


Tuning Optical Excitations of Graphene Quantum Dots Through Selective Oxidation: Effect of Epoxy Groups



Abstract

1. Introduction
2. Materials and Methods
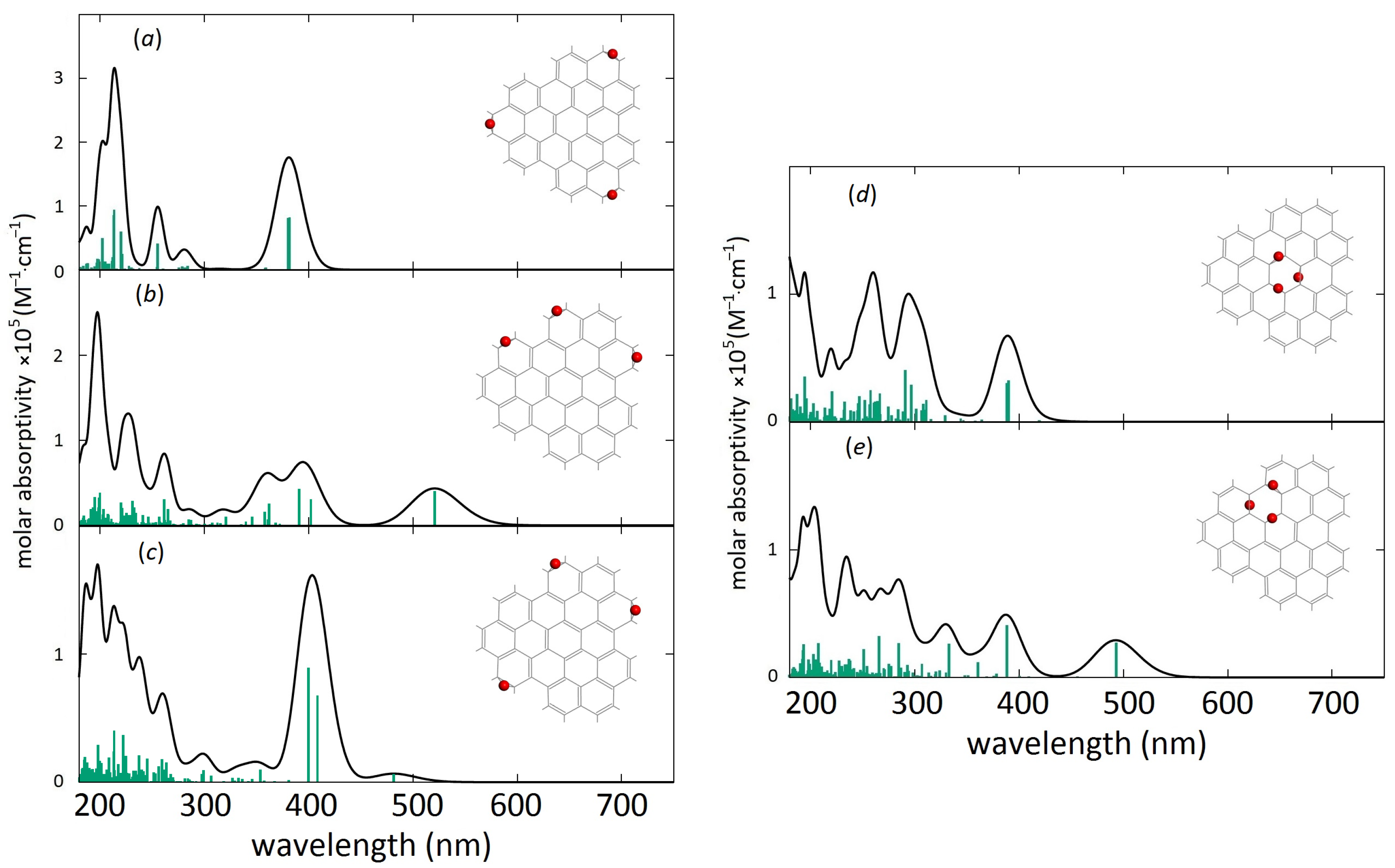
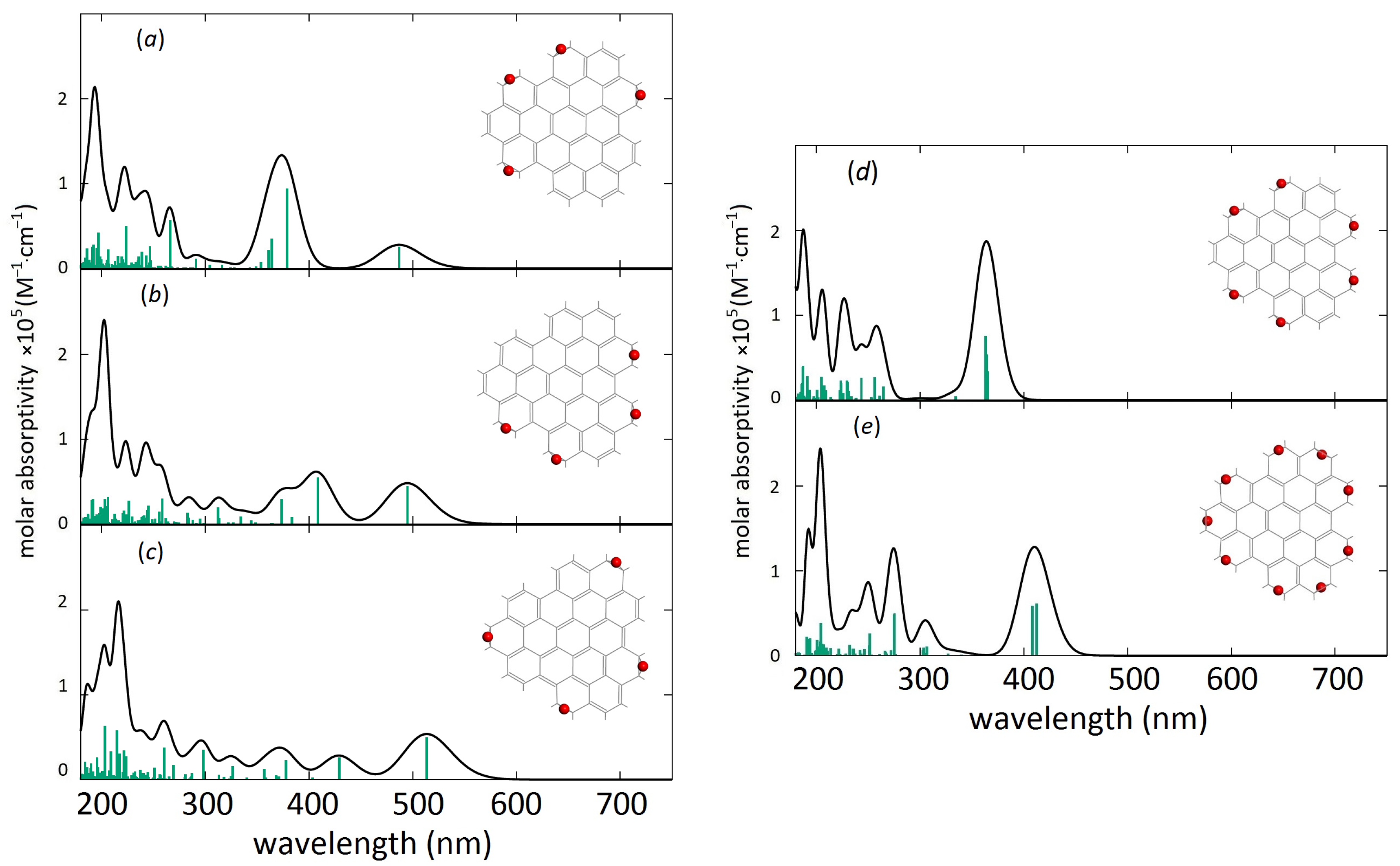
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GQD | Graphene quantum dot |
| DFT | Density functional theory |
| LED | Light-emitting diode |
| PAH | Polycyclic aromatic hydrocarbons |
| GGA | Generalized gradient approximation |
| sTD-DFT | Simplified time-dependent DFT |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
References
- Zhou, J.; Zhou, H.; Tang, J.; Deng, S.; Yan, F.; Li, W.; Qu, M. Carbon dots doped with heteroatoms for fluorescent bioimaging: A review. Microchim. Acta 2017, 184, 343–368. [Google Scholar] [CrossRef]
- Essner, J.B.; Baker, G.A. The emerging roles of carbon dots in solar photovoltaics: A critical review. Environ. Sci. Nano 2017, 4, 1216–1263. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, W.; Gong, Y.; Liu, Q.; Chen, Z. Graphene/Quantum Dot Heterostructure Photodetectors: From Material to Performance. Adv. Opt. Mater. 2022, 10, 2201889. [Google Scholar] [CrossRef]
- Yuan, F.; Li, S.; Fan, Z.; Meng, X.; Fan, L.; Yang, S. Shining carbon dots: Synthesis and biomedical and optoelectronic applications. Nano Today 2016, 11, 565–586. [Google Scholar] [CrossRef]
- Hola, K.; Zhang, Y.; Wang, Y.; Giannelis, E.P.; Zboril, R.; Rogach, A.L. Carbon dots—Emerging light emitters for bioimaging, cancer therapy and optoelectronics. Nano Today 2014, 9, 590–603. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Xu, T.; Liao, H.; Yao, C.; Liu, Y.; Li, Z.; Chen, Z.; Pan, D.; Sun, L.; et al. Gram-scale synthesis of single-crystalline graphene quantum dots with superior optical properties. Nat. Commun. 2014, 5, 5357. [Google Scholar] [CrossRef]
