Electrode Surface Composition of Dual-Intercalation, All-Graphite Batteries


Abstract
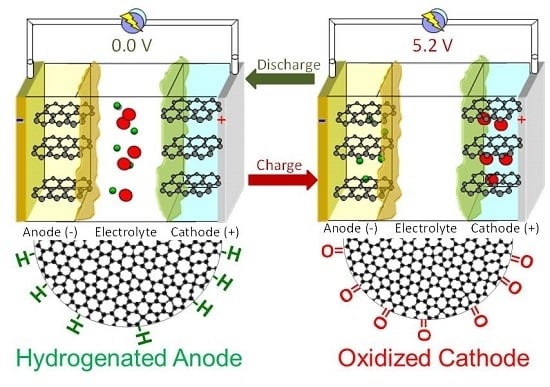
:
1. Introduction
2. Results and Discussion
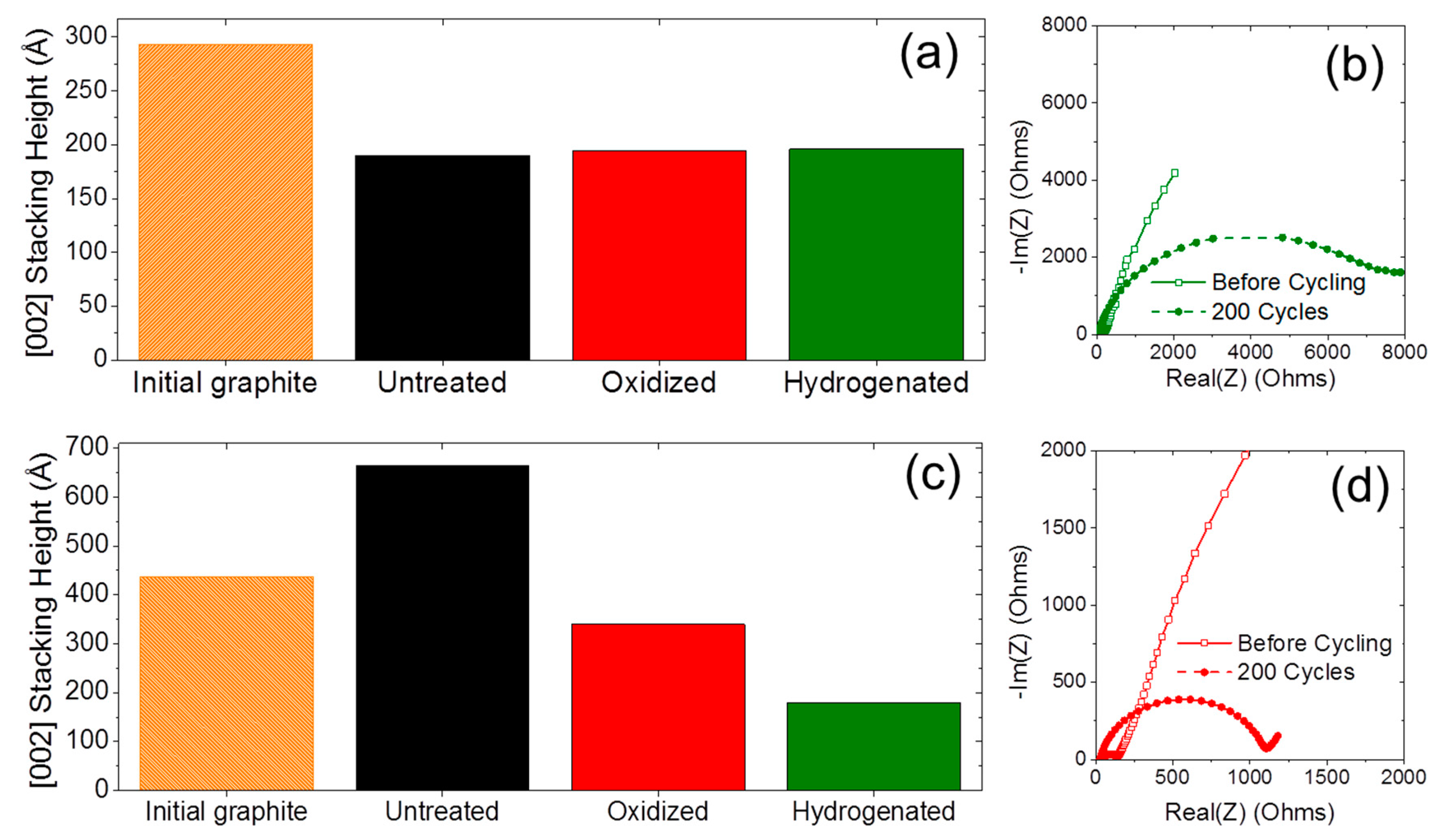
2.1. Material Properties
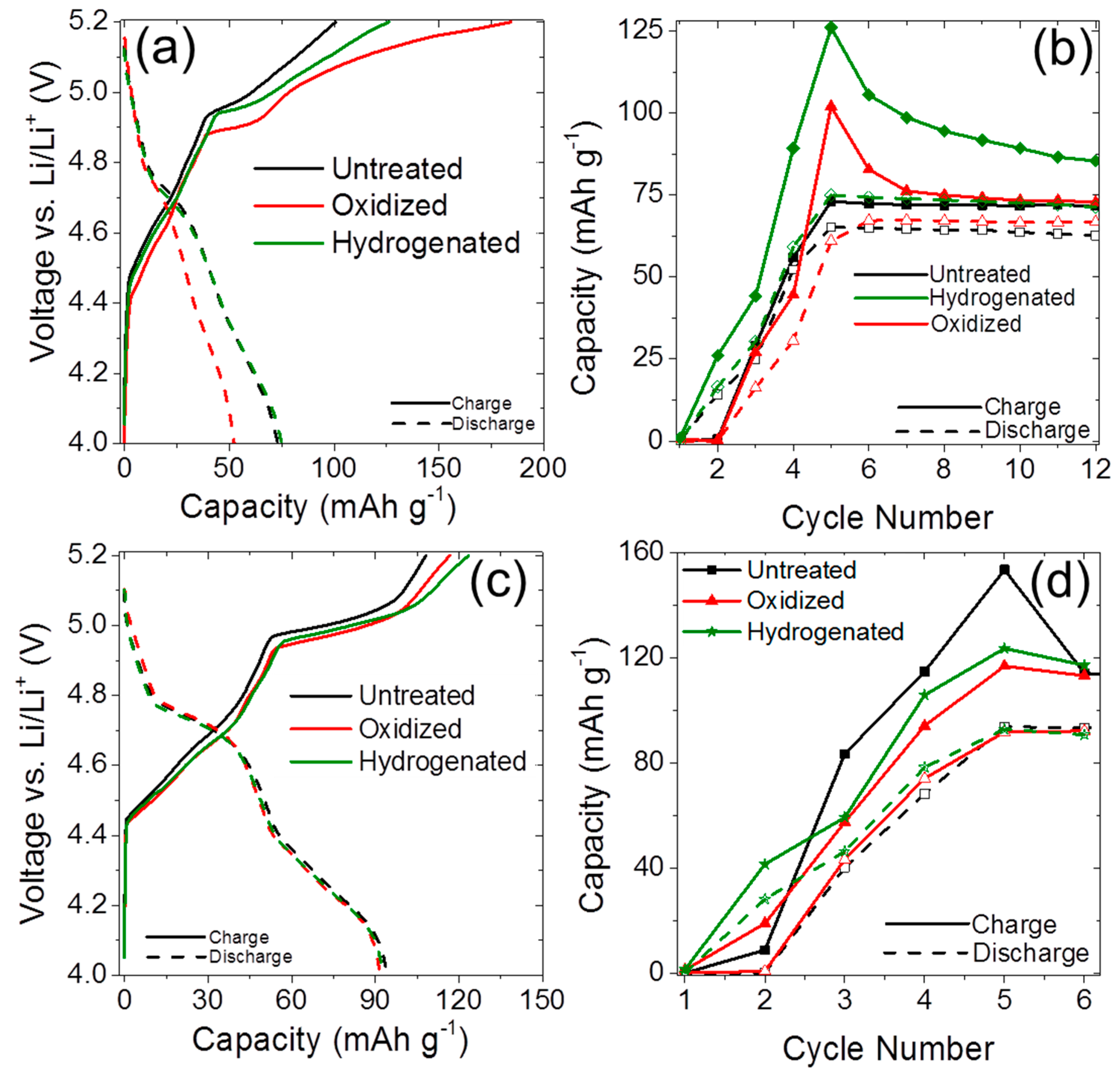
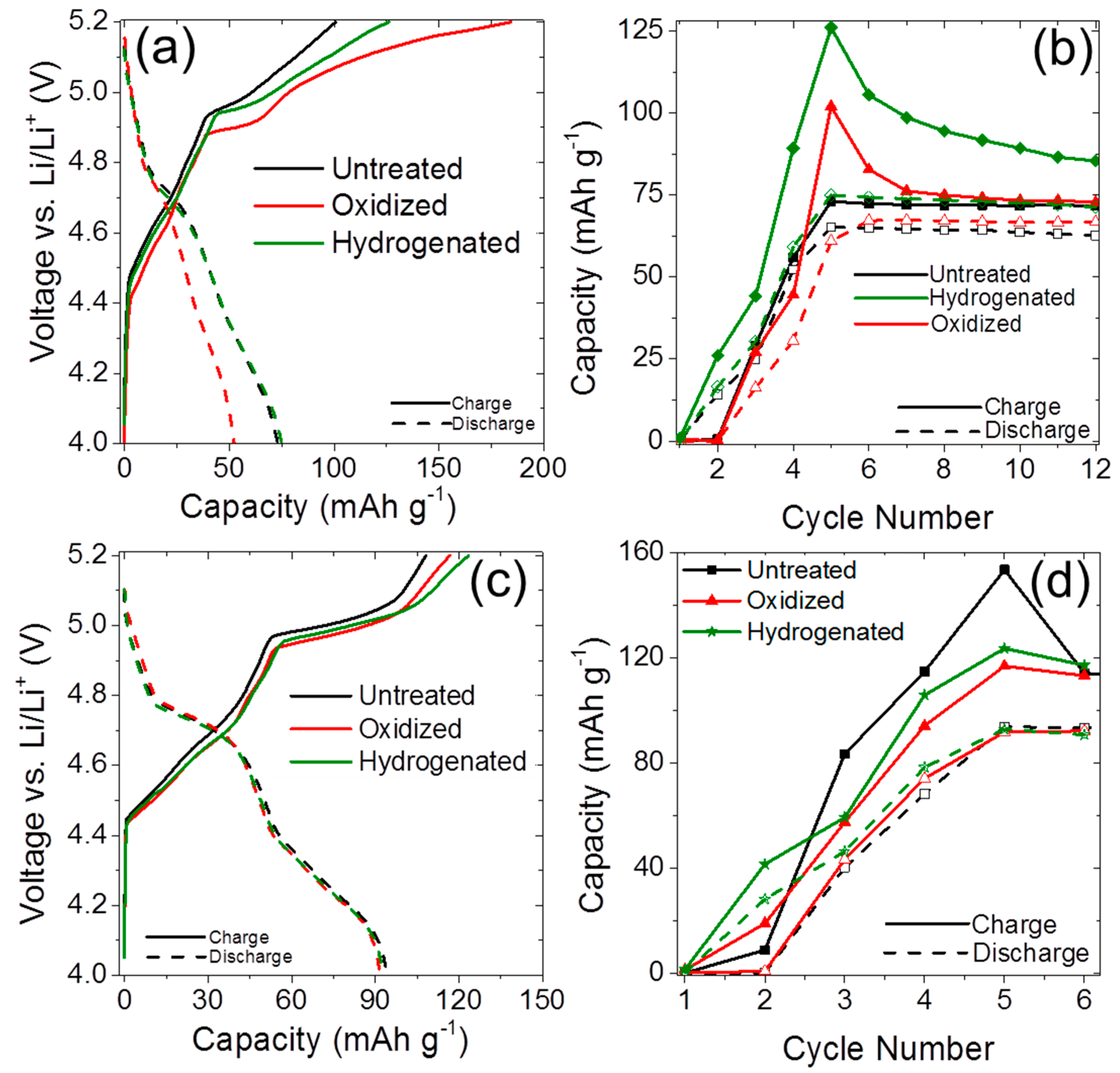
