–3353. [Google Scholar] [CrossRef]
- Dharaskar, S.A.; Wasewar, K.L.; Varma, M.N.; Shende, D.Z.; Yoo, C. Synthesis, Characterization and Application of 1-Butyl-3-Methylimidazolium Tetrafluoroborate for Extractive Desulfurization of Liquid Fuel. Arab. J. Chem. 2016, 9, 578–587. [Google Scholar] [CrossRef]
- Burrell, A.K.; Warner, B.P.; McCleskey, T.M.; Agrawal, A. Preparation and Purification of Ionic Liquids and Precursors. U.S. Patent 7,763,186 B2, 27 July 2010. [Google Scholar]
- Dupont, J.; Consorti, C.S.; Suarez, P.A.Z.; De Souza, R.F.; Fulmer, S.L.; Richardson, D.P.; Smith, T.E.; Wolff, S. Preparation of 1-butyl-3-methyl imidazolium-based room temperature ionic liquids. Org. Synthe. 2002, 79, 236–243. [Google Scholar]
- Oh, W.S. Synthesis and Applications of Imidazolium-Based Ionic Liquids and Their Polymer Derivatives. Ph.D. Thesis, Missouri University of Science and Technology, Rolla, MO, USA, 2012. Available online: https://scholarsmine.mst.edu/doctoral_dissertations/1958/ (accessed on 30 June 2020).
- Creary, X.; Willis, E.D. Preparation of 1-butyl-3-methylimidazolium tetrafluoroborate. Org. Synth. 2005, 82, 166–169. [Google Scholar]
- Ganesh, I. Li2O-ZnO-Co3O4-TiO2 Composite Thin-Film Electrocatalyst for Efficient Water Oxidation Catalysis. Mater. Manuf. Process. 2017, 32, 431–441. [Google Scholar] [CrossRef]
- Trasatti, S. (Ed.) Studies in Physical and Theoretical Chemistry. In Electrodes of Conductive Metallic Oxides; Elsevier Scientific Publishing Co.: Amsterdam, The Netherlands, 1981; Volume 11. [Google Scholar]
- Lewandowski, A.; Waligora, L.; Galinski, M. Ferrocene as a Reference Redox Couple for Aprotic Ionic Liquids. Electroanalysis 2009, 21, 2221–2227. [Google Scholar] [CrossRef]
- Hönisch, B.; Ridgwell, A.; Schmidt, D.N.; Thomas, E.; Gibbs, S.J.; Sluijs, A.; Zeebe, R.; Kump, L.; Martindale, R.C.; Greene, S.E.; et al. The geological record of ocean acidification. Science 2012, 335, 1058–1063. [Google Scholar] [CrossRef]
- Ennis, E.; Handy, S. The Chemistry of the C2 Position of Imidazolium Room Temperature Ionic Liquids. Curr. Org. Synth. 2007, 4, 381–389. [Google Scholar] [CrossRef]
- Chen, S.; Vijayaraghavan, R.; MacFarlane, D.R.; Izgorodina, E.I. Ab initio prediction of proton NMR chemical shifts in imidazolium ionic liquids. J. Phys. Chem. B 2013, 117, 3186–3197. [Google Scholar] [CrossRef]
- Leugers, R.A.; Putzing, C.L.; Leugers, M.A. The Handbook of Infrared and Raman Spectra of Inorganic Compounds and Organic Salts, Four Volume Set. In The Dow Chemical Company; Academic Press: San Diego, CA, USA, 1997; p. 1151. [Google Scholar]
- Haas, T.; Krause, R.; Weber, R.; Demler, M.; Schmid, G. Technical Photosynthesis Involving CO2 Electrolysis and Fermentation. Nat. Catal. 2018, 1, 32–39. [Google Scholar] [CrossRef]
- Kanan, M.W.; Surendranath, Y.; Nocera, D.G. Cobalt-Phosphate Oxygen-Evolving Compound. Chem. Soc. Rev. 2009, 38, 109–114. [Google Scholar] [CrossRef]
- Palishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Chen, Y.; Kanan, M.W. Tin Oxide Dependence of the CO2 Reduction Efficiency on Tin Electrodes and Enhanced Activity for Tin/Tin Oxide Thin-Film Catalysts. J. Am. Chem. Soc. 2012, 134, 1986–1989. [Google Scholar] [CrossRef] [PubMed]
- FT-IR Spectrum of NaHCO3; National Institute of Standards and Technology, U.S. Department of Commerce. NIST Chemistry WebBook SRD 69. Available online: https://webbook.nist.gov/cgi/cbook.cgi?ID=B6007910&Units=SI&Mask=80 (accessed on 22 October 2019).