- Yoon, H.; Park, M.; Kim, J.; Novak, T.G.; Lee, S.; Jeon, S. Toward highly efficient luminescence in graphene quantum dots for optoelectronic applications. Chem. Phys. Rev. 2021, 2, 031303. [Google Scholar] [CrossRef]
- Shen, J.; Zhu, Y.; Yang, X.; Zong, J.; Zhang, J.; Li, C. One-pot hydrothermal synthesis of graphene quantum dots surface-passivated by polyethylene glycol and their photoelectric conversion under near-infrared light. New J. Chem. 2012, 36, 97–101. [Google Scholar] [CrossRef]
- Tetsuka, H.; Asahi, R.; Nagoya, A.; Okamoto, K.; Tajima, I.; Ohta, R.; Okamoto, A. Optically Tunable Amino-Functionalized Graphene Quantum Dots. Adv. Mater. 2012, 24, 5333–5338. [Google Scholar] [CrossRef]
- Qu, D.; Zheng, M.; Zhang, L.; Zhao, H.; Xie, Z.; Jing, X.; Haddad, R.E.; Fan, H.; Sun, Z. Formation mechanism and optimization of highly luminescent N-doped graphene quantum dots. Sci. Rep. 2014, 4, 5294. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, P.; De Corato, M.; Ruini, A.; Zhao, S.; Meggendorfer, F.; Straasø, L.A.; Rondin, L.; Simon, P.; Li, J.; et al. Bandgap Engineering of Graphene Nanoribbons by Control over Structural Distortion. J. Am. Chem. Soc. 2018, 140, 7803–7809. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, H.; Elhaes, H.; Ibrahim, M.A. Tuning electronic properties in graphene quantum dots by chemical functionalization: Density functional theory calculations. Chem. Phys. Lett. 2018, 695, 138–148. [Google Scholar] [CrossRef]
- Biroju, R.K.; Rajender, G.; Giri, P. On the origin and tunability of blue and green photoluminescence from chemically derived graphene: Hydrogenation and oxygenation studies. Carbon 2015, 95, 228–238. [Google Scholar] [CrossRef]
- Facure, M.H.M.; Schneider, R.; Mercante, L.A.; Correa, D.S. A review on graphene quantum dots and their nanocomposites: From laboratory synthesis towards agricultural and environmental applications. Environ. Sci. Nano 2020, 7, 3710–3734. [Google Scholar] [CrossRef]
