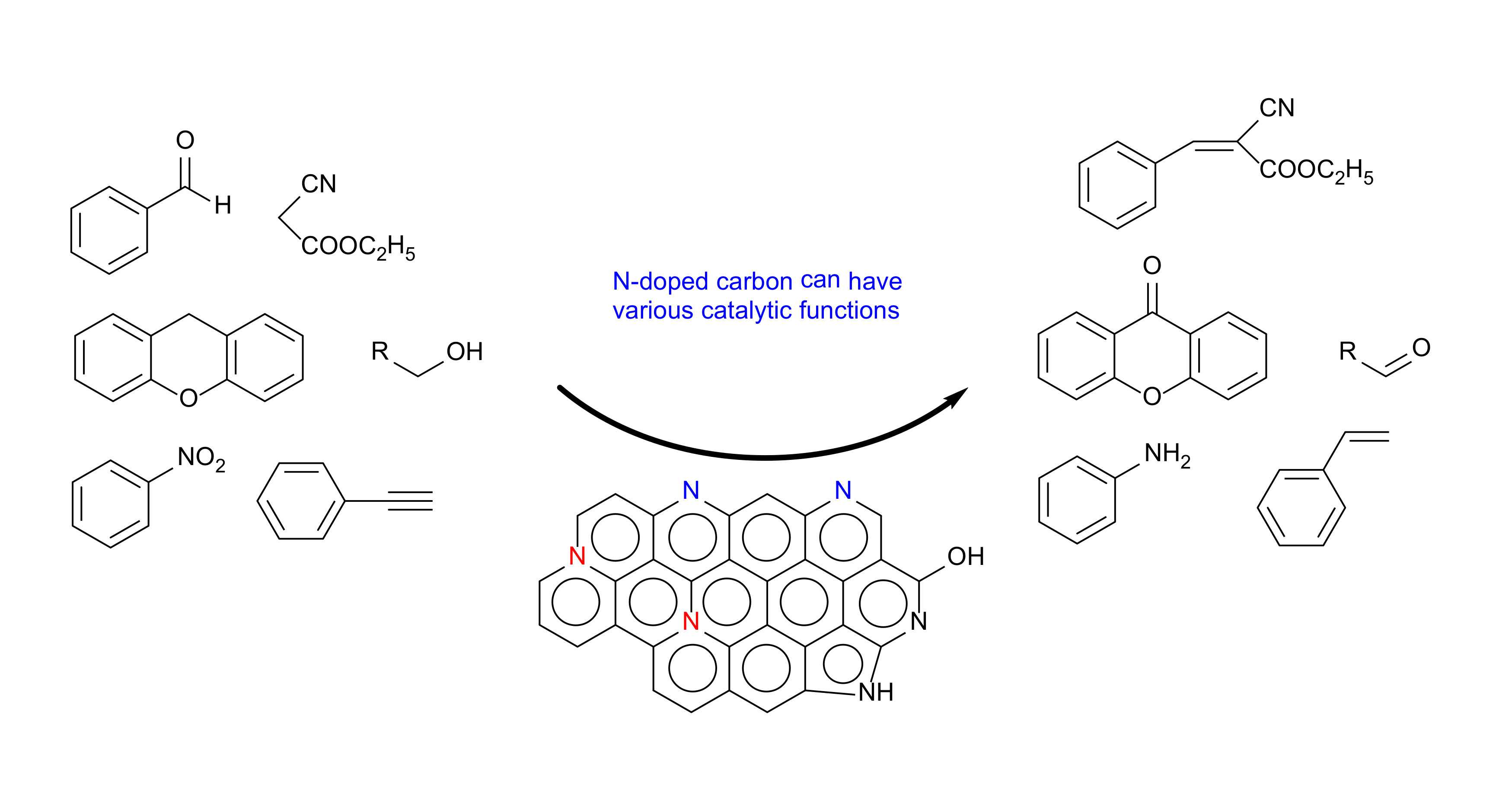
1. Introduction
Various catalysts are used in industry [
1], in which suitable active components are selected and tailored into an effective catalyst for a target reaction under optimized conditions. That is, different catalyst components are selected and used according to target production processes. It is interesting to design and prepare a catalyst, single or multi-component, that can have different active sites on its surface and act as a multifunctional catalyst: the same catalyst can be effectively used for different types of synthetic reaction. One of potential components for such a multifunctional catalyst may be carbon.
Carbon is an interesting material, due to its physicochemical and electrochemical properties and has been used as one of the industrial functional materials. It is also important as either a carrier or a catalyst in the field of catalysis. The carbon materials have a few different functional groups on their surface, depending on their sources and carbonization processes, and these are catalytically active in chemical reactions like oxidation [
2]. Their surface functionalization can further be made by doping foreign species, such as nitrogen [
3]. Nitrogen-doped carbon and carbon nitride would be interesting functional materials. Those modified carbon materials are prepared by different methods, including chemical vapor deposition, well-designed organic synthesis, and doping of nitrogen to parent bulk carbon materials (say, activated carbon). In addition to their effectiveness in electrochemical applications, those carbon-based materials will serve as metal-free catalysts in synthetic organic reactions. It is interesting to indicate, again, that the surface-modified carbon materials including C as a major component and a few other minor ones, such as nitrogen, boron, phosphorous and so on, could catalyze different organic reactions.
Nitrogen-doped carbon and carbon nitride are interesting functional materials that may serve as electrochemical and chemical catalysts and supports for transition metal catalysts, as reported in the literature. For example, Bitter et al. [
4] and Wang et al. [
5] used chemical vapor deposition with nitrogen-containing compounds (nitriles, pyridine, amines) to prepare nitrogen-doped carbon nanotubes. These carbon materials are active for base-catalyzed Knoevenagel condensation. Bitter et al. suggest the importance of pyridine type doped nitrogen species for the genesis of catalytic activity [
4]. A theoretical study indicates that carbon atoms at edge sites of graphite sheets may interact with oxygen molecules with the aid of adjacent doped nitrogen atoms [
6]; then, the nitrogen-doped carbon can show a good performance as a Pt-free electrode in proton-exchange membrane fuel cells [
7]. Thomas et al. show that their well-designed synthetic carbon nitride materials can catalyze Friedel−Craft and cyclization reactions [
8]. Jin et al. [
9] and Xu et al. [
10] indicate porous carbon nitride materials, prepared using porous silica materials as templates, to be active for Knoevenagel and transesterification reactions. The present authors doped nitrogen to an activated carbon (AC) by treating in a mixture of ammonia and air at temperatures of 400–800 °C. The nitrogen-doped AC (N-AC) materials, so prepared, are active for such several liquid phase reactions as Knoevenagel condensation [
11], transesterification [
11], aerobic oxidation of xanthene [
12] and alcohols [
13], and chemo-selective reduction of nitrobenzene, styrene, 3-nitrostyrene, and phenylacetylene with hydrazine [
14,
15], which are ordinarily catalyzed by bases, metals, and metal oxides [
16,
17,
18,
19,
20,
21,
22]. X-ray photoelectron spectroscopy (XPS) surface analysis indicates that different types of nitrogen dopants are involved in the formation of catalytically active sites for those different reactions. In the present work, the authors will review those interesting results to demonstrate the applicability of nitrogen-doped carbon materials as metal-free multifunctional catalysts for several organic synthetic reactions.
2. Preparation of Nitrogen-Doped Activated Carbon
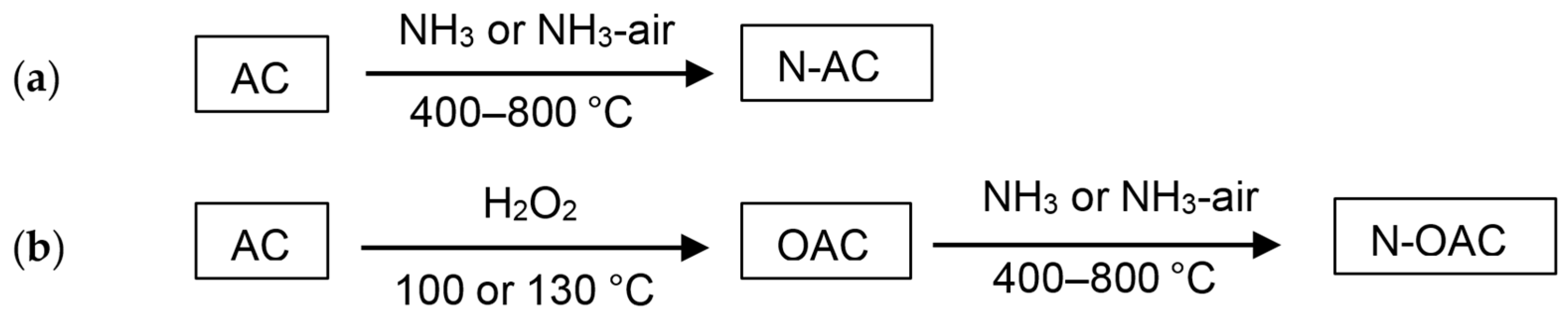
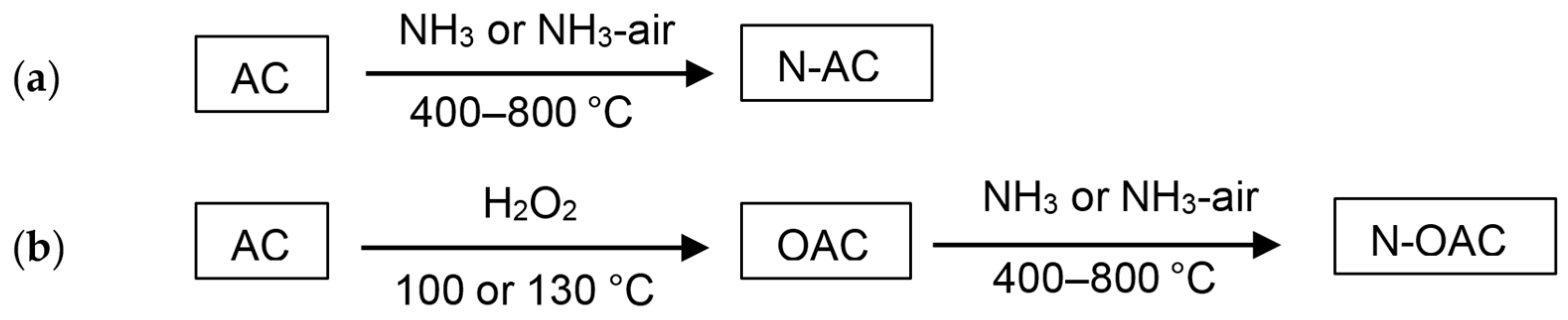
In 1991, Störh et al. reported the preparation of N-ACs from a commercial AC by thermal treatments with ammonia and hydrogen cyanide [
3]. Shortly later, Jansen and Bekkum also reported the preparation of N-ACs from AC by amination (ammonia treatment) and ammoxidation (treatment with an ammonia and air mixture) at elevated temperatures [
23]. Taking the easiness of the procedures into account, our group employed the amination and ammoxidation methods for the preparation of N-AC from a commercial AC (
Figure 1a) [
11,
13]. The total amount of nitrogen doped to AC with the ammoxidation tended to be larger than those with the amination. It was also observed that the amount of doped nitrogen was increased with the increasing doping temperature [
11]. Oxidized AC samples (OAC), which were prepared by H
2O
2 treatments, were also used as the source carbon materials for the nitrogen doping (N-OAC) (
Figure 1b) [
13]. When the same conditions were applied for the nitrogen doping, the amount of nitrogen doped over N-OAC was larger than that over N-AC. Interestingly, the total amount of surface oxygen species was decreased after the nitrogen doping over both AC and OAC [
24]. These observations suggest that surface oxygen species are involved in the nitrogen doping. Possible roles of carboxylic oxygen species for the nitrogen doping were proposed by Jansen and Bekkum [
23]. Of course, other type oxygen species, like ketones and ethers, could also contribute to the nitrogen doping. Although the detailed mechanism of the nitrogen doping is not clear at present, one of the important roles of oxygen species would be abstracting hydrogen atoms from carbon surface and/or ammonia.