- Li, C.W.; Kanan, M.W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y. Electrolytic Reduction of Carbon Dioxide to Hydrocarbons. Kagaku Kogyo 1990, 41, 560–564. [Google Scholar]
- Ogura, K. Fixation of Carbon Dioxide by Electrolytic Reduction. Kagaku 1991, 46, 307–309. [Google Scholar]
- Viswanathan, B. Chapter 10—Electro-Catalytic Reduction of Carbon Dioxide. In New and Future Developments in Catalysis; Steven, L.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 275–295. [Google Scholar]
- Wang, Y.; Hatakeyama, M.; Ogata, K.; Wakabayashi, M.; Jin, F.; Nakamura, S. Activation of CO2 by Ionic Liquid EMIM–BF4 in the Electrochemical System: A Theoretical Study. Phys. Chem. Chem. Phys. 2015, 17, 23521–23531. [Google Scholar] [CrossRef]
- Gennaro, A.; Isse, A.A.; Severin, M.G.; Vianello, E.; Bhugun, I.; Savéant, J.M. Mechanism of the Electrochemical Reduction of Carbon Dioxide at Inert Electrodes in Media of Low Proton Availability. J. Chem. Soc. Faraday Trans. 1996, 92, 3963–3968. [Google Scholar] [CrossRef]
- Lamy, E.; Nadjo, L.; Saveant, J.M. Standard Potential and Kinetic Parameters of the Electrochemical Reduction of Carbon Dioxide in Dimethylformamide. J. Electroanal. Chem. Interfacial Electrochem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- Fischer, J.; Lehmann, T.; Heitz, E. The Production of Oxalic Acid from CO2 and H2O. J. Appl. Electrochem. 1981, 11, 743–750. [Google Scholar] [CrossRef]
- Ikeda, S.; Takagi, T.; Ito, K. Selective Formation of Formic Acid, Oxalic Acid, and Carbon Monoxide by Electrochemical Reduction of Carbon Dioxide. Bull. Chem. Soc. Jpn. 1987, 60, 2517–2522. [Google Scholar] [CrossRef]
- Gamsjager, H.; Sangster, T.G.J.; Saxena, S.K. (Eds.) Chemical Thermodynamics. In Chemical Thermodynamics of Tin; OECD Nuclear Energy Agency: Paris, France, 2012; Volume 12. [Google Scholar]