- Zhu, X.; Su, H. Exciton characteristics in graphene epoxide. ACS Nano 2014, 8, 1284–1289. [Google Scholar] [CrossRef]
- Hossain, Z.; Johns, J.E.; Bevan, K.H.; Karmel, H.J.; Liang, Y.T.; Yoshimoto, S.; Mukai, K.; Koitaya, T.; Yoshinobu, J.; Kawai, M.; et al. Chemically homogeneous and thermally reversible oxidation of epitaxial graphene. Nat. Chem. 2012, 4, 305–309. [Google Scholar] [CrossRef]
- Johns, J.E.; Hersam, M.C. Atomic covalent functionalization of graphene. Acc. Chem. Res. 2013, 46, 77–86. [Google Scholar] [CrossRef]
- Vinogradov, N.A.; Schulte, K.; Ng, M.L.; Mikkelsen, A.; Lundgren, E.; Mårtensson, N.; Preobrajenski, A.B. Impact of Atomic Oxygen on the Structure of Graphene Formed on Ir(111) and Pt(111). J. Phys. Chem. C 2011, 115, 9568–9577. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, W.; Luo, Y.; Yang, J.; Hou, J.G. How Graphene Is Cut upon Oxidation? J. Am. Chem. Soc. 2009, 131, 6320–6321. [Google Scholar] [CrossRef]
- Sheely, A.; Gifford, B.; Tretiak, S.; Bishop, A. Tunable Optical Features of Graphene Quantum Dots from Edge Functionalization. J. Phys. Chem. C 2021, 125, 9244–9252. [Google Scholar] [CrossRef]
- Yan, J.-A.; Chou, M.Y. Oxidation functional groups on graphene: Structural and electronic properties. Phys. Rev. B 2010, 82, 125403. [Google Scholar] [CrossRef]
- Rosas, J.J.H.; Gutiérrez, R.E.R.; Escobedo-Morales, A.; Anota, E.C. First principles calculations of the electronic and chemical properties of graphene, graphane, and graphene oxide. J. Mol. Model. 2011, 17, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Bongiorno, A. Density Functional Theory Modeling of Multilayer “Epitaxial” Graphene Oxide. Acc. Chem. Res. 2014, 47, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Chou, C.-P.; Pham, B.Q.; Witek, H.A.; Irle, S.; Morokuma, K. Quantum chemical investigation of epoxide and ether groups in graphene oxide and their vibrational spectra. Phys. Chem. Chem. Phys. 2013, 15, 3725–3735. [Google Scholar] [CrossRef]