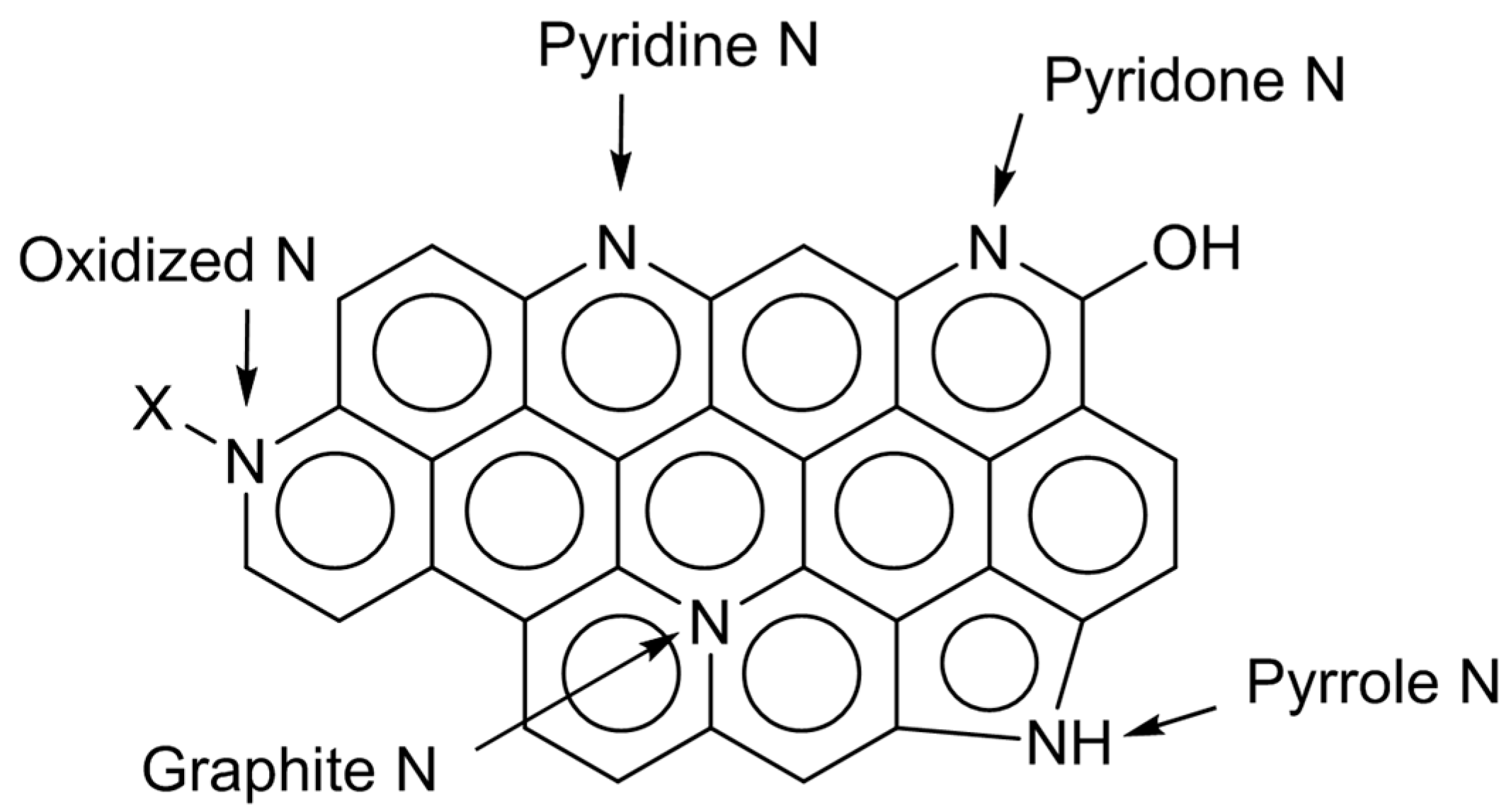
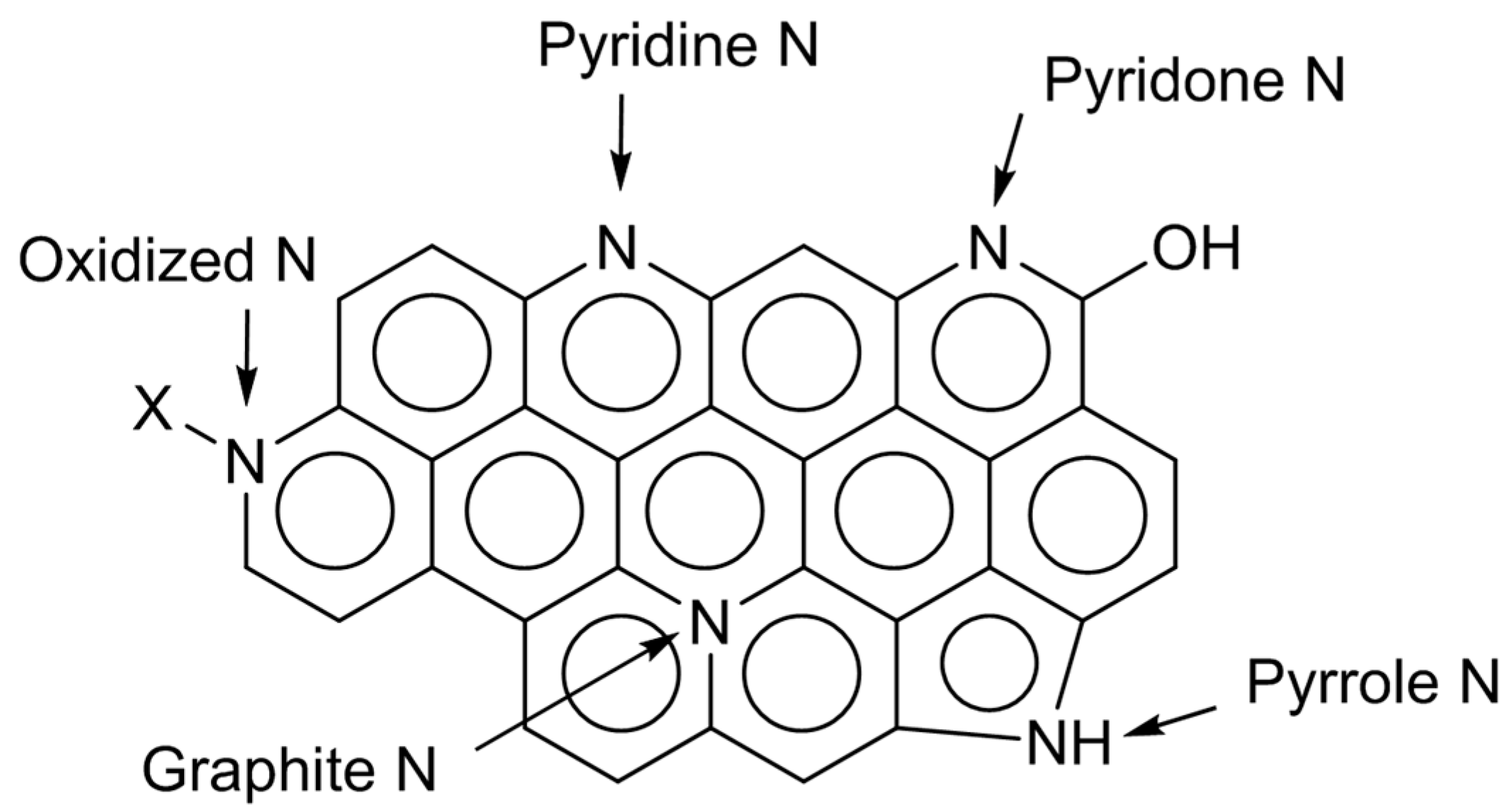
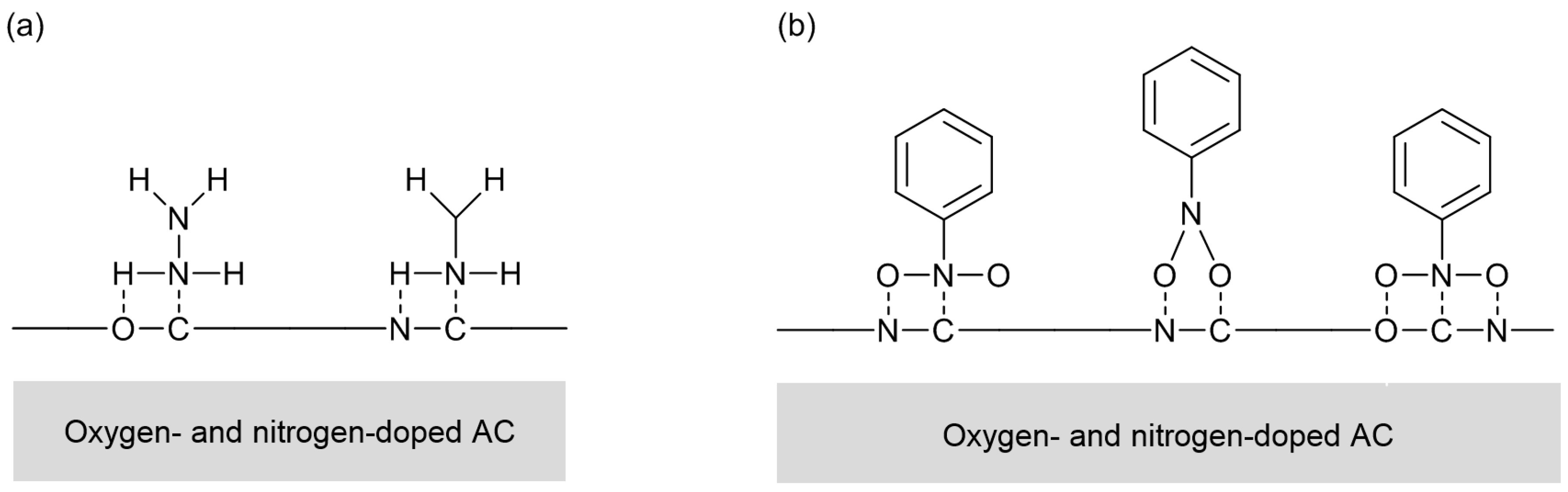
Over nitrogen-doped carbon, there can exist several types of nitrogen species, which are pyridine-type, pyrrole/pyridone-type, graphite-type, and oxidized ones (
Figure 2). The electronic states of those species are different. So, their relative amounts can be estimated by deconvolution of XPS N
1s spectra. In the cases of N-AC, the distribution of nitrogen species was not largely changed by the doping conditions [
11]. On the other hand, N-OAC prepared by the amination showed a larger fraction of graphite-type nitrogen species than N-AC [
13]. Significance of surface oxygen species for nitrogen doping is again suggested. The following sections will describe the use of N-AC and N-OAC thus prepared for several test reactions.