- Shi, J.; Li, Q.-Y.; Shi, F.; Song, N.; Jia, Y.-J.; Hu, Y.-Q.; Shen, F.-X.; Yang, D.-W.; Dai, Y.-N. Design of a Two-Compartment Electrolysis Cell for the Reduction of CO2 to CO in Tetrabutylammonium Perchlorate/Propylene Carbonate for Renewable Electrical Energy Storage. J. Electrochem. Soc. 2016, 163, G82–G87. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IL Synthesized | Reactants | Reaction Procedure | Ref. |
|---|---|---|---|
| Bmim[Br] | MI and BB | Refluxing reactants mixture (RM) at 70 °C for 48 h | [33] |
| Bmim[Br] | MI and BB | Refluxing RM at <40 °C for 24 h | [34] |
| Bmim[Cl] | MI and CB | Refluxing RM at 70 °C for 7 days | [35] |
| Bmim[Cl] | MI and CB | Refluxing RM in acetonitrile (MeCN) at 80 °C for 48 h | [35,36] |
| Bmim[BF4] | Bmim[Br] and NaBF4 | Refluxing RM in acetone at 40 °C for 10 h and extraction with DCM | [33] |
| Bmim[BF4] | Bmim[Cl] and KBF4 | Stirred RM in H2O at RT † for 2 h and vacuum separation with DCM | [35,36] |
| Bmim[BF4] | Bmim[Cl] and NaBF4 | Stirred RM in H2O at 45 °C for 15 min, and distillation with DCM | [37] |
| Bmim[BF4] | Bmim[Br] and NaBF4 | Refluxing RM at <25 °C for 3 h and continuous liquid–liquid extraction with DCM | [34] |
| Bmim[PF6] | Bmim[Cl] and KPF6 | Stirred RM in H2O at RT for 2 h and vacuum separation with DCM | [35] |
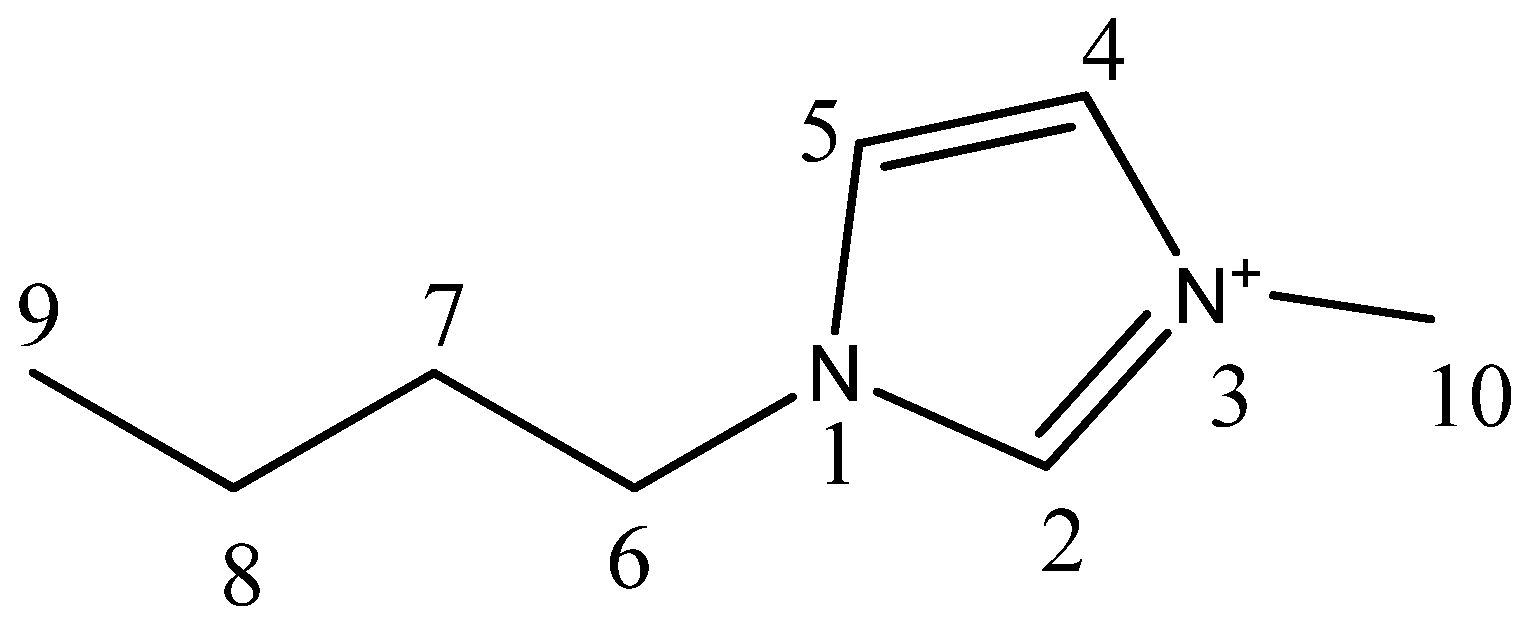
| Compound | Analysis | NMR Signal Position δ in Ppm) of [bmim]+ Group ¥ (Multiplicity ‡) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| C2 (1H) | C4 (1H) | C5 (1H) | C6 (2H) | C7 (2H) | C8 (2H) | C9 (3H) | C10 (3H) | ||
| bmim[Br] | 1H-NMR | 9.708 (s) | 7.306 (s) | 7.454 (s) | 4.288 (t) | 1.874 (quint) | 1.425 (sext) | 0.958 (t) | 4.063 (s) |
| bmim[BF4] | 1H-NMR | 9.40 (s) | 7.40 (s) | 7.46 (s) | 4.25 (t) | 1.87 (quint) | 1.36 (sext) | 0.95 (t) | 4.02 (s) |
| bmim[BF4] ‡ | 1H-NMR | 8.838 (s) | 7.22 (s) | 7.32 (s) | 4.20 (t) | 1.866 (quint) | 1.36 (sext) | 0.96 (t) | 3.962 (s) |
| bmim[Br] | 13C-NMR | 137.001 | 121.975 (d) | 123.564 (d) | 49.759 (d) | 32.002 | 19.344 | 13.329 | 36.545 |
