Activated Carbons Derived from High-Temperature Pyrolysis of Lignocellulosic Biomass

 , ,
, ,
Abstract
1. Introduction
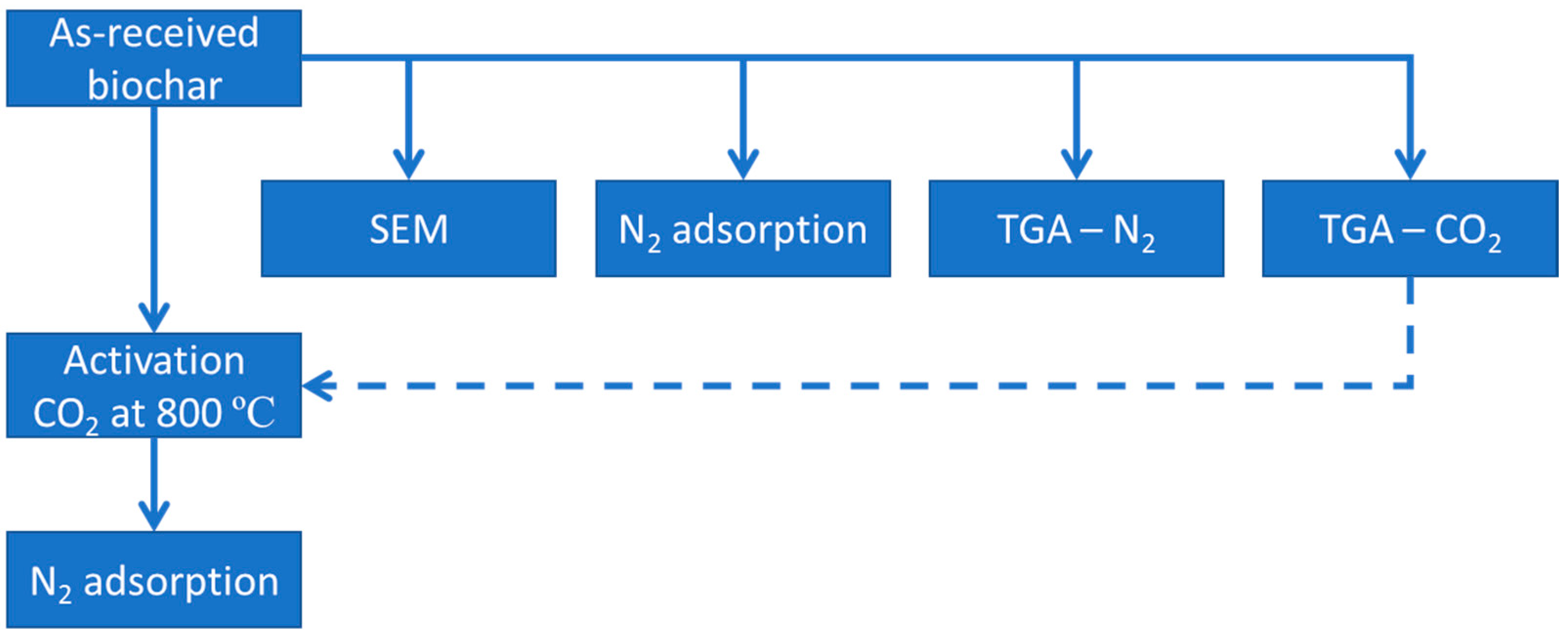
2. Materials and Experimental Details
2.1. Materials
2.2. Charring by Fast Biomass Pyrolysis
2.3. Initial Thermogravimetric Analyzes
2.4. Activation
2.5. Characterization
3. Results
3.1. Morphology of Charred Materials
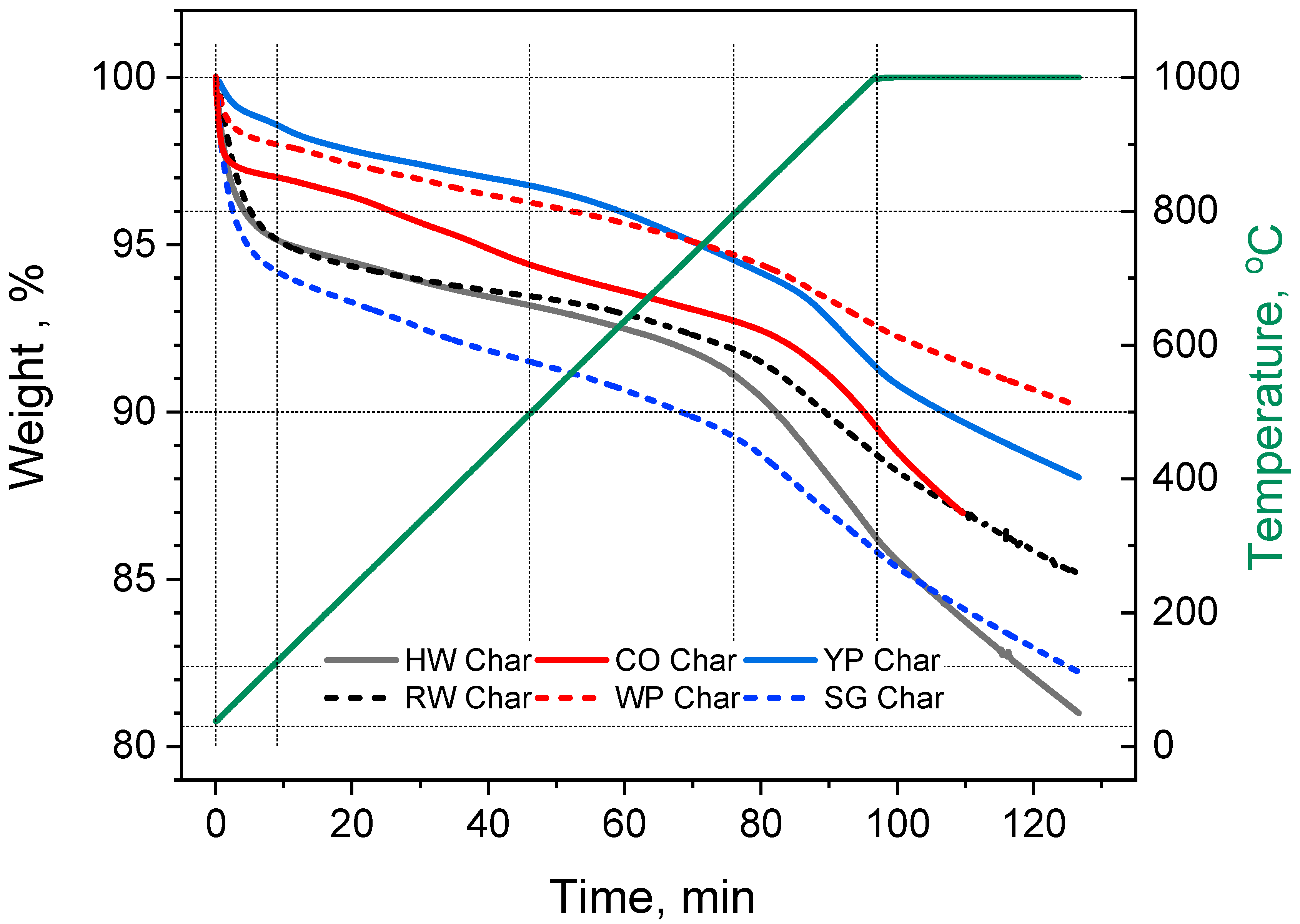
3.2. Char Devolatilization
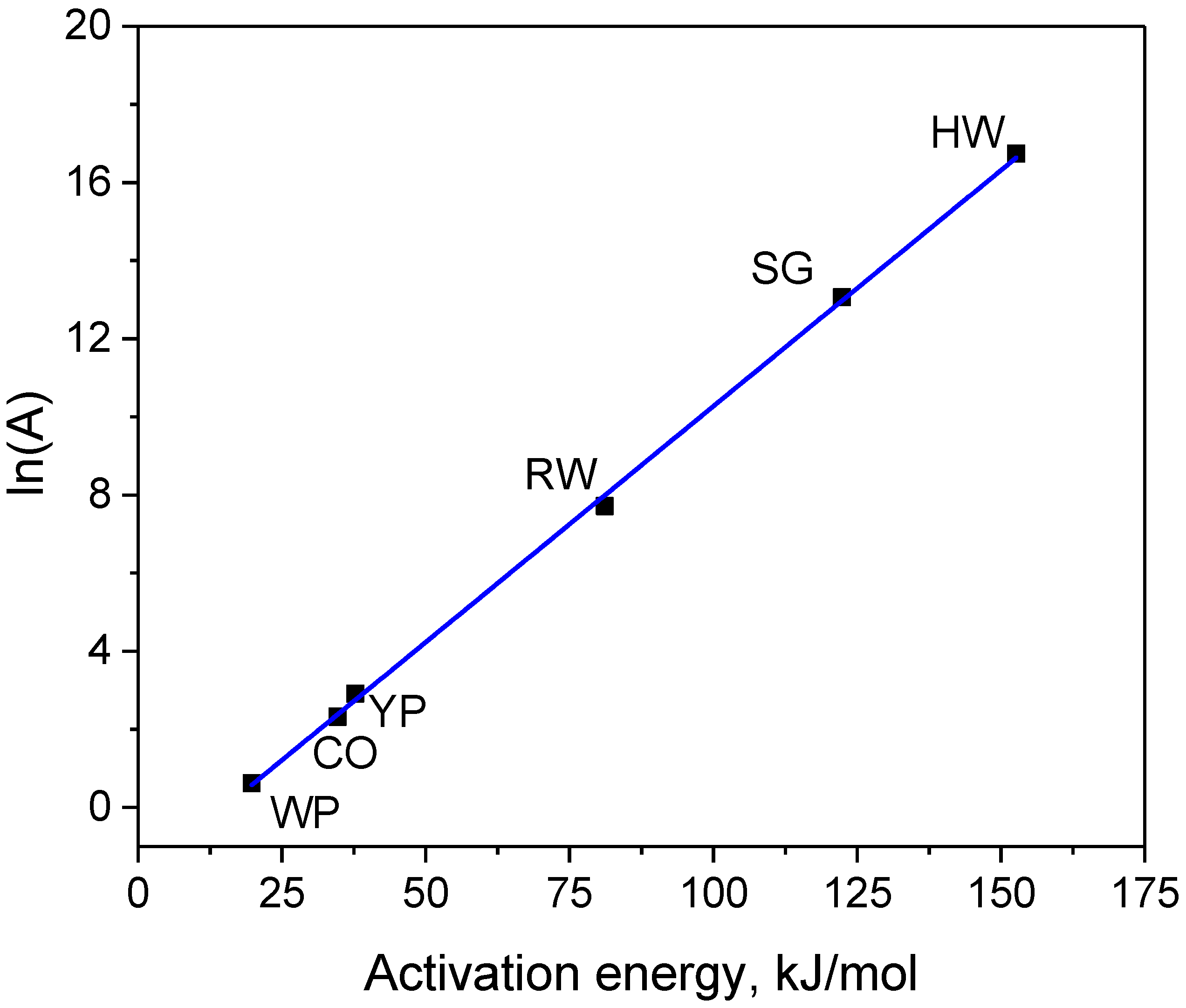
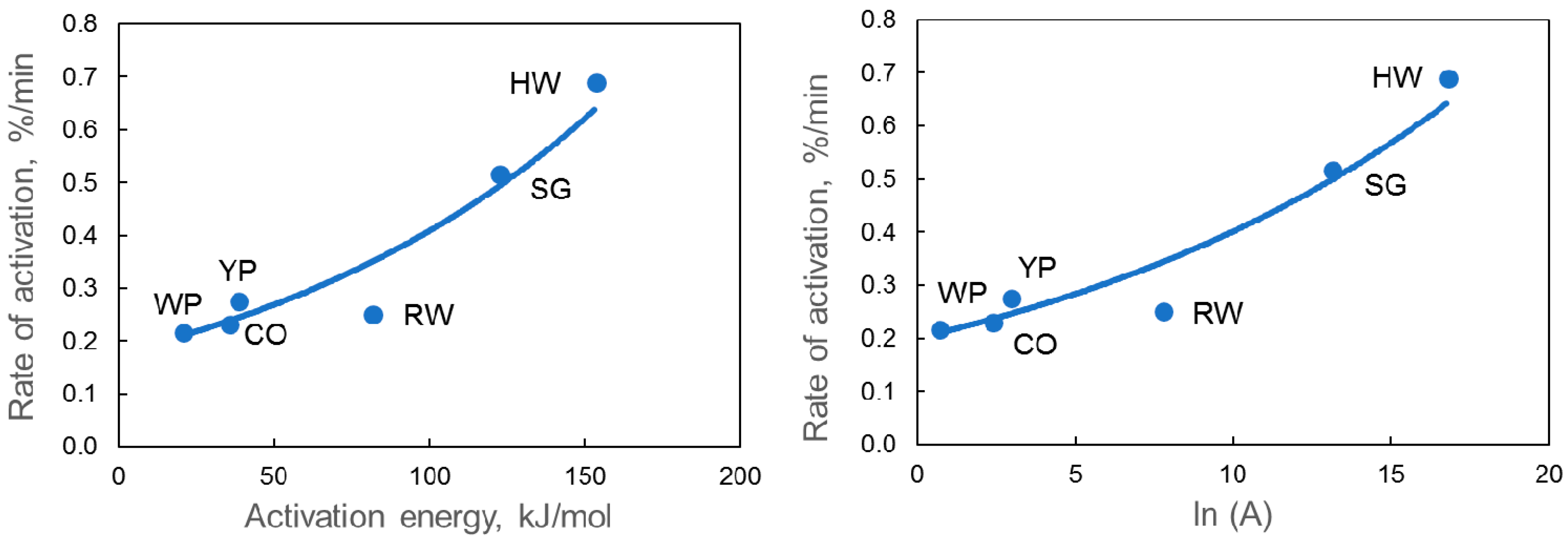
3.3. Oxidation Kinetics in Carbon Dioxide
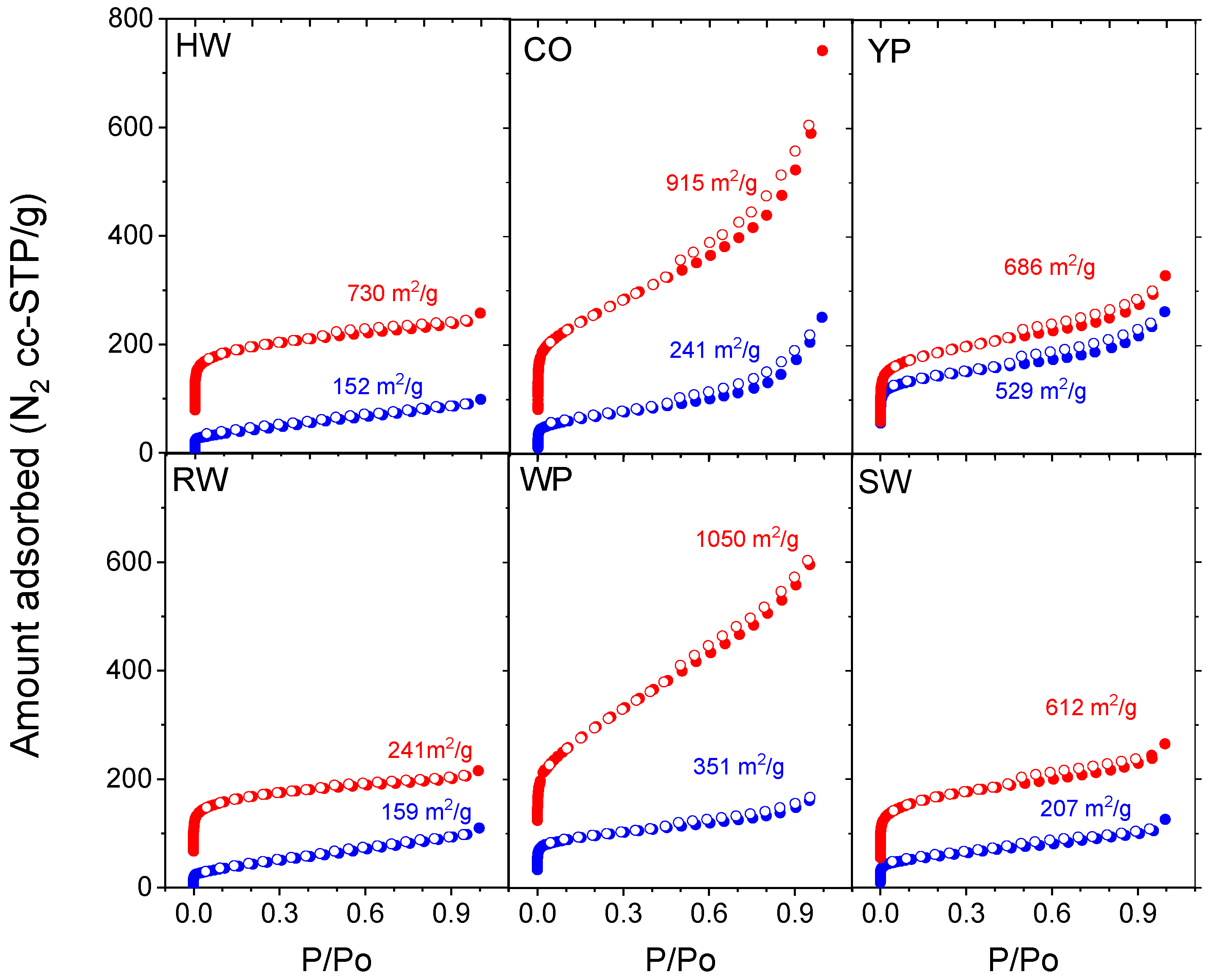
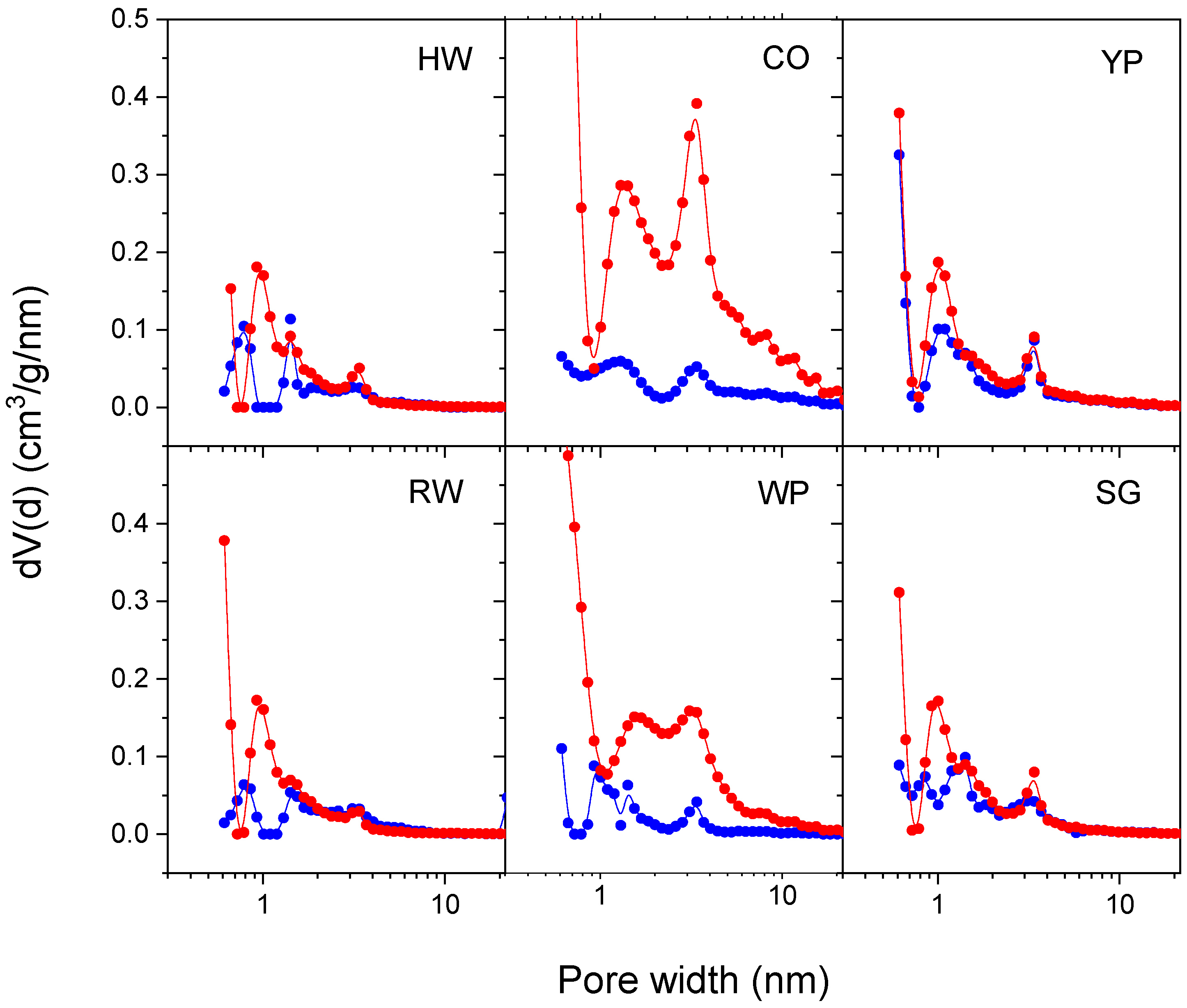
3.4. Adsorption Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- González-García, P. Activated carbon from lignocellulosics precursors: A review of the synthesis methods, characterization techniques and applications. Renew. Sustain. Energy Rev. 2018, 82, 1393–1414. [Google Scholar] [CrossRef]
- Nor, N.M.; Lau, L.C.; Lee, K.T.; Mohamed, A.R. Synthesis of activated carbon from lignocellulosic biomass and its applications in air pollution control—A review. J. Environ. Chem. Eng. 2013, 1, 658–666. [Google Scholar] [CrossRef]
- Vilella, P.C.; Lira, J.A.; Azevedo, D.C.S.; Bastos-Neto, M.; Stefanutti, R. Preparation of biomass-based activated carbons and their evaluation for biogas upgrading purposes. Ind. Crop. Prod. 2017, 109, 134–140. [Google Scholar] [CrossRef]