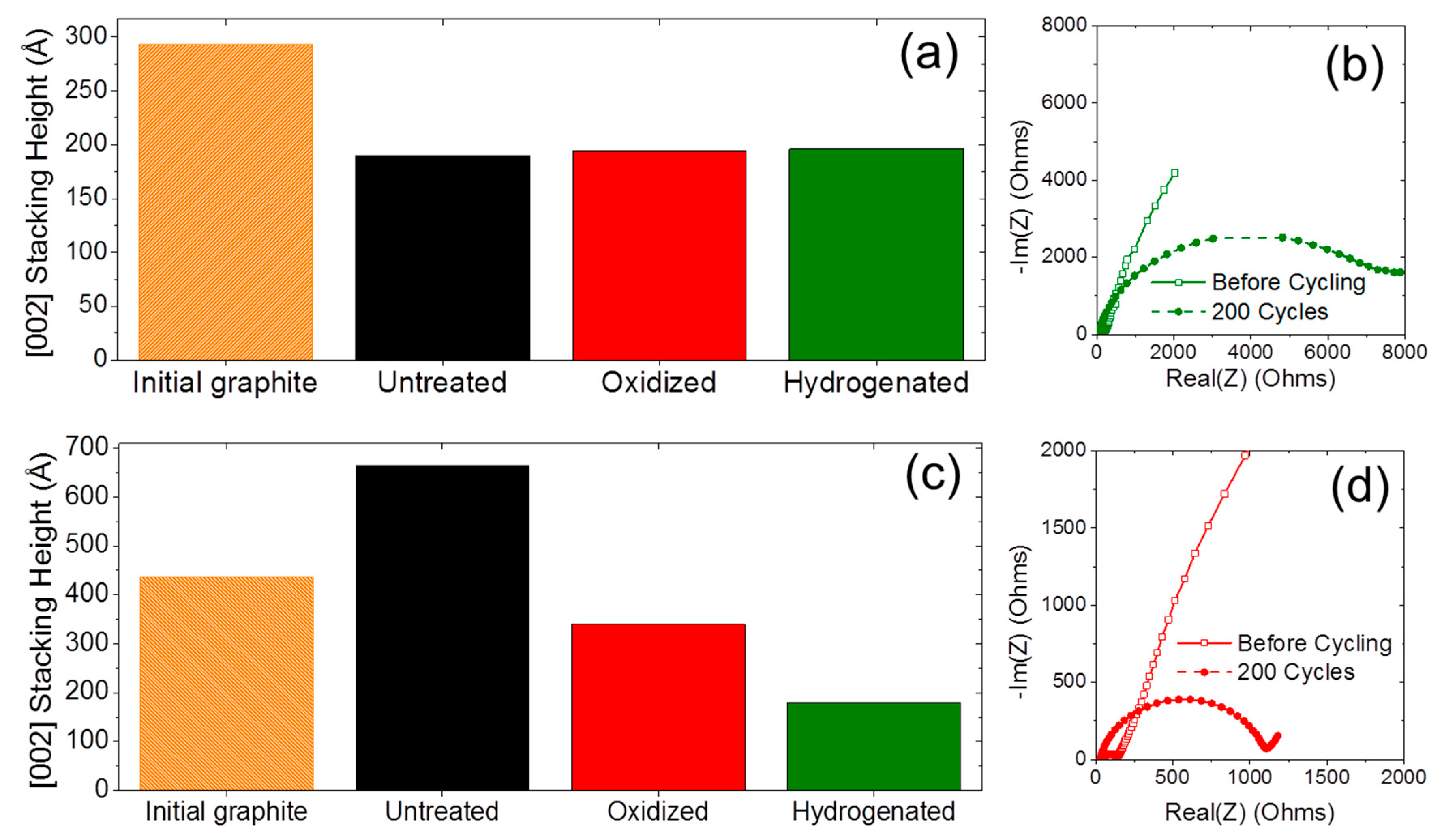
2.2. Anion Intercalation
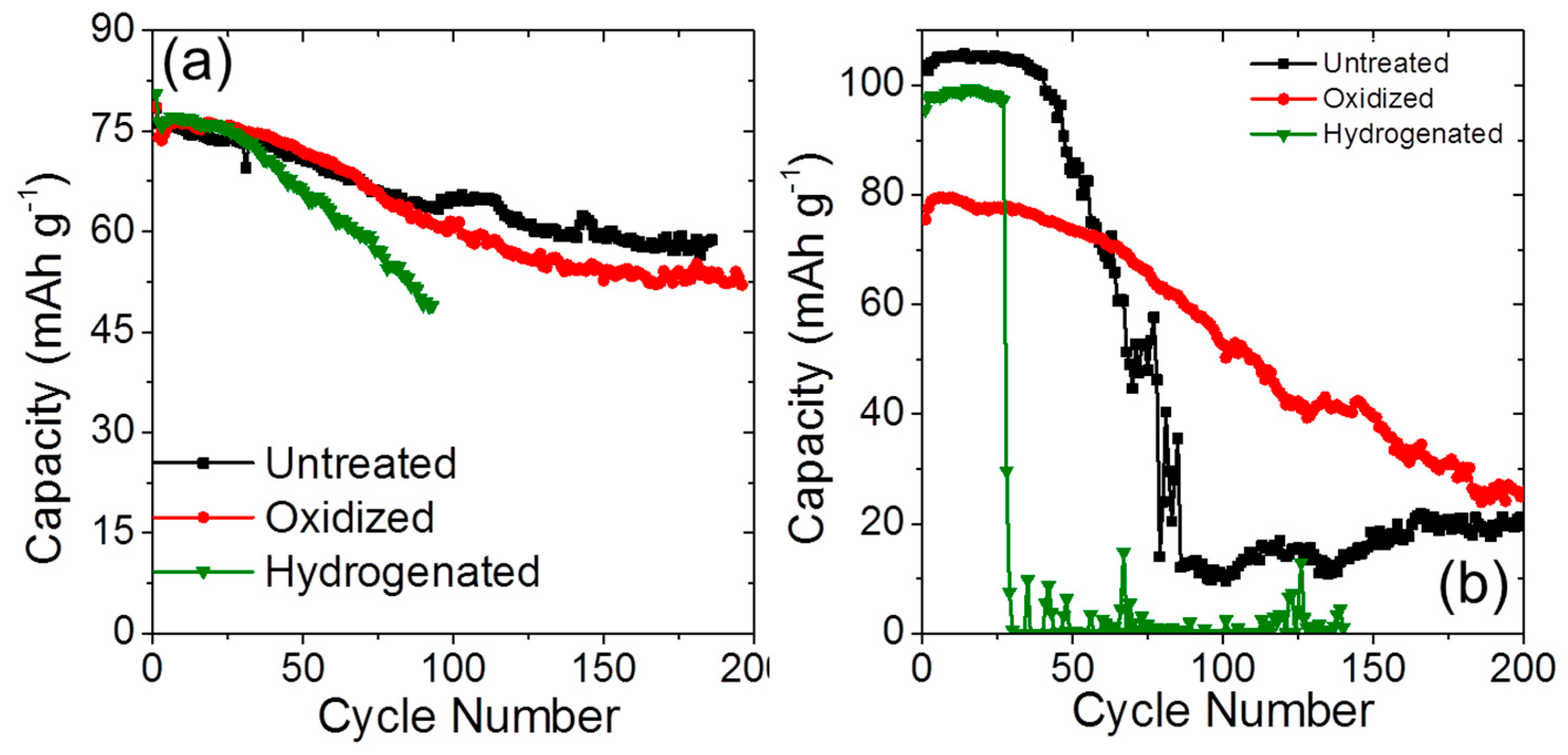
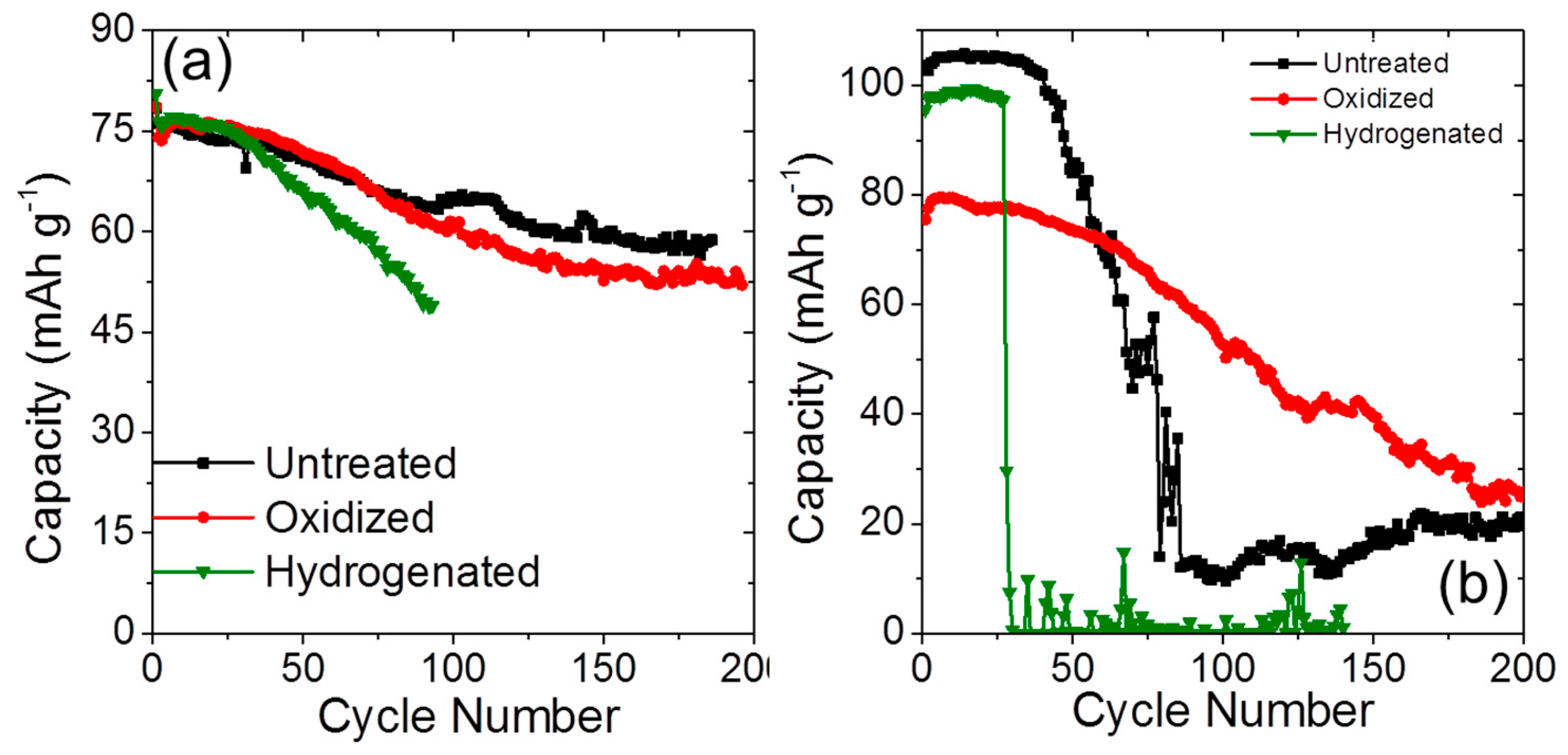
2.3. Long-Term Cyclability