3. Use of Nitrogen-Doped Activated Carbon for Base Catalyzed Reactions
Research on solid base catalysts is still an important issue in the field of catalysis, because they have advantages over homogeneous bases, particularly in the catalyst separation and recovery [
16,
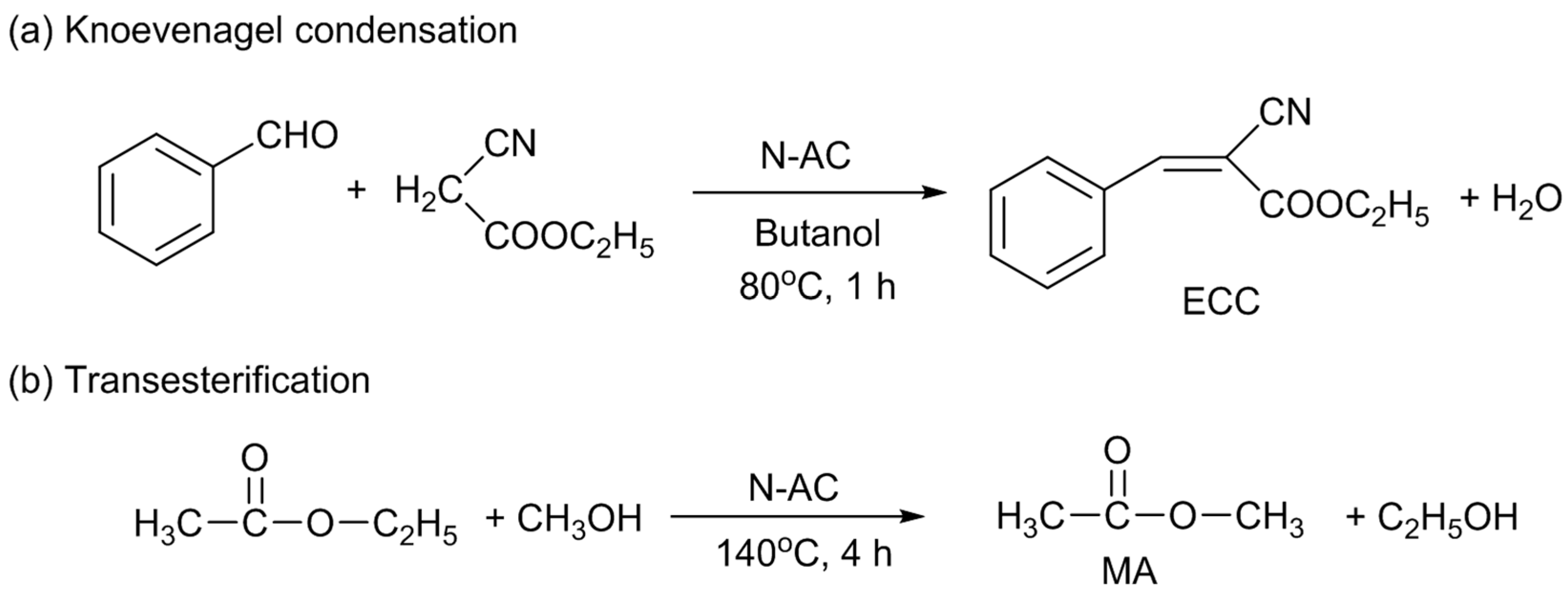
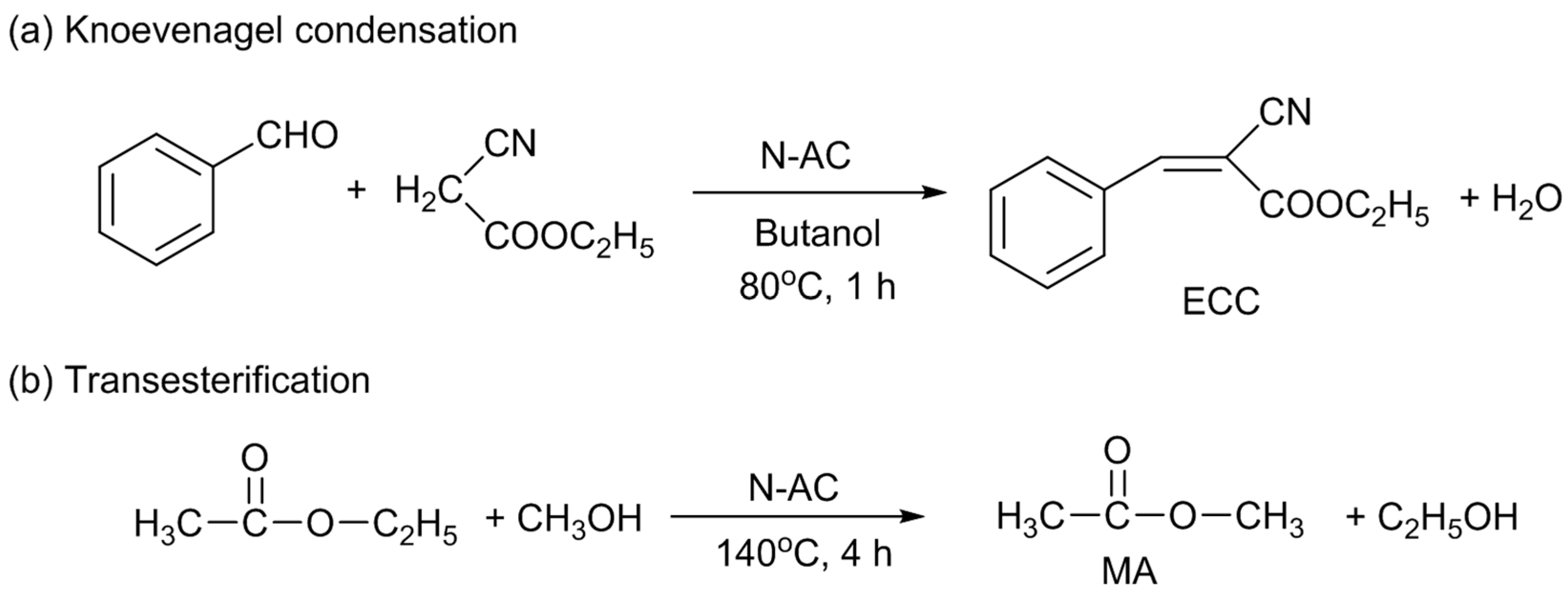
17]. So, at first, Knoevenagel condensation of benzaldehyde and ethyl cyanoacetate and transesterification of ethyl acetate and methanol were selected as the model base catalyzed reactions for the use of N-AC [
10]. The former and latter reactions produce ethyl cyanocinnamate (ECC) and methyl acetate (MA), respectively (
Scheme 1). Both the ECC and MA yields increased with the total amount of nitrogen doped; however, as described above, the distribution of the doped N species did not largely change. So, it could not be identified which of the nitrogen species was responsible for the catalytic activity at that time. N-AC was also used for Knoevenagel condensation reactions of ethyl cyanoacetate with another aldehyde of butyraldehyde and with ketones of acetophenone and benzophenone. N-AC was also active for butyraldehyde, but inactive for the ketones, because of lower reactivities of ketones than those of aldehydes.
In a following study [
25], our group also prepared nitrogen-doped carbon materials from polyacrylonitrile (PAN) and used them for the same Knoevenagel reaction. Nitrogen-doped carbon (N-PAN) was obtained by calcination of PAN in air at a few hundred degrees centigrade, and subsequent ammoxidation at higher temperatures. The resulting N-PAN catalysts had larger amounts of pyridine-type nitrogen species than N-AC, and the catalytic activity of N-PAN was also higher than N-AC. Furthermore, the activity of N-PAN was almost linearly increased with the amount of pyridine-type nitrogen species. On the basis of these observations, it was concluded that pyridine-type nitrogen species are involved in the sites active for the reaction. The same conclusion was made by Bitter et al. for the same reaction with nitrogen-containing carbon nanotubes [
4].
For comparison, liquid pyridine molecules were used for the Knoevenagel condensation and the transesterification reactions [
11]. Their activities for the reactions were much lower than N-AC. Thus, the pyridine-type nitrogen atoms are unlikely to be the active sites by themselves and the presence of them in a large conjugated graphene-like structure would probably be requisite. Kondo et al. showed that nitrogen atoms doped on graphene could modify the electronic conjugated structure of the graphene surface and, furthermore, pyridine-type nitrogen atoms could give Lewis basic nature to neighboring carbon atoms whose number could be more than 10 [
26]. It is well known that base catalysts abstract proton from the methylene group of ethyl cyanoacetate, initiating the Knoevenagel reaction [
17]. Probably, such Lewis basic sites, including both pyridine-type nitrogen and neighboring C atoms, would activate the methylene group of ethyl cyanoacetate, which should be responsible for the higher activity in the presence of the larger amount of pyridine-type nitrogen species.
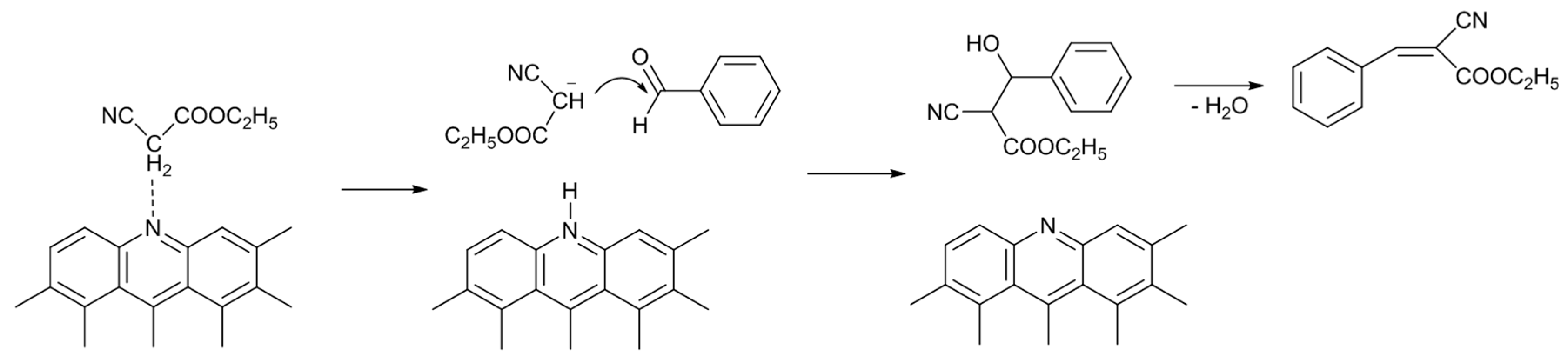
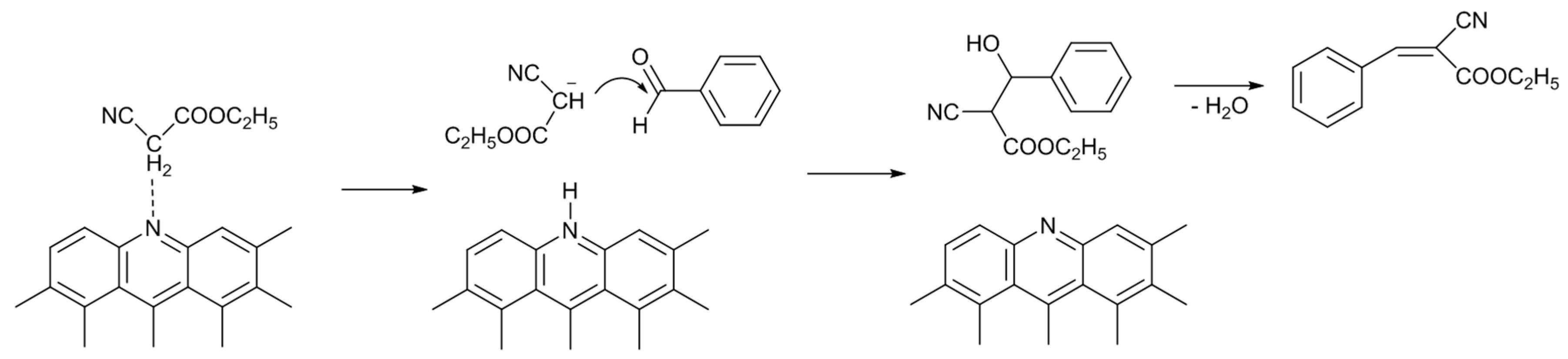
Scheme 2 illustrates a possible reaction mechanism of the Knoevenagel condensation reaction catalyzed by N-AC. A pyridine-type nitrogen atom (and neighboring carbon atoms) abstracts a proton from the methylene group of ethyl cyanoacetate, producing carbocation. Then, the carbocation attacks the carbon atom of the carbonyl group of benzaldehyde, producing ECC through the dehydration of an alcohol intermediate compound.