- Radovic, L.R.; Silva, I.F.; Ume, J.I.; Menéndez, J.A.; Leon, C.A.L.Y.; Scaroni, A.W. An experimental and theoretical study of the adsorption of aromatics possessing electron-withdrawing and electron-donating functional groups by chemically modified activated carbons. Carbon 1997, 35, 1339–1348. [Google Scholar] [CrossRef]
- Figueiredo, J.; Pereira, M.F.; Freitas, M.M.; Órfão, J.J. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Cagnon, B.; Py, X.; Guillot, A.; Stoeckli, F.; Chambat, G. Contributions of hemicellulose, cellulose and lignin to the mass and the porous properties of chars and steam activated carbons from various lignocellulosic precursors. Bioresour. Technol. 2009, 100, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, O.; Zabaniotou, A. Agricultural residues as precursors for activated carbon production—A review. Renew. Sustain. Energy Rev. 2007, 11, 1966–2005. [Google Scholar] [CrossRef]
- Suhas; Carrott, P.J.M.; Carrott, M.M.L.R. Lignin—From natural adsorbent to activated carbon: A review. Bioresour. Technol. 2007, 98, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Gratuito, M.K.B.; Panyathanmaporn, T.; Chumnanklang, R.-A.; Sirinuntawittaya, N.; Dutta, A. Production of activated carbon from coconut shell: Optimization using response surface methodology. Bioresour. Technol. 2008, 99, 4887–4895. [Google Scholar] [CrossRef] [PubMed]