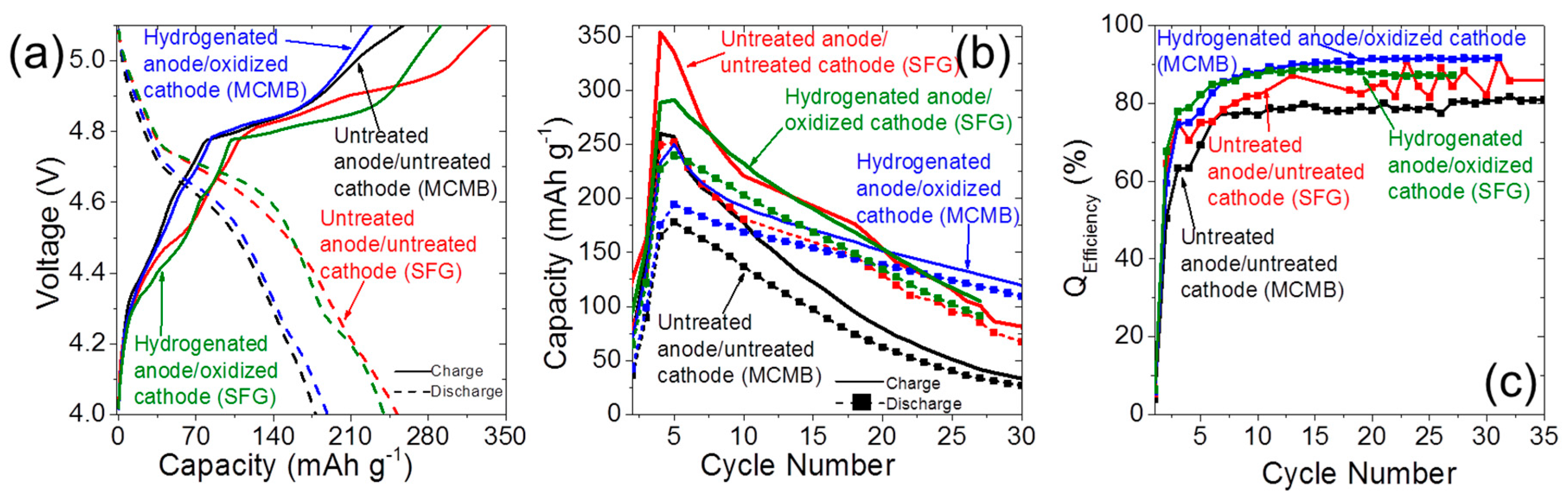
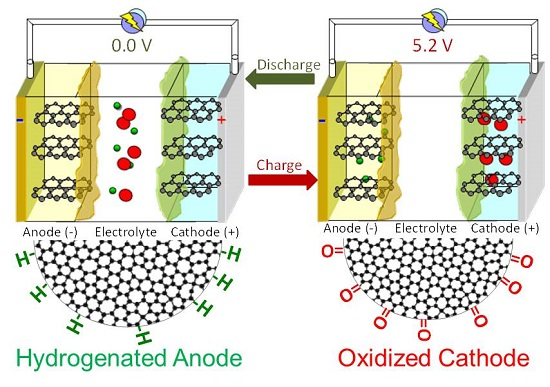
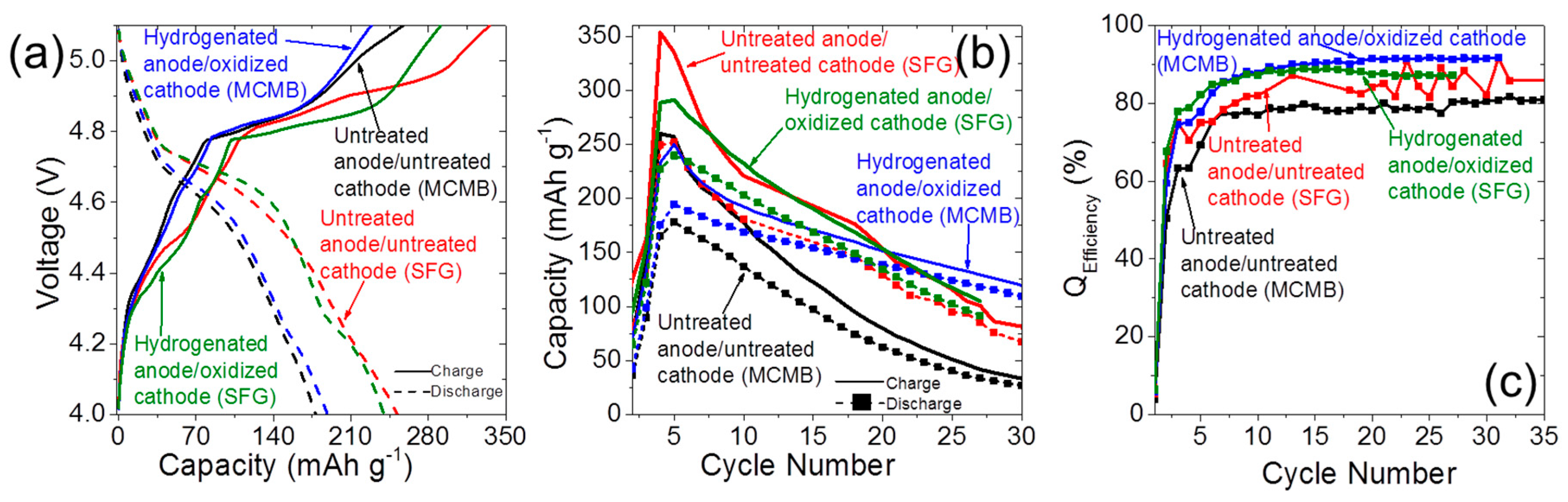
2.4. Dual-Ion Intercalation Configuration
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kelly, R.L.; Oriti, G.; Julian, A.L. Reducing fuel consumption in a forward operating base using an energy management system. In Proceedings of the IEEE 2013 Energy Conversion Congress and Exposition, Denver, CO, USA, 15–19 September 2013; pp. 1330–1336.