- Chen, S.; Ullah, N.; Wang, T.; Zhang, R. Tuning the optical properties of graphene quantum dots by selective oxidation: A theoretical perspective. J. Mater. Chem. C 2018, 6, 6875–6883. [Google Scholar] [CrossRef]
- Cui, P.; Xue, Y. Tuning nonradiative recombination loss by selective oxidation patterns of epoxy groups bound to different sites of graphene quantum dots. Chem. Eng. J. 2022, 431, 134052. [Google Scholar] [CrossRef]
- Feng, J.; Dong, H.; Yu, L.; Dong, L. The optical and electronic properties of graphene quantum dots with oxygen-containing groups: A density functional theory study. J. Mater. Chem. C 2017, 5, 5984–5993. [Google Scholar] [CrossRef]
- Yan, X.; Cui, X.; Li, L.-S. Synthesis of Large, Stable Colloidal Graphene Quantum Dots with Tunable Size. J. Am. Chem. Soc. 2010, 132, 5944–5945. [Google Scholar] [CrossRef]
- Kastler, M.; Schmidt, J.; Pisula, W.; Sebastiani, D.; Müllen, K. From Armchair to Zigzag Peripheries in Nanographenes. J. Am. Chem. Soc. 2006, 128, 9526–9534. [Google Scholar] [CrossRef]
- Rieger, R.; Müllen, K. Forever young: Polycyclic aromatic hydrocarbons as model cases for structural and optical studies. J. Phys. Org. Chem. 2010, 23, 315–325. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Zhang, M.; Yao, B.; Li, Y.; Huang, L.; Li, C.; Shi, G. Water-enhanced oxidation of graphite to graphene oxide with controlled species of oxygenated groups. Chem. Sci. 2016, 7, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mat. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- McNellis, E.R.; Meyer, J.; Reuter, K. Azobenzene at coinage metal surfaces: Role of dispersive van der Waals interactions. Phys. Rev. B 2009, 80, 205414. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S. A simplified Tamm-Dancoff density functional approach for the electronic excitation spectra of very large molecules. J. Chem. Phys. 2013, 138, 244104. [Google Scholar] [