- Yahya, M.A.; Al-Qodah, Z.; Ngah, C.W.Z. Agricultural bio-waste materials as potential sustainable precursors used for activated carbon production: A review. Renew. Sustain. Energy Rev. 2015, 46, 218–235. [Google Scholar] [CrossRef]
- Yakout, S.M.; El-Deen, G.S. Characterization of activated carbon prepared by phosphoric acid activation of olive stones. Arab. J. Chem. 2016, 9, S1155–S1162. [Google Scholar] [CrossRef]
- Li, W.; Peng, J.; Zhang, L.; Yang, K.; Xia, H.; Zhang, S.; Guo, S. Preparation of activated carbon from coconut shell chars in pilot-scale microwave heating equipment at 60 kW. Waste Manag. 2009, 29, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Daud, W.M.A.W.; Ali, W.S.W.; Sulaiman, M.Z. The effects of carbonization temperature on pore development in palm-shell-based activated carbon. Carbon 2000, 38, 1925–1932. [Google Scholar] [CrossRef]
- Paethanom, A.; Yoshikawa, K. Influence of pyrolysis temperature on rice husk char characteristics and its tar adsorption capability. Energies 2012, 5, 4941–4951. [Google Scholar] [CrossRef]
- Correa, C.R.; Stollovsky, M.; Hehr, T.; Rauscher, Y.; Rolli, B.; Kruse, A. Influence of the carbonization process on activated carbon properties from lignin and lignin-rich biomasses. ACS Sustain. Chem. Eng. 2017, 5, 8222–8233. [Google Scholar] [CrossRef]