- Reddy, M.V.; Subba Rao, G.V.; Chowdari, B.V.R. Metal oxides and oxysalts as anode materials for Li ion batteries. Chem. Rev. 2013, 113, 5364–5457. [Google Scholar] [CrossRef] [PubMed]
- Schmuelling, G.; Placke, T.; Kloepsch, R.; Fromm, O.; Meyer, H.-W.; Passerini, S.; Winter, M. X-ray diffraction studies of the electrochemical intercalation of bis(trifluoromethanesulfonyl)imide anions into graphite for dual-ion cells. J. Power Sour. 2013, 239, 563–571. [Google Scholar] [CrossRef]
- Borodin, O.; Behl, W.; Jow, T.R. Oxidative stability and initial decomposition reactions of carbonate, sulfone, and alkyl phosphate-based electrolytes. J. Phys. Chem. C 2013, 117, 8661–8682. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y.; Dunn, B. Where do batteries end and supercapacitors begin? Science 2014, 343, 1210–1211. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Gogotsi, Y. Capacitive energy storage in nanostructured carbon–electrolyte systems. Acc. Chem. Res. 2013, 46, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Dyatkin, B.; Presser, V.; Heon, M.; Lukatskaya, M.R.; Beidaghi, M.; Gogotsi, Y. Development of a green supercapacitor composed entirely of environmentally friendly materials. ChemSusChem 2013, 6, 2269–2280. [Google Scholar] [CrossRef] [PubMed]
- Dahn, J.R.; Seel, J.A. Energy and capacity projections for practical dual-graphite cells. J. Electrochem. Soc. 2000, 147, 899–901. [Google Scholar] [CrossRef]
- Santhanam, R.; Noel, M. Electrochemical intercalation of ionic species of tetrabutylammonium perchlorate on graphite electrodes. A potential dual-intercalation battery system. J. Power Sour. 1995, 56, 101–105. [Google Scholar] [CrossRef]
- Tasaki, K. Density functional theory study on structural and energetic characteristics of graphite intercalation compounds. J. Phys. Chem. C 2013, 118, 1443–1450. [Google Scholar] [CrossRef]
- Read, J.A.; Cresce, A.V.; Ervin, M.H.; Xu, K. Dual-graphite chemistry enabled by a high voltage electrolyte. Energy Environ. Sci. 2014, 7, 617–620. [Google Scholar] [CrossRef]
- Read, J.A. In-situ studies on the electrochemical intercalation of hexafluorophosphate anion in graphite with selective cointercalation of solvent. J. Phys. Chem. C 2015, 119, 8438–8446. [Google Scholar] [CrossRef]
- Er, D.; Li, J.; Naguib, M.; Gogotsi, Y.; Shenoy, V.B. Ti3C2 mxene as a high capacity electrode material for metal (Li, Na, K, Ca) ion batteries. ACS Appl. Mater. Interfaces 2014, 6, 11173–11179. [Google Scholar] [CrossRef] [PubMed]
- Panero, S.; Spila, E.; Scrosati, B. A new type of a rocking-chair battery family based on a graphite anode and a polymer cathode. J. Electrochem. Soc. 1996, 143, L29–L30. [Google Scholar] [CrossRef]
- Kim, H.; Park, K.-Y.; Hong, J.; Kang, K. All-graphene-battery: Bridging the gap between supercapacitors and lithium ion batteries. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, Y.; Zhang, F.; Lee, C.-S. A novel aluminum–graphite dual-ion battery. Adv. Energy Mater. 2016. [Google Scholar] [CrossRef]
- Tasaki, K. Solvent decompositions and physical properties of decomposition compounds in Li-ion battery electrolytes studied by DFT calculations and molecular dynamics simulations. J. Phys. Chem. B 2005, 109, 2920–2933. [Google Scholar] [CrossRef] [PubMed]
- Dyatkin, B.; Zhang, Y.; Mamontov, E.; Kolesnikov, A.I.; Cheng, Y.; Meyer, H.M.; Cummings, P.T.; Gogotsi, Y. Influence of surface oxidation on ion dynamics and capacitance in porous and non-porous carbon electrodes. J. Phys. Chem. C 2016, 120, 8730–8741. [Google Scholar] [CrossRef]