It was also reported that, for the Knoevenagel reaction, the catalytic performances of the most active N-AC and N-PAN were comparable with, or higher than those of some solid base catalysts reported so far [
25]. Thus, N-AC and N-PAN could be alternatives to conventional solid base catalysts.
4. Use of Nitrogen-Doped Activated Carbon for Aerobic Oxidation Reactions
Oxidation is one of the fundamental transformations in organic chemistry, and for it, the use of molecular oxygen is strongly desired. So, as the second step, N-AC and N-OAC were applied for the aerobic oxidation of xanthene and alcohols. For these reactions, Hayashi et al. had already reported the use of untreated commercial AC samples [
2,
27,
28,
29]. Because the AC catalysts required rather high reaction temperatures and/or long reaction time to get reasonable product yields, these reactions were supposed to be suitable ones to examine the catalytic performance of N-AC. At first, the oxidation of xanthene (XT) to xanthone (XO) was carried out (
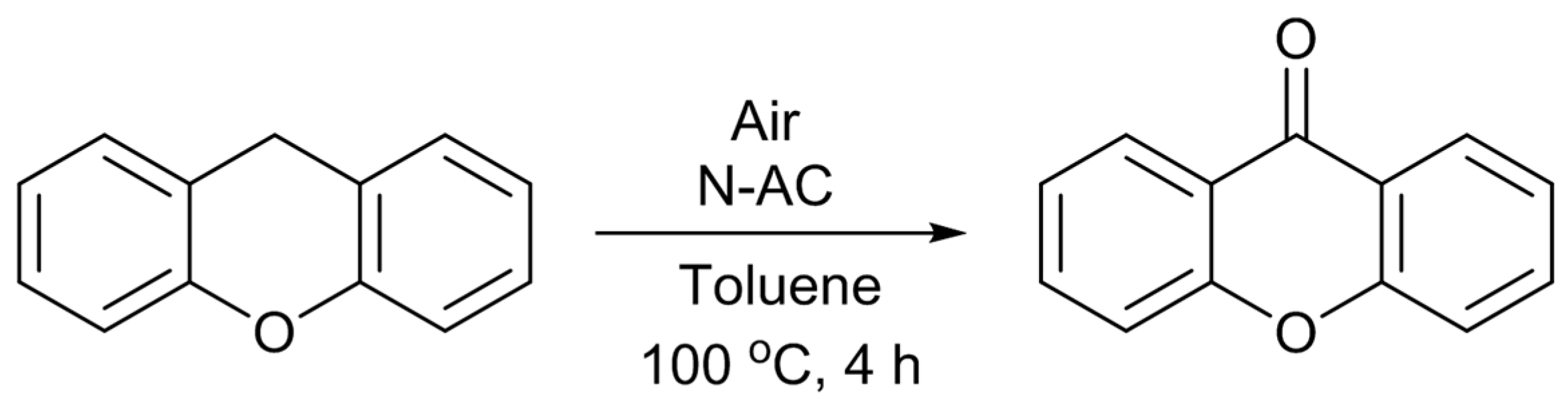
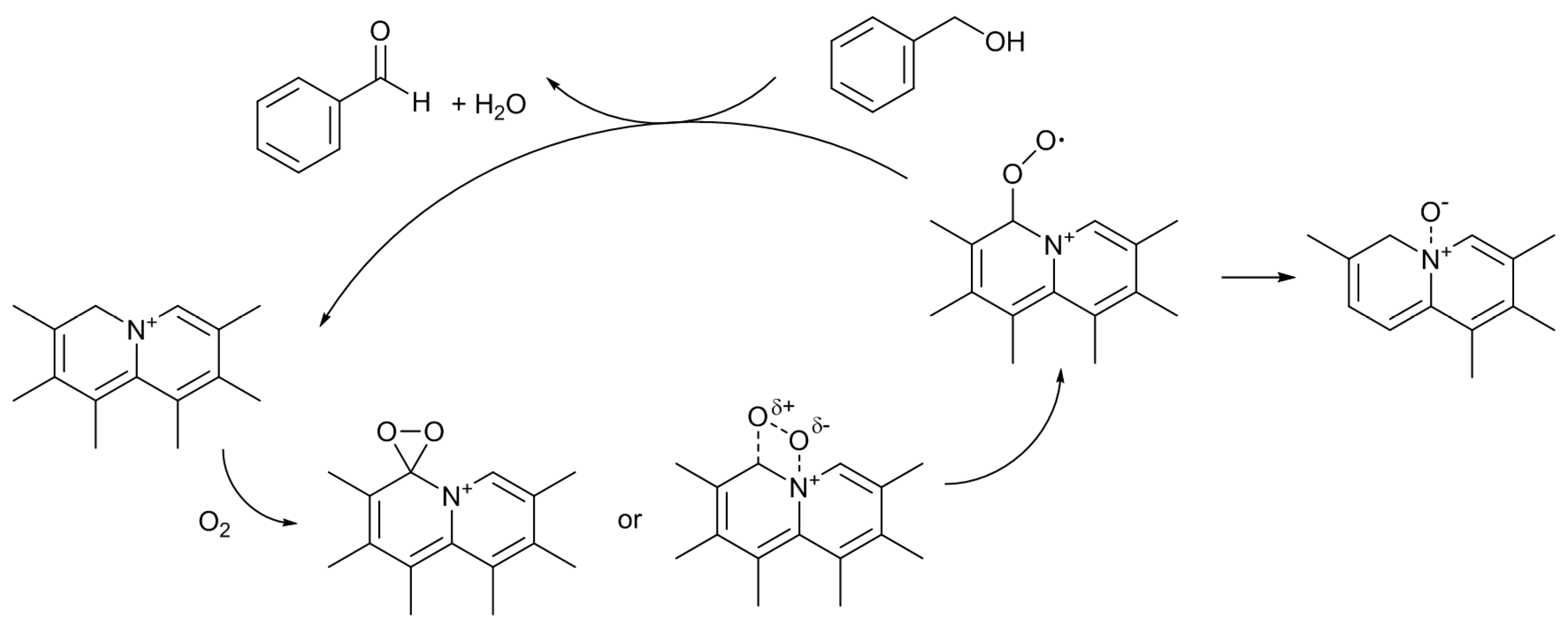
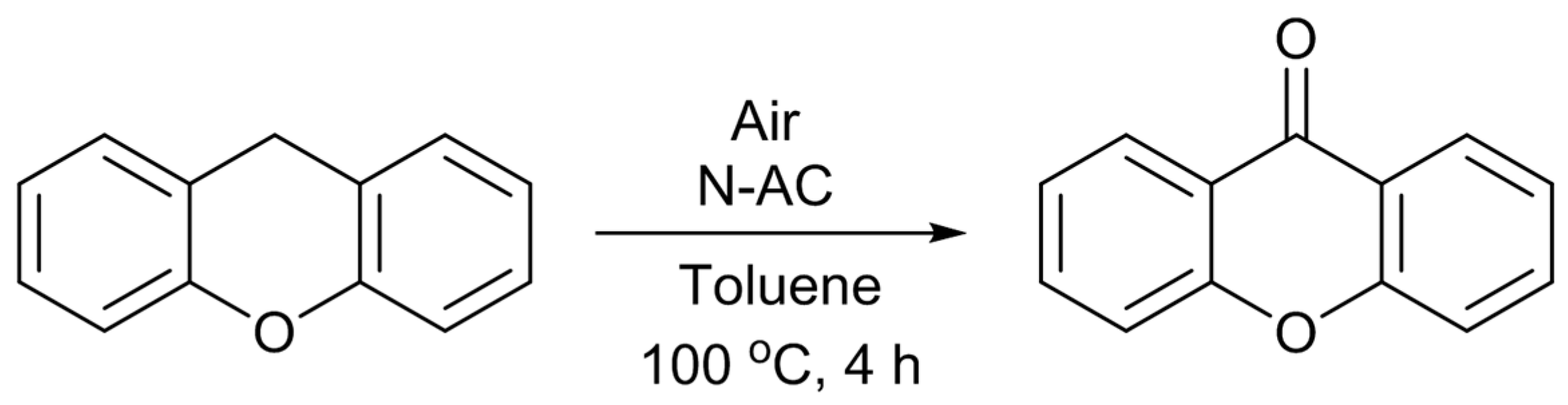
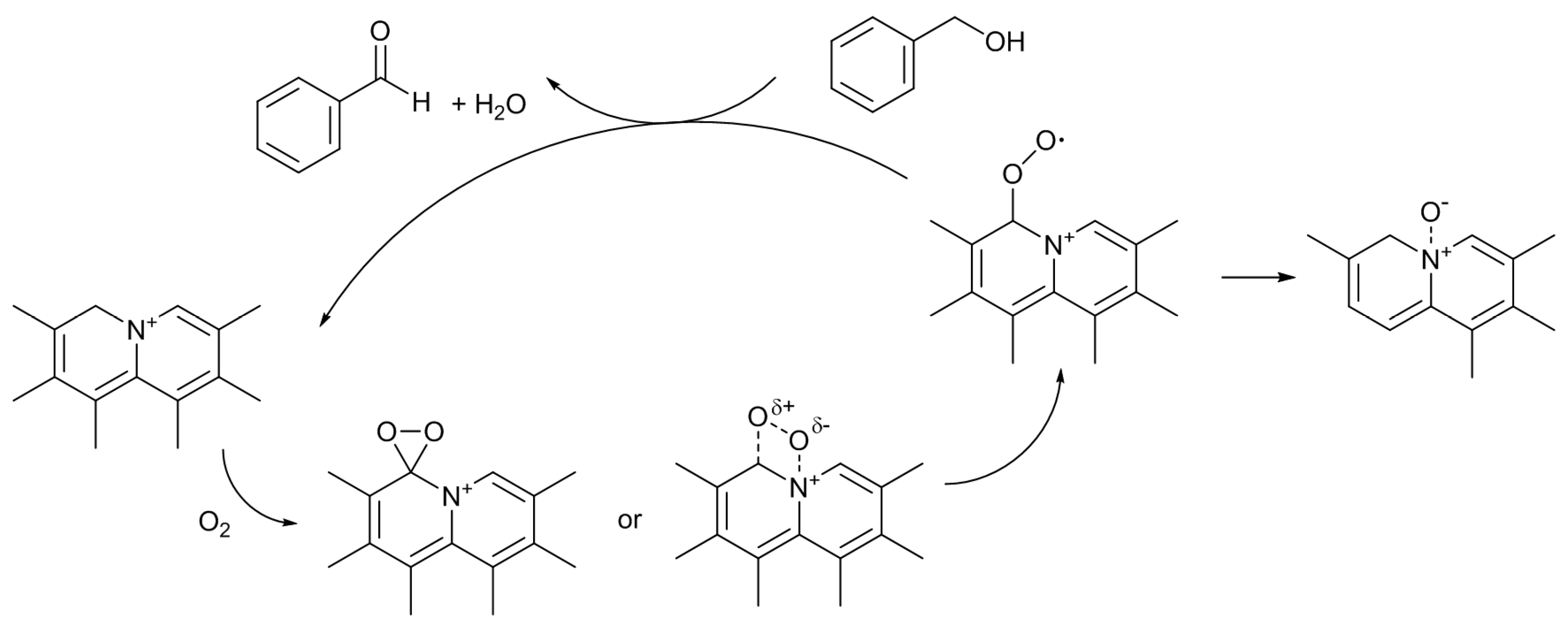
Scheme 3) [
12]. Under the reaction conditions employed, only XO was formed as the product. The catalytic activity of the untreated AC was very low, and it gave only 4% of the XO yield. As expected, the catalytic activity was largely enhanced by the nitrogen-doping. The most active N-AC gave an XO yield of 38%. The yield was found to increase with the amount of doped pyridine-type nitrogen species, although there was some scatter in the plot, suggesting that this type of nitrogen species is involved in the active sites for the reaction. However, similar to the case of Knoevenagel reaction, liquid pyridine molecules had no activity for the oxidation reaction. The significance of the presence of pyridine-type nitrogen atoms in a large conjugated graphitic structure was again suggested. As described above, doped pyridine-type nitrogen and surrounding carbon atoms have Lewis basic nature. Similar to the cases of the Knoevenagel condensation, the methylene group of XT could also be activated by such Lewis basic sites. Furthermore, Nishida and Hayashi have proposed that the cleavage of the benzylic C–H bond of XT initiates the reaction, and XO is formed through the formation of a hydroperoxide compound [