- Lehmann, J.; Gaunt, J.; Rondon, M. Bio-char Sequestration in terrestrial ecosystems—A review. Mitig. Adapt. Strat. Glob. Chang. 2006, 11, 403–427. [Google Scholar] [CrossRef]
- McFarlane, Z.D.; Myer, P.R.; Cope, E.R.; Evans, N.D.; Bone, T.C.; Biss, B.E.; Mulliniks, J.T. Effect of biochar type and size on in vitro rumen fermentation of orchard grass hay. Agric. Sci. 2017, 8, 316–325. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Chen, W.; Yang, H.; Chen, H. The structure evolution of biochar from biomass pyrolysis and its correlation with gas pollutant adsorption performance. Bioresour. Technol. 2017, 246, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Inguanzo, M.; Menendez, J.A.; Fuente, E.; Pis, J.J. Reactivity of pyrolyzed sewage sludge in air and CO2. J. Anal. Appl. Pyrolysis 2001, 58, 943–954. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Sarath, G.; Mitchell, R.B.; Sattler, S.E.; Funnell, D.; Pedersen, J.F.; Graybosch, R.A.; Vogel, K.P. Opportunities and roadblocks in utilizing forages and small grains for liquid fuels. J. Ind. Microbiol. Biotechnol. 2008, 35, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Rouquerol, J.; Avnir, D.; Fairbridge, C.W.; Everett, D.H.; Haynes, J.M.; Pernicone, N.; Ramsay, J.D.F.; Sing, K.S.W.; Unger, K.K. Recommendations for the characterization of porous solids (Technical Report). Pure Appl. Chem. PAC 1994, 66, 1739–1758. [Google Scholar] [CrossRef]