- Dyatkin, B.; Mamontov, E.; Cook, K.C.; Gogotsi, Y. Capacitance, charge dynamics, and electrolyte-surface interactions in functionalized carbide-derived carbon electrodes. Prog. Nat. Sci. Mater. Int. 2015, 25, 631–641. [Google Scholar] [CrossRef]
- Osswald, S.; Portet, C.; Gogotsi, Y.; Laudisio, G.; Singer, J.P.; Fischer, J.E.; Sokolov, V.V.; Kukushkina, J.A.; Kravchik, A.E. Porosity control in nanoporous carbide-derived carbon by oxidation in air and carbon dioxide. J. Solid State Chem. 2009, 182, 1733–1741. [Google Scholar] [CrossRef]
- Punckt, C.; Muckel, F.; Wolff, S.; Aksay, I.A.; Chavarin, C.A.; Bacher, G.; Mertin, W. The effect of degree of reduction on the electrical properties of functionalized graphene sheets. Appl. Phys. Lett. 2013, 102, 023114. [Google Scholar] [CrossRef]
- Kostecki, R.; Richardson, T.; Boesenberg, U.; Pollak, E.; Lux, S. Modified carbon black materials for lithium-ion batteries. U.S. Patent US9368798 B2, 14 June 2016. [Google Scholar]
- Isono, Y.; Yoshida, A.; Hishiyama, Y.; Kaburagi, Y. Carbonization and graphitization of shavings filed away from kapton. Carbon 2004, 42, 1799–1805. [Google Scholar] [CrossRef]
- Mochalin, V.N.; Shenderova, O.; Ho, D.; Gogotsi, Y. The properties and applications of nanodiamonds. Nat. Nano 2012, 7, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Taberna, P.L.; Portet, C.; Simon, P. Electrode surface treatment and electrochemical impedance spectroscopy study on carbon/carbon supercapacitors. Appl. Phys. A 2006, 82, 639–646. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface Chemistry | MCMB | SFG | CGP | |||
|---|---|---|---|---|---|---|
| Specific Surface Area | Functional Group Content | Specific Surface Area | Functional Group Content | Specific Surface Area | Functional Group Content | |
| Untreated | 1.99 m2·g−1 | 0.52 wt % | 1.34 m2·g−1 | 0.95 wt % | 1.48 m2·g−1 | 0.41 wt % |
| Oxidized | 2.49 m2·g−1 | 1.72 wt % | 4.64 m2·g−1 | 11.92 wt % | 6.21 m2·g−1 | 3.54 wt % |
| Hydrogenated | 2.26 m2·g−1 | 0.44 wt % | 2.72 m2·g−1 | 2.05 wt % | 6.26 m2·g−1 | 0.21 wt % |
| Element | MCMB | SFG | |
|---|---|---|---|
| Oxidized | Hydrogenated | Oxidized | |
| Carbon (at %) | 98.2 | 99.4 | 98.2 |
| Oxygen (at %) | 1.8 | 0.6 | 1.8 |
| Material | Surface Chemistry | Cycle 1 Efficiency | Steady-State Efficiency | Peak Capacity | Polarization Potential |
|---|---|---|---|---|---|
| MCMB Cathode | Untreated | 72.5% | 88.1% | 73.1 mAh·g−1 | 4.94 V |
| Oxidized | 74.1% | 87.7% | 92.4 mAh·g−1 | 4.92 V | |
| Hydrogenated | 59.5% | 81.4% | 75.0 mAh·g−1 | 5.03 V | |
| SFG Cathode | Untreated | 79.1% | 82.0% | 94.2 mAh·g−1 | 5.04 V |
| Oxidized | 78.5% | 88.2% | 94.1 mAh·g−1 | 5.02 V | |
| Hydrogenated | 75.1% | 83.8% | 92.7 mAh·g−1 | 5.02 V | |
| CGP Anode | Untreated | 88.9% | 99.5% | 309.5 mAh·g−1 | |
| Oxidized | 86.4% | 99.1% | 340.5 mAh·g−1 | ||
| Hydrogenated | 87.7% | 99.2% | 268.7 mAh·g−1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyatkin, B.; Halim, J.; Read, J.A. Electrode Surface Composition of Dual-Intercalation, All-Graphite Batteries. C 2017, 3, 5. https://doi.org/10.3390/c3010005
Dyatkin B, Halim J, Read JA. Electrode Surface Composition of Dual-Intercalation, All-Graphite Batteries. C. 2017; 3(1):5. https://doi.org/10.3390/c3010005
Chicago/Turabian StyleDyatkin, Boris, Joseph Halim, and Jeffrey A. Read. 2017. "Electrode Surface Composition of Dual-Intercalation, All-Graphite Batteries" C 3, no. 1: 5. https://doi.org/10.3390/c3010005
APA StyleDyatkin, B., Halim, J., & Read, J. A. (2017). Electrode Surface Composition of Dual-Intercalation, All-Graphite Batteries. C, 3(1), 5. https://doi.org/10.3390/c3010005