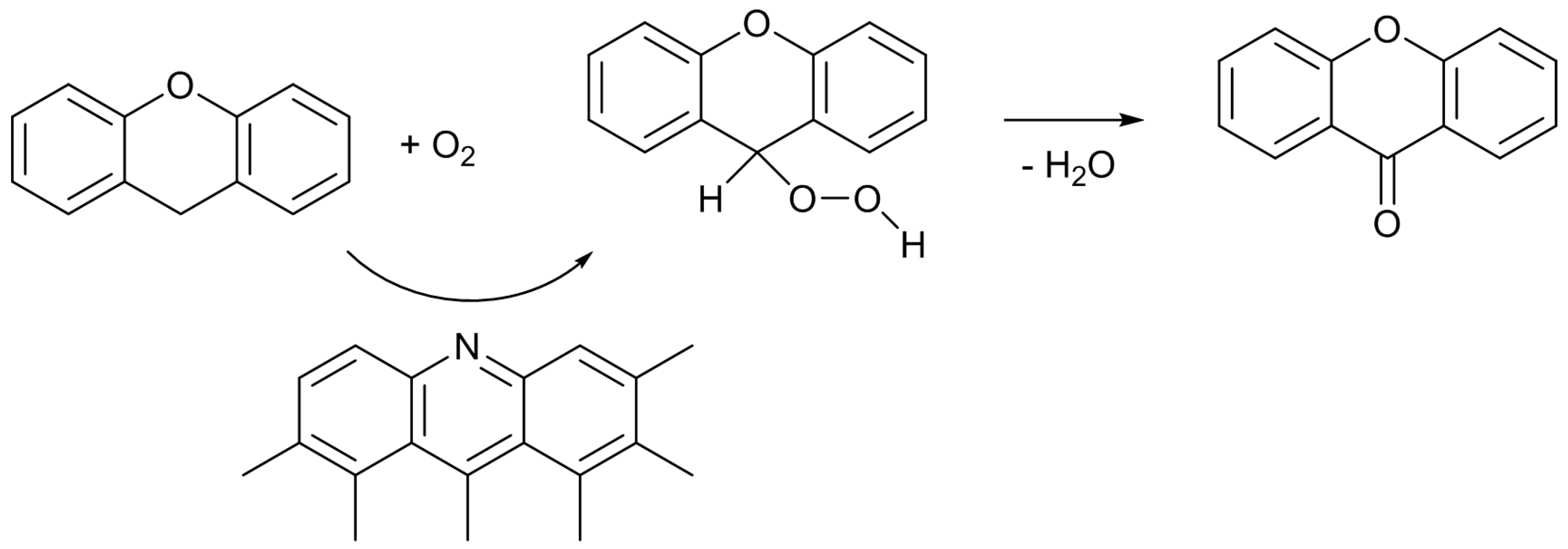
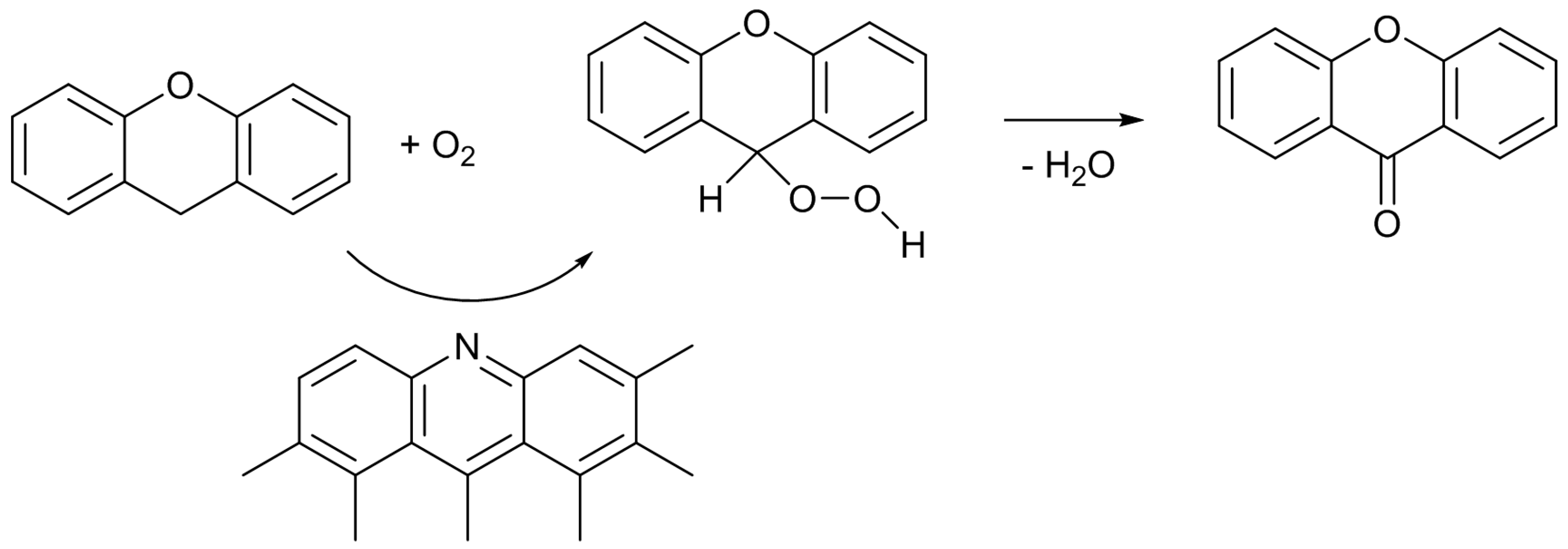
29]. Hence, a plausible reaction mechanism can be illustrated as
Scheme 4. Although the details are not clear at present, the interactions among the surface Lewis basic sites involving pyridine-type nitrogen, the methylene group of XT, and oxygen molecule, would result in the formation of the hydroperoxide, which is converted to XO by dehydration.
The recyclability of N-AC was examined [
12]. The activity of N-AC slightly decreased after the first recycling, but it did not further change during the following two-time recycling runs. To elucidate the reason of the decrease in the activity of N-AC, XPS spectra of the recycled N-AC were measured. It was found that oxidized nitrogen species were formed by the catalyst recycling. This suggested that a part of doped nitrogen species was oxidized during the first reaction run, resulting in the decrease in activity. But further formation of the oxidized species would not occur during the following reaction runs, and so the deactivation was not observed during the following recycling runs.
For comparison, commercial Ru/AC and Pd/AC catalysts were also used for the reaction [
12]. The XO yield was in the order of Ru/AC (XO yield = 98%) > N-AC (38%) > Pd/AC (25%). Thus, N-AC needs a longer reaction time than Ru/AC to get good conversion levels; however, one can say that the metal-free N-AC could be a less costly substituent for Ru/AC. Pd/AC was also subjected to catalyst recycling experiments. Similar to the cases of N-AC, the activity of Pd/AC slightly decreased after the first recycling, but further change of the activity was not observed during the following two-time recycling runs.
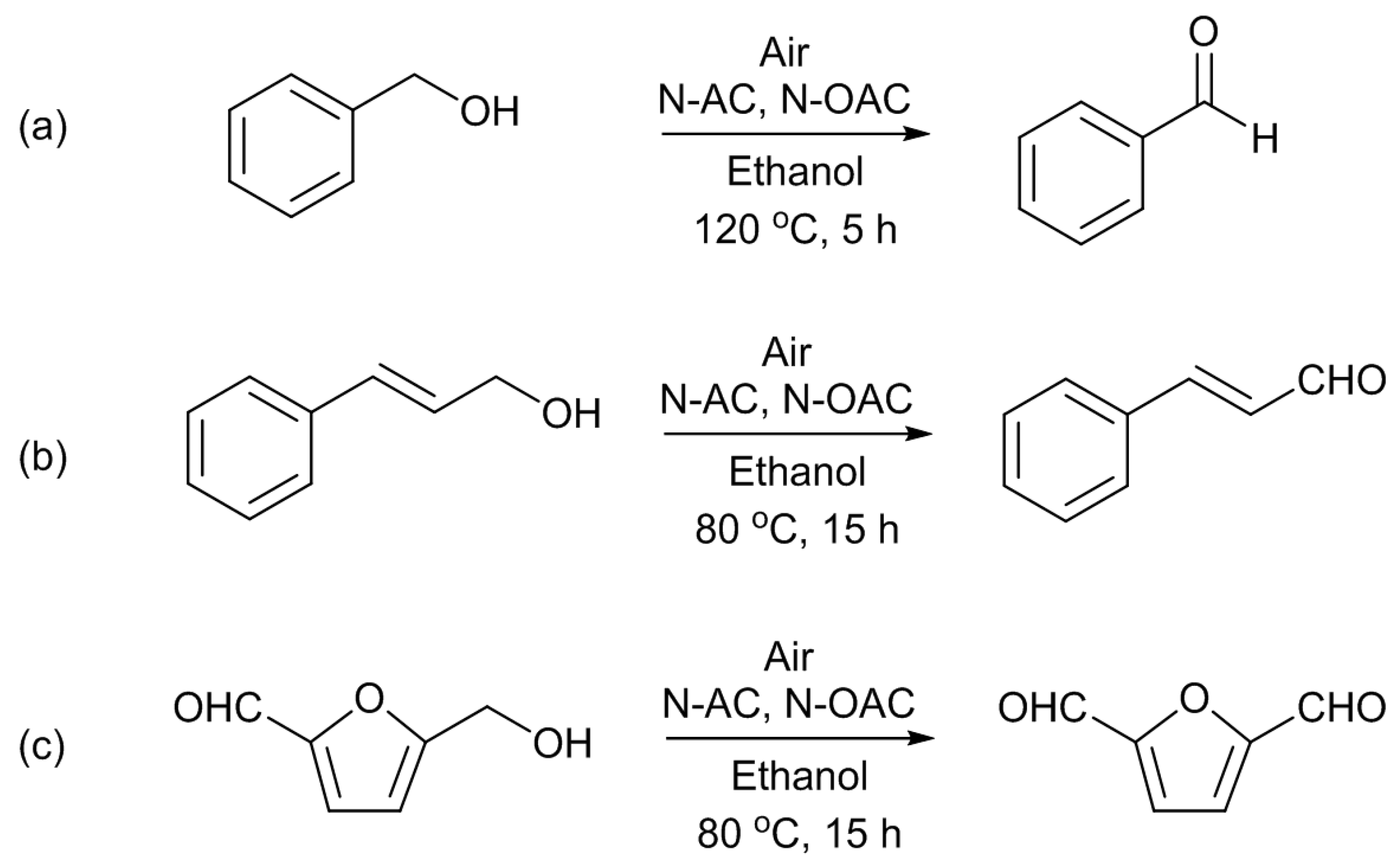
Next, the aerobic oxidation of alcohols was conducted using AC, OAC, N-AC, and N-OAC [
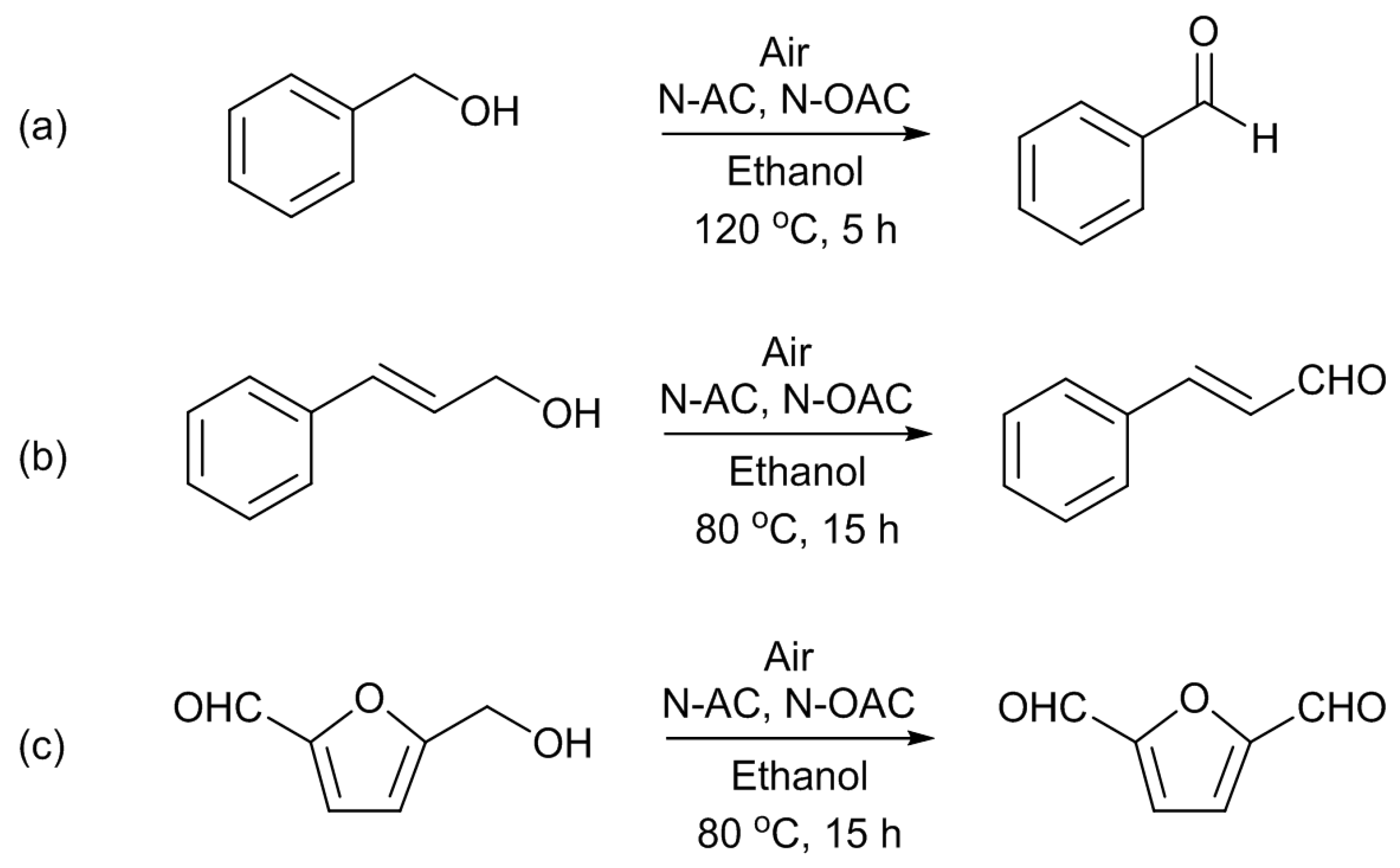
13]. The substrates used were benzyl alcohol (BAL), cinnamyl alcohol (CAL), and 5-hydroxymethyl-2-furaldehyde (HMF) (