- Bahadur, J.; Contescu, C.I.; Rai, D.K.; Gallego, N.C.; Melnichenko, Y.B. Clustering of water molecules in ultramicroporous carbon: In-situ small-angle neutron scattering. Carbon 2017, 111, 681–688. [Google Scholar] [CrossRef]
- Zhao, C.; Jiang, E.; Chen, A. Volatile production from pyrolysis of cellulose, hemicellulose and lignin. J. Energy Inst. 2017, 90, 902–913. [Google Scholar] [CrossRef]
- Gorgulho, H.F.; Mesquita, J.P.; Gonçalves, F.; Pereira, M.F.R.; Figueiredo, J.L. Characterization of the surface chemistry of carbon materials by potentiometric titrations and temperature-programmed desorption. Carbon 2008, 46, 1544–1555. [Google Scholar] [CrossRef]
- Agrawal, R. The compensation effect: A fact or a fiction. J. Therm. Anal. Calorim. 1989, 35, 909–917. [Google Scholar] [CrossRef]
- Conner, W.C. A general explanation for the compensation effect: The relationship between ΔS‡ and activation energy. J. Catal. 1982, 78, 238–246. [Google Scholar] [CrossRef]
- Feates, F.S.; Harris, P.S.; Reuben, B.G. Compensation effect in the kinetics of the catalysed oxidation of carbon. J. Chem. Soc. Faraday Trans. 1974, 70, 2011–2020. [Google Scholar] [CrossRef]
- Yip, K.; Ng, E.; Li, C.-Z.; Hayashi, J.-I.; Wu, H. A mechanistic study on kinetic compensation effect during low-temperature oxidation of coal chars. Proc. Combust. Inst. 2011, 33, 1755–1762. [Google Scholar] [CrossRef]