| bmim[BF4] | 13C-NMR | 135.47 | 121.722 (d) | 123.075 (d) | 48.90 (d) | 31.193 | 18.542 | 12.591 | 35.521 |
| bmim[BF4] ‡ | 13C-NMR | 136.59 | 121.931 (d) | 123.514 (d) | 49.875 (d) | 31.865 | 19.374 | 13.294 | 36.315 |
| Entry | CPBE Time (min) | Current Density (Ma/Cm2) | Charge Density (C/Cm2) | FE (%; ±0.2) | Selectivity (%; ±1) | Mixture of CO + H2 | Yield of CO (%) | Gases Formed (μMoles/Cm2/H; ±1) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | H2 | CO | TOF (1/s) | H2 | CO | |||||
| 1. ‡ | 60 | 21.8 | 46.3 | 97.0 | 0 | >98 | 0 | 0.75 | 0 | 232.7 | 0 |
| 2. ¥ | 160 | 7.80 | 35.0 | 5.1 | 79.9 | 6 | 94 | 0.75 | 75.10 | 3.468 | 54.33 |
| 3. | 180 | 27.5 | 180.0 | 3.8 | 91.2 | 4 | 96 | 0.75 | 87.55 | 11.81 | 283.52 |
| 4. § | 160 | 16.8 | 258.0 | 4.62 | 87.87 | 5 | 95 | 0.75 | 83.47 | 23.18 | 440.50 |
| 5. ϕ | 160 | 14.7 | 39.5 | 6.67 | 76.63 | 8 | 92 | 0.75 | 70.49 | 3.667 | 51.37 |
| 6. ‽ | 20 | 16.5 | 44.6 | 3.8 | 91.2 | 4 | 96 | 0.75 | 87.55 | 3.401 | 54.66 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganesh, I. BMIM-BF4 RTIL: Synthesis, Characterization and Performance Evaluation for Electrochemical CO2 Reduction to CO over Sn and MoSi2 Cathodes. C 2020, 6, 47. https://doi.org/10.3390/c6030047
Ganesh I. BMIM-BF4 RTIL: Synthesis, Characterization and Performance Evaluation for Electrochemical CO2 Reduction to CO over Sn and MoSi2 Cathodes. C. 2020; 6(3):47. https://doi.org/10.3390/c6030047
Chicago/Turabian StyleGanesh, Ibram. 2020. "BMIM-BF4 RTIL: Synthesis, Characterization and Performance Evaluation for Electrochemical CO2 Reduction to CO over Sn and MoSi2 Cathodes" C 6, no. 3: 47. https://doi.org/10.3390/c6030047
APA StyleGanesh, I. (2020). BMIM-BF4 RTIL: Synthesis, Characterization and Performance Evaluation for Electrochemical CO2 Reduction to CO over Sn and MoSi2 Cathodes. C, 6(3), 47. https://doi.org/10.3390/c6030047