Scheme 5). The last substrate is one of platform chemicals from biomass resources. BAL required a higher temperature (120 °C) to get reasonable product yields than CAL and HMF (80 °C). The selectivity to aldehydes was 100% for the BAL and CAL oxidations, but it was slightly lower for HMF, because of the formation of the acetal from the substrate and the solvent of ethanol. Other alcohols of cyclohexylmethanol (CHM) and 3-phenyl-1-propanol (HCAL), which are analogous to BAL and CAL, respectively, were also used as the organic reactants for the oxidation; however, it was found that they were inactive under the reaction conditions employed. The difference between the active alcohol group (BAL, CAL, HMF) and the inactive one (CHM, HCAL) was ascribed to the presence of a conjugated system adjacent to the carbon atom bonding to the hydroxyl group.
For all the alcohol substrates shown in
Scheme 5, the product yield with the parent AC and OAC were low, and were largely enhanced by nitrogen doping, depending on the doping conditions. The H
2O
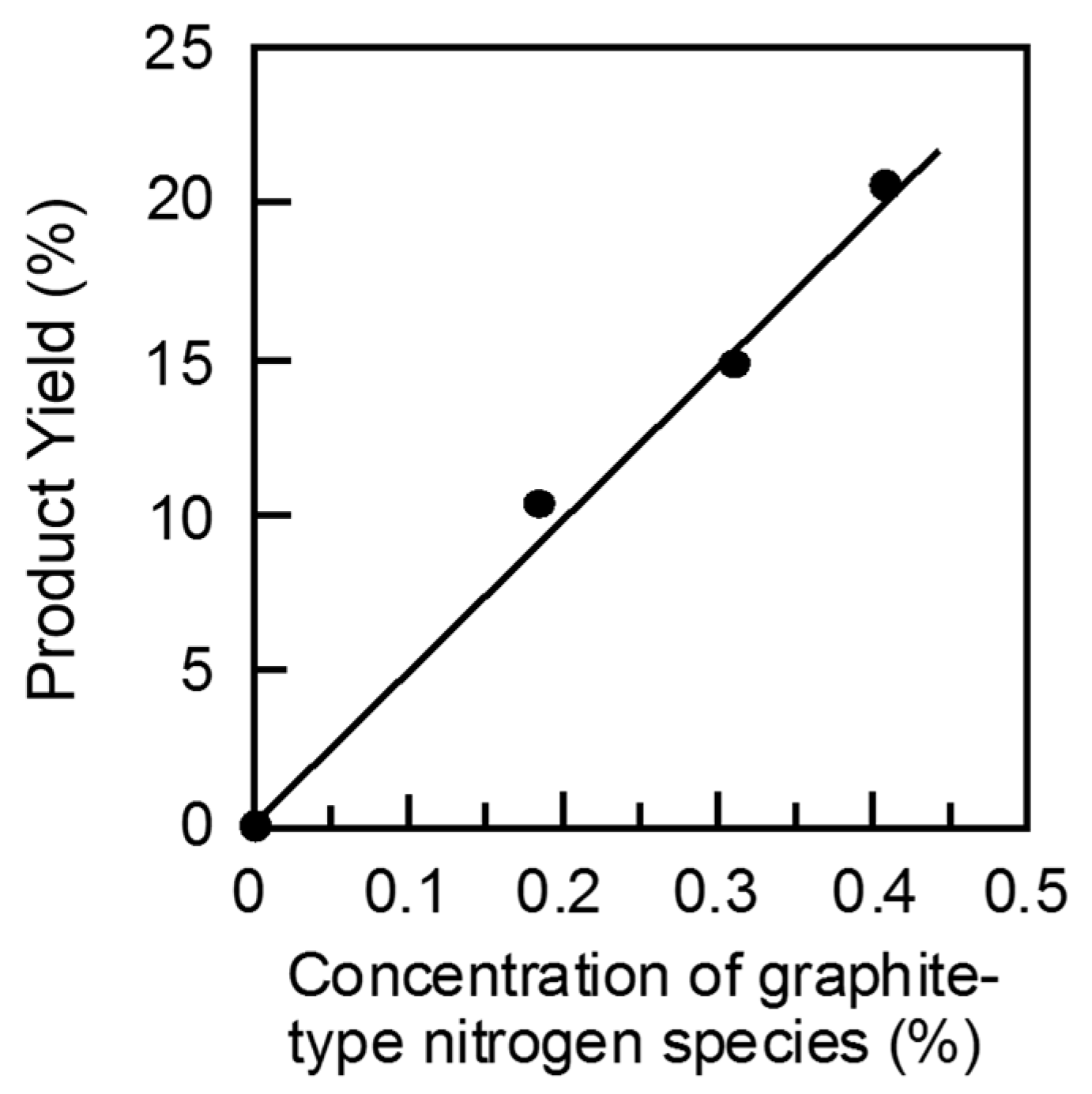
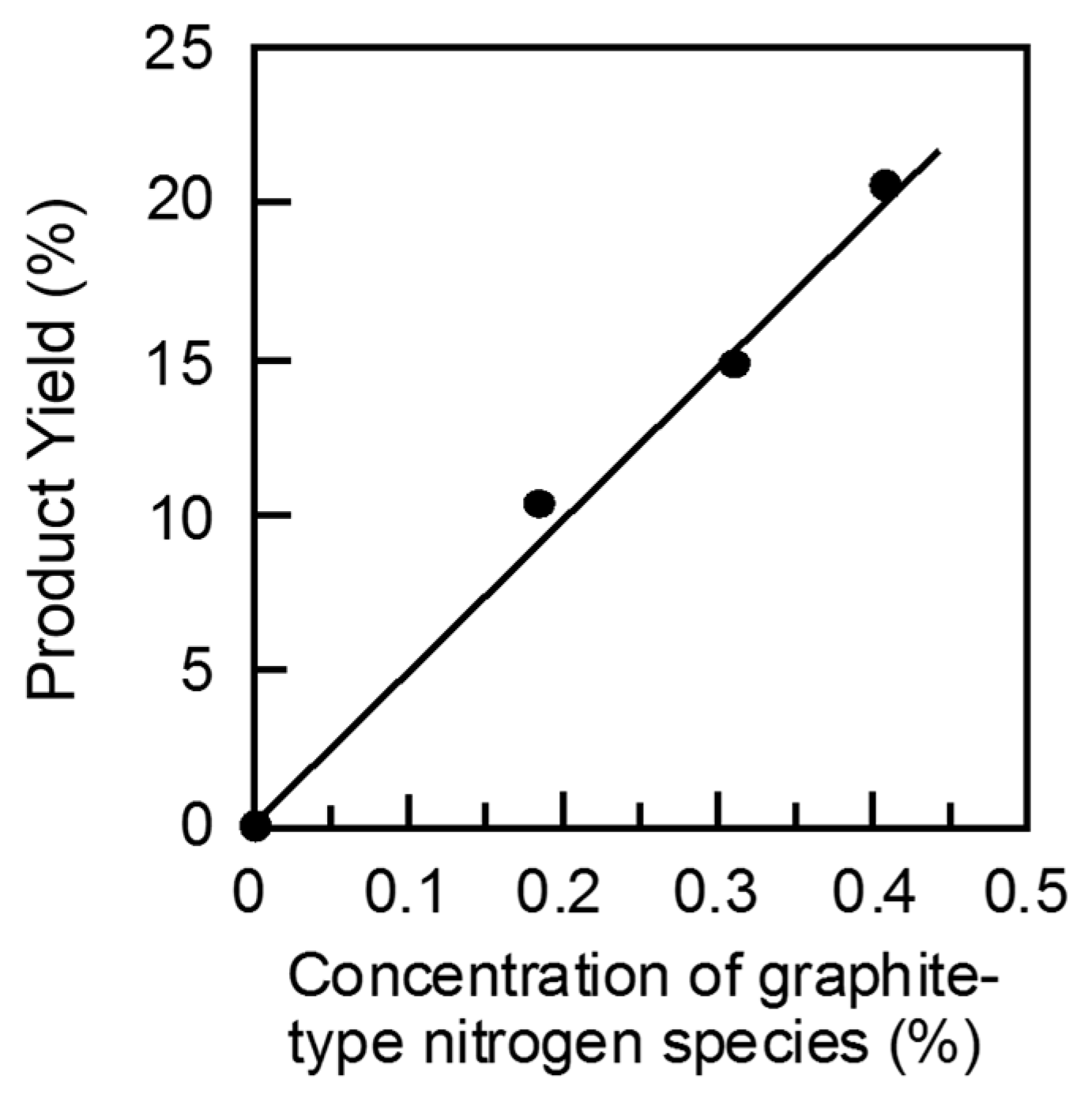
2 treatment of AC at 130 °C and successive amination of the resulting OAC at 800 °C gave the most active N-OAC catalyst. The relationship between the yield and the amount of each different nitrogen species was examined. As shown in
Figure 3, a good linear relationship is seen between the product yield of the HMF oxidation and the amount of graphite-type nitrogen species, but no good correlation could not be found for the other two types of nitrogen species, pyridine-type and pyrrole-type nitrogen species [
30]. Similar results were obtained for the BAL and CAL oxidation reactions [
13], although there was a scatter of the plot at small amounts of graphite-type nitrogen species for the former reaction. These observations strongly suggest that graphite-type nitrogen species contribute to the genesis of active sites on the surface of AC for the oxidation of the alcohols. This is in accordance with the results reported by Long et al., who used nitrogen-doped graphene nano-sheet catalysts for the same BAL oxidation reaction [
31].
Thus, graphite-type nitrogen species would be responsible for the catalytic activity of N-AC (and N-OAC) for the alcohol oxidation. On the other hand, as described above, the active sites for the oxidation of XT were suggested to involve pyridine-type nitrogen species. For these reactions, the activation of oxygen molecules is an important factor. For oxygen reduction reaction (ORR) with nitrogen-doped carbon materials, the type of nitrogen dopants creating active sites are still controversial, which is an important issue for the use of such carbon materials for fuel cells as alternatives to high-cost Pt catalysts. For example, a model calculation of Ikeda et al. [
6] indicates that an oxygen molecule may be activated on a carbon atom bonded to a graphite-type doped nitrogen atom at edge sites, while the ORR study of Guo et al. [
32], using model nitrogen-doped carbon catalysts, showed that such activation would occur at a carbon atom bonded to a pyridine-type doped nitrogen atom. There is also a possibility that both graphite-type and pyridine-type doped nitrogen atoms contribute to the oxygen activation, but their effectiveness would be different. Of course, the activation of the organic substrates (XT and alcohols) should also be taken into account. Further computational and atomic level studies are required to discuss the difference in the active sites between the XT oxidation and the alcohol oxidation reactions in more detail.
The most active N-OAC was subjected to recycling runs. The activity of N-OAC gradually decreased during the first two-time recycling, but not during the following recycling runs. XPS measurements of the fresh and recycled N-OAC revealed that oxidized nitrogen species were formed by the catalyst recycling. These observations are very similar to those for the XT oxidation mentioned above. When the BAL oxidation was carried out in the presence of a radical scavenger of
p-benzoquinone, the reaction was almost stopped, revealing that radical species are included in the oxidation processes. On the basis of these results, possible reaction pathways were proposed (
Scheme 6) [
13].
The catalytic performance of the most active N-OAC was compared with those of conventional carbon-supported noble metal catalysts, Ru/AC and Pt/AC [
13]. Ru/AC was more active than Pt/AC, and a smaller amount of Ru/C could give product yields similar to those with N-OAC for a shorter reaction time. Also, Ru/C could oxidize CHM and HCAL, which were inactive with N-OAC. Thus, Ru/C was much more active than N-OAC; however, the selectivity values for aldehydes obtained with Ru/C were much lower than those with N-OAC because of the formation of acetal. The formation of acetal from aldehyde and alcohol is known to be catalyzed by acid. Probably, nitrogen-doping on AC and OAC reduced the number of the acid sites existing on their surfaces, resulting in the high selectivity to aldehyde. From the viewpoint of the selectivity, N-OAC would be more preferable than Ru/AC.
5. Use of Nitrogen-Doped Activated Carbon for Transfer Hydrogenation with Hydrazine
The last example of the utilization of N-AC and N-OAC is the transfer hydrogenation with hydrazine. Hydrogenation is one of the important organic synthesis reactions. A number of chemicals are produced by hydrogenation processes. Among several reducing reagents, hydrazine is an effective one, as well as gaseous hydrogen. Huang et al. had reported interesting results of 3-nitrostyrene hydrogenation with a hydroxyapatite supported rhodium catalyst [
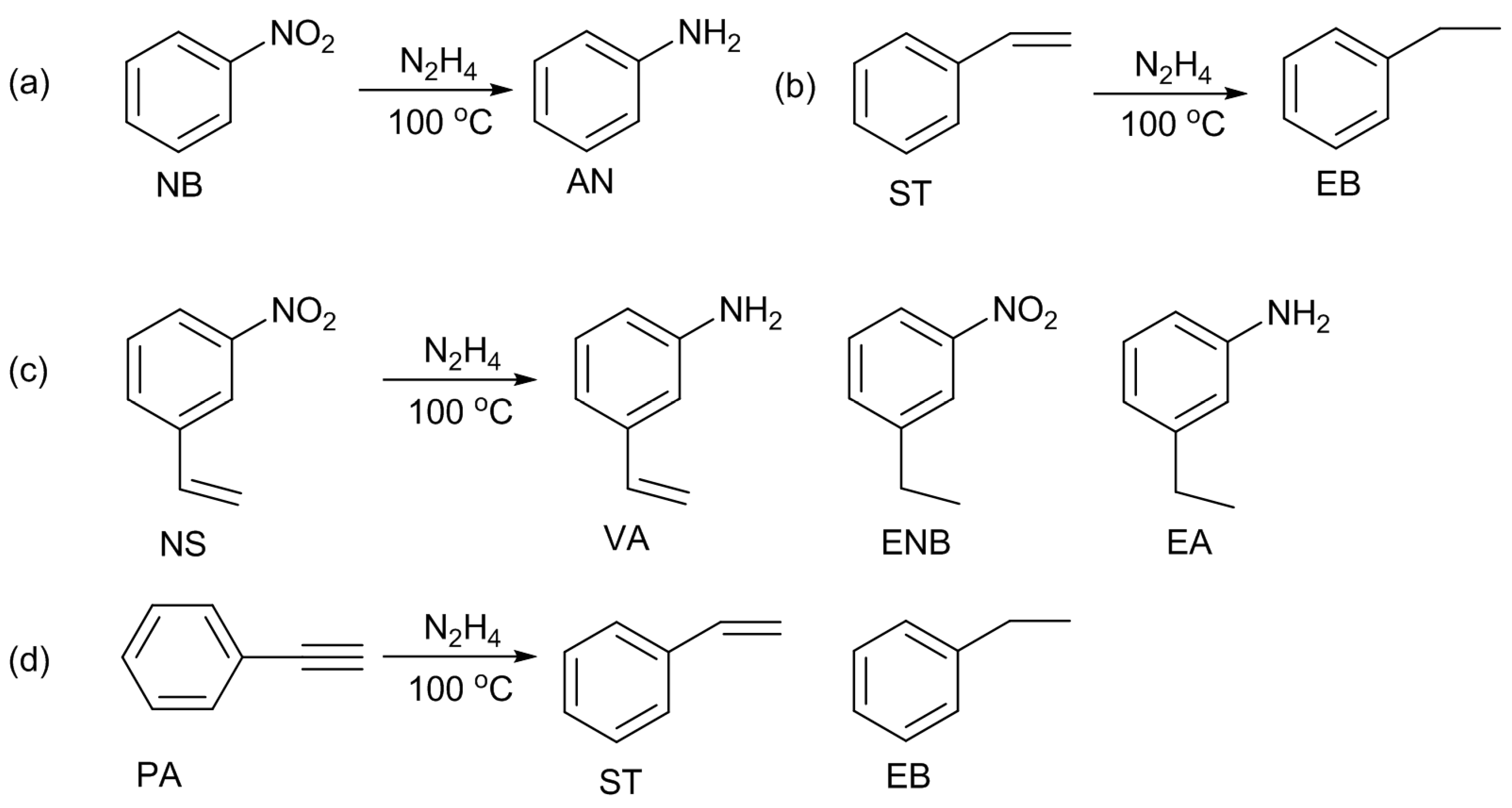
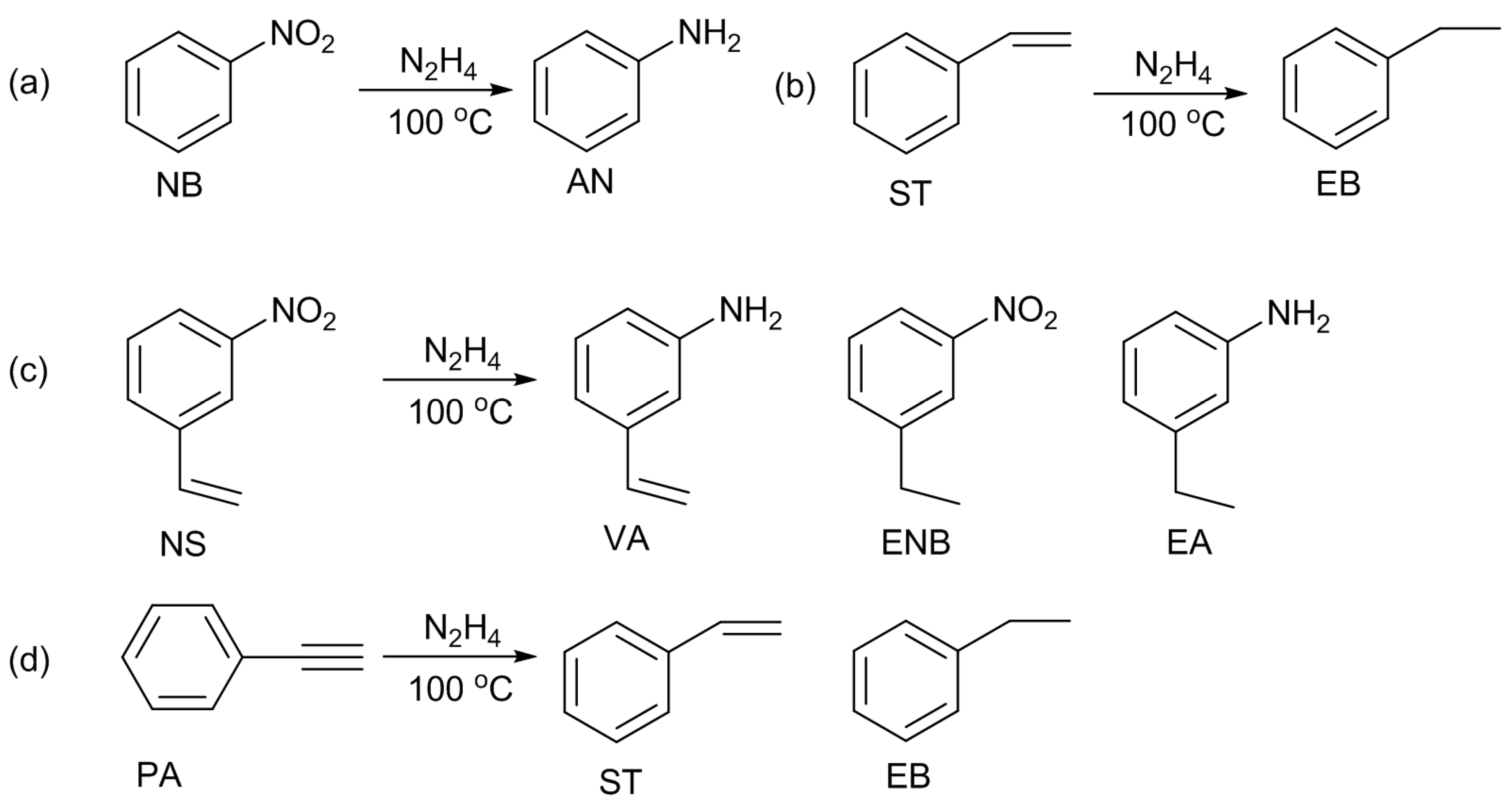
22]. When hydrazine was used as the reducing reagent, 3-vinylaniline was selectively produced. On the other hand, 3-ethylnitrobenzene was selectively formed when gaseous hydrogen was used. The selectivity of the hydrogenation of nitro or alkene groups can be tunable by selecting the reducing reagent. Under these circumstances, our group employed N-AC and N-OAC for the hydrogenation of nitrobenzene (NB), styrene (ST), and 3-nitrostyrene (NS) with hydrazine (
Scheme 7a–c) [
14].
Under the reaction conditions used, the NB hydrogenation selectively produced aniline. This reaction proceeded without the carbon catalysts, and gave a NB conversion of 26%. The parent AC was active, and gave 40% conversion. The H
2O
2 treatment of AC significantly enhanced the activity. The un-doped OAC showed 80% conversion. Such enhancement was cancelled by the nitrogen-doping. N-OAC gave lower NB conversion than OAC. On the other hand, the conversion obtained with N-AC was higher than that with AC. These results imply that both surface oxygen and nitrogen species contribute to the formation of active sites. Actually, the NB conversion increased linearly with the total amount of surface oxygen and nitrogen species [
14]. Probably, those sites would activate both or either of hydrazine and NB molecules, as illustrated in
Scheme 8 [
14].
For comparison, one selected N-AC was employed for the NB hydrogenation using gaseous hydrogen at 2 MPa. No reaction occurred. Probably, the ability of N-AC to activate molecular hydrogen would be insufficient.
The ST hydrogenation also proceeded in the absence of the catalysts at a conversion level of 11%. The conversion was improved by using the carbon catalysts (AC, OAC, N-AC, N-OAC); however, the extent of the improvements were very small (<7%), revealing that the activity of those catalysts for the ST hydrogenation was very low. The activation of the vinyl group in ST molecule might be difficult over the carbon catalysts.
Those results of NB and ST hydrogenation reactions led us to investigate the use of the carbon catalysts for the hydrogenation of NS, which has two reducible substituents of nitro and vinyl groups. Selective hydrogenation of a nitro group is an important issue, when another reducible group is present in the same molecule. Considering the higher NB conversions than the ST conversions, we expected the selective formation of 3-vinylaniline (VA) from NS with hydrazine in the presence of the carbon catalyst. This product is the most valuable compared with the other hydrogenated products of ethylnitrobenzene (ENB) and ethylaniline (EA). In the absence of catalyst, the NS conversion was 55%; this value was much higher than those of the NB (26%) and ST (11%) hydrogenations. AC and N-AC gave similar NS conversions (62% and 56%). In contrast, OAC and N-OAC yielded much higher NS conversions of 97% and 93%, respectively. Thus, in every case, the NS conversion was higher than NB and ST conversions, suggesting that the nitro group of NS molecule promoted the hydrogenation of the vinyl group.
Product distribution was changed by the catalyst. In the absence of catalyst, the selectivity to the most desired product of VA was 16%. The selectivity to VA was in a region between 20% and 29% with AC, N-AC, and OAC, while N-OAC gave the highest VA selectivity of 54%. Thus, being different from our expectations, the selectivity to VA was not so high. This would result from the above-mentioned promotional effect of the nitro group on the hydrogenation of the vinyl group. However, one can say that the presence of surface nitrogen species suppressed the hydrogenation of the vinyl group of NS and/or VA.
The promotional effect of the nitro group was further investigated for the hydrogenation of the ethynyl group of phenylacetylene (PA) (
Scheme 7d) by using one selected N-OAC [
15]. In the absence of catalyst, the hydrogenation of PA alone occurred at a small extent (4%). Even in the presence of N-OAC, the same PA conversion was obtained, revealing that N-OAC was inactive for the hydrogenation of PA alone. When NB was added to the reaction mixture, the PA conversion was enhanced, and the enhancement became larger in the presence of N-OAC. Thus, N-OAC is active for the PA hydrogenation assisted by NB co-existing in the reaction mixture. On this NB-assisted reaction, the NB/PA molar ratio had no effect above 2.
The PA hydrogenation was carried out by adding other organic compounds of benzaldehyde, benzyl cyanide, and aniline; however, these compounds had no effect on the hydrogenation reaction. Thus, NB has a specific nature for assisting the hydrogenation of ethynyl group.
In the hydrogenation of acetylene compounds, the selective production of olefins at high conversion levels is a challenging task. In the cases of using N-OAC for the NB-assisted PA hydrogenation, the selectivity to ST was decreased with the reaction time, i.e., the PA conversion.
As described above, the surface nitrogen and oxygen species are involved in the generation of active sites for the reduction of NB with hydrazine. The reduction of NB should start with the adsorption on the catalyst through its nitro group, as illustrated in
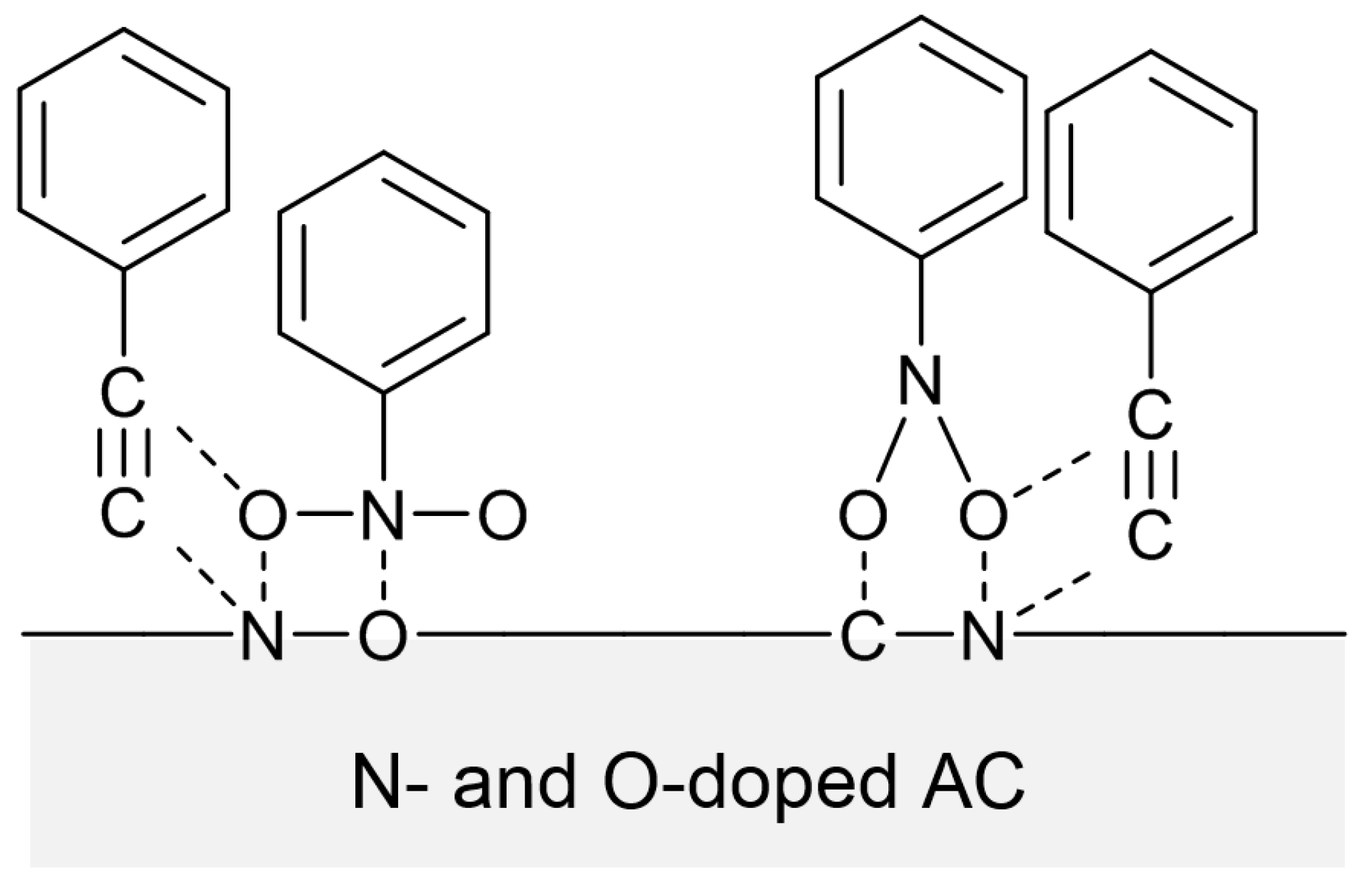
Scheme 8b. PA should also be adsorbed in the presence of NB. It is highly probable that interactions between PA and NB occur, allowing PA to be adsorbed and reduced on the surface of N-OAC. Possible interactions were proposed to occur between C≡C bond and O–N bonds (
Scheme 9) [
15], in which O and N could originate from both NB and the surface species on N-OAC. Interactions between PA and adsorbed hydrazine might also contribute to the PA hydrogenation assisted by NB. To elucidate the mechanism of the NB-assisted PA hydrogenation, further computational and/or atomic level spectroscopic studies would be required.
6. Summary
In this article, recent studies by our group on the utilization of N-AC (and N-OAC) for Knoevenagel condensation, transesterification, aerobic oxidation of XT and alcohols, and transfer hydrogenation of NB, ST, NS, and PA with hydrazine has been reviewed. It is demonstrated that the nitrogen- and oxygen-doped activated carbon (AC) catalysts can be metal-free active catalysts for several organic synthetic reactions being usually catalyzed by base, metal, and metal oxide catalysts. Doped-nitrogen species existed on the AC surface in different structures. Among them, pyridine-type nitrogen species was suggested to be involved in the active sites for Knoevenagel condensation (
Scheme 2), and for the oxidation of XT (
Scheme 4), while graphite-type nitrogen species was suggested for the oxidation of alcohols (
Scheme 6). For the hydrogenation of NB, both surface nitrogen and oxygen species would be involved in the active sites (
Scheme 8). N-AC is practically inactive for the transfer hydrogenation of vinyl and ethynyl groups, but it can catalyze those hydrogenation reactions assisted by co-existing NB (
Scheme 9). Comparison of N-AC with conventional catalysts shows that N-AC can alternate with conventional solid base catalysts and supported metal catalysts for the Knoevenagel condensation and oxidation reactions.
At present, the number of reactions tested for the use of N-AC is not so much. It is thus required to further investigate the catalyst structure and activity relationship for other organic reactions, from experimental and theoretical points of view. To prepare more effective carbon catalysts, co-doping boron or phosphorous with nitrogen may be also interesting.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}