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- McDougall, G.J. The physical nature and manufacture of activated carbon. J. S. Afr. Inst. Min. Metall. 1991, 91, 109–120. [Google Scholar]
- Marsh, H.; Rodríguez-Reinoso, F. Characterization of activated carbon. In Activated Carbon; Elsevier Science Ltd. Press: Oxford, UK, 2006; pp. 143–242. [Google Scholar] [CrossRef]
- Pendleton, P.; Wong, S.H.; Schumann, R.; Levay, G.; Denoyel, R.; Rouquero, J. Properties of activated carbon controlling 2-Methylisoborneol adsorption. Carbon 1997, 35, 1141–1149. [Google Scholar] [CrossRef]
- Al-Degs, Y.S.; El-Barghouthi, M.I.; Khraisheh, M.A.; Ahmad, M.N.; Allen, S.J. Effect of surface area, micropores, secondary micropores, and mesopores volumes of activated carbons on reactive dyes adsorption from solution. Sep. Sci. Technol. 2005, 39, 97–111. [Google Scholar] [CrossRef]
- Gisi, S.D.; Lofrano, G.; Grassi, M.; Notarnicola, M. Characteristics and adsorption capacities of low-cost sorbents for wastewater treatment: A review. Sustain. Mater. Technol. 2016, 9, 10–40. [Google Scholar] [CrossRef]
- Rutherford, D.W. Changes in Composition and Porosity Occurring During the Thermal Degradation of Wood and Wood Components; U.S. Dept. of the Interior, U.S. Geological Survey: Reston, VA, USA, 2005.
- Cha, J.S.; Park, S.H.; Jung, S.C.; Ryu, C.; Jeon, J.K.; Shin, M.C.; Park, Y.K. Production and utilization of biochar: A review. J. Ind. Eng. Chem. 2016, 40, 1–15. [Google Scholar] [CrossRef]
- Xie, X.; Goodell, B.; Zhang, D.; Nagle, D.C.; Qian, Y.; Peterson, M.L.; Jellison, J. Characterization of carbons derived from cellulose and lignin and their oxidative behavior. Bioresour. Technol. 2009, 100, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.J.; Woodley, R.E.; Halas, D.D.R. Gas-Graphite systems. In Nuclear Graphite; Nightingale, R.E., Ed.; Academic Press: New York, NY, USA, 2013; pp. 387–444. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Properties | Hardwood | Softwood | Grass | |||
|---|---|---|---|---|---|---|---|
| HW | CO | YP | RW | WP | SG | ||
| Raw material | Moisture, % | 40.85 | 38.72 | 46.80 | 60.21 | 61.07 | 10.28 |
| Carbon, % | 44.82 | 47.79 | 47.67 | 25.99 | 49.30 | 43.22 | |
| Hydrogen, % | 6.06 | 5.82 | 6.33 | 7.71 | 6.48 | 5.22 | |
| Nitrogen, % | 0.07 | 0.25 | 0.30 | 1.35 | 0.15 | 0.59 | |
| Dried biomass | Moisture, % | <10 | 0.67 | 3.24 | <10 | 2.49 | 10.28 |
| Total volatile matter, % | 81.8 | 89.26 | 91.72 | 85.3 | 90.88 | 79.4 | |
| Fixed carbon, % | 7.66 | 8.15 | 3.78 | 4.53 | 6.07 | 8.40 | |
| Total ash, % | 0.54 | 1.76 | 1.26 | 0.17 | 0.57 | 1.80 | |
| Biochar | Moisture (<120 °C), % | 4.39 | 2.69 | 2.01 | 5.23 | 2.03 | 5.94 |
| Light volatiles (120–500 °C), % | 2.02 | 2.67 | 1.63 | 1.56 | 1.74 | 2.76 | |
| Volatiles (500–800 °C), % | 1.84 | 1.25 | 2.01 | 1.57 | 1.47 | 1.94 | |
| Heavy volatiles (800–1000 °C), % | 4.74 | 3.66 | 3.47 | 3.15 | 2.21 | 3.58 | |
| Volatiles (isotherm 1000 °C), % | 5.45 | 2.97 | 3.23 | 3.64 | 2.40 | 3.60 | |
| Carbon yield, % | 80.73 | 87.10 | 88.70 | 83.75 | 90.73 | 82.21 | |
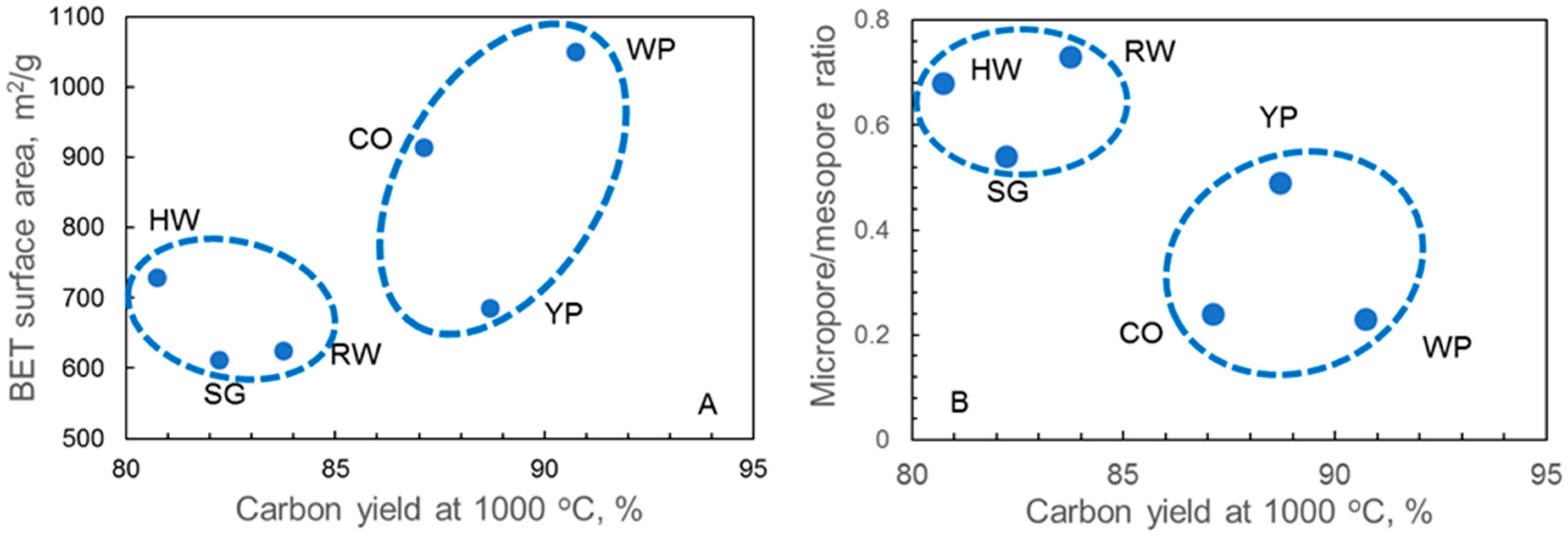
| BET surface area, m2/g | 152 | 241 | 529 | 159 | 351 | 207 | |
| Pore volume (<300 nm), cm3/g | 0.15 | 0.39 | 0.40 | 0.17 | 0.26 | 0.19 | |
| Micropore volume (<2 nm), cm3/g | 0.05 | 0.07 | 0.19 | 0.07 | 0.13 | 0.08 | |
| Mesopore volume (2–50 nm), cm3/g | 0.09 | 0.32 | 0.21 | 0.11 | 0.13 | 0.11 | |
| Activated carbon | Moisture (<120 °C), % | 4.39 | 2.69 | 2.01 | 5.23 | 2.03 | 8.94 |
| Volatiles (120–800 °C), % | 3.86 | 3.92 | 3.64 | 3.13 | 3.21 | 4.70 | |
| Gasified (800 °C), % | 31.01 | 21.32 | 43.75 | 37.27 | 35.21 | 25.25 | |
| Activated product (total yield), % | 60.74 | 72.07 | 50.60 | 54.37 | 59.55 | 61.11 | |
| BET surface area, m2/g | 730 | 915 | 686 | 625 | 1050 | 612 | |
| Pore volume (<300 nm), cm3/g | 0.40 | 1.15 | 0.51 | 0.33 | 1.44 | 0.41 | |
| Micropore volume (<2 nm), cm3/g | 0.27 | 0.28 | 0.25 | 0.24 | 0.33 | 0.22 | |
| Mesopore volume (2–50 nm), cm3/g | 0.10 | 0.85 | 0.25 | 0.07 | 1.06 | 0.18 | |
| Micropore volume (<2 nm), % | 67.5 | 24.3 | 49.0 | 72.7 | 22.9 | 53.7 | |
| Mesopore volume (2–50 nm), % | 25.0 | 73.9 | 49.0 | 21.2 | 73.6 | 43.9 | |
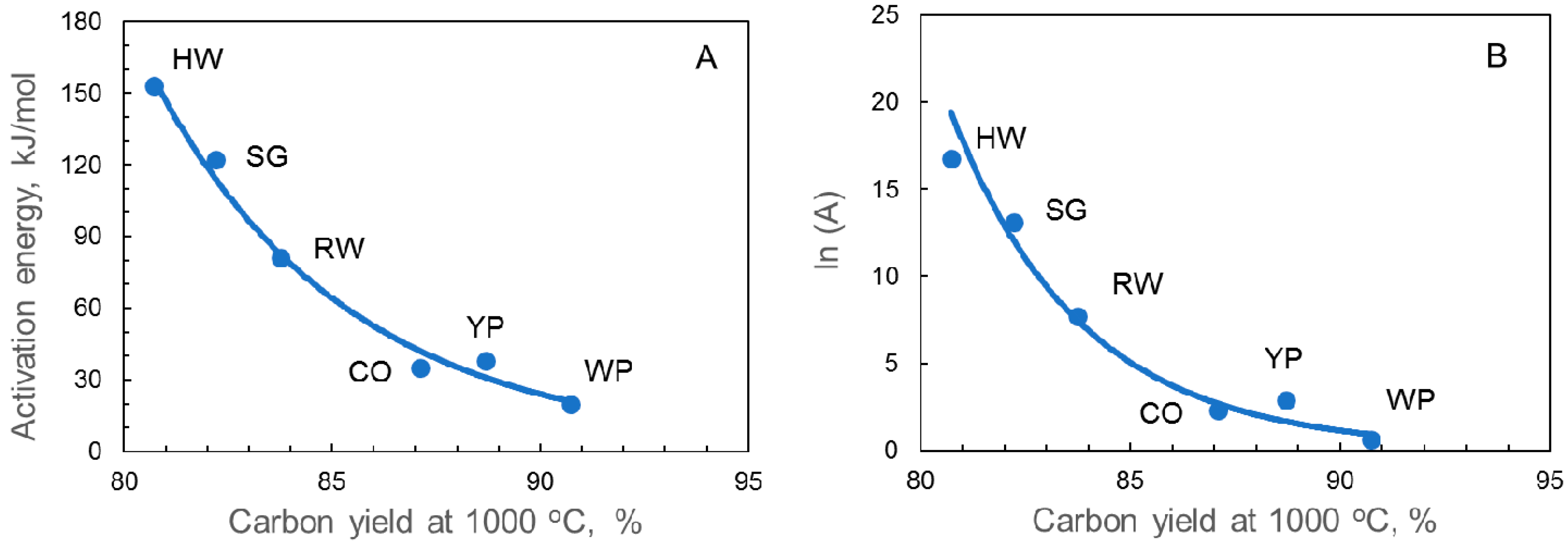
| Samples | Time at 800 °C, min | Eact, kJ/mol | A, min−1 | ln(A) | Weight Loss % | ||
|---|---|---|---|---|---|---|---|
| Global | Moisture-Corrected | Volatiles-Corrected | |||||
| HW char | 43 | 153 | 1.9 × 107 | 16.74 | 38 | 33 | 31 |
| CO char | 143 | 35 | 1.0 × 101 | 2.32 | 27 | 24 | 21 |
| YP char | 113 | 38 | 1.8 × 101 | 2.91 | 47 | 45 | 43 |
| RW char | 119 | 81 | 2.2 × 103 | 7.71 | 44 | 39 | 37 |
| WP char | 148 | 20 | 1.9 × 100 | 0.63 | 39 | 37 | 35 |
| SG char | 58 | 122 | 4.7 × 105 | 13.07 | 34 | 28 | 25 |
| Average weight loss % | 32 ± 7 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contescu, C.I.; Adhikari, S.P.; Gallego, N.C.; Evans, N.D.; Biss, B.E. Activated Carbons Derived from High-Temperature Pyrolysis of Lignocellulosic Biomass. C 2018, 4, 51. https://doi.org/10.3390/c4030051
Contescu CI, Adhikari SP, Gallego NC, Evans ND, Biss BE. Activated Carbons Derived from High-Temperature Pyrolysis of Lignocellulosic Biomass. C. 2018; 4(3):51. https://doi.org/10.3390/c4030051
Chicago/Turabian StyleContescu, Cristian I., Shiba P. Adhikari, Nidia C. Gallego, Neal D. Evans, and Bryan E. Biss. 2018. "Activated Carbons Derived from High-Temperature Pyrolysis of Lignocellulosic Biomass" C 4, no. 3: 51. https://doi.org/10.3390/c4030051
APA StyleContescu, C. I., Adhikari, S. P., Gallego, N. C., Evans, N. D., & Biss, B. E. (2018). Activated Carbons Derived from High-Temperature Pyrolysis of Lignocellulosic Biomass. C, 4(3), 51. https://doi.org/10.3390/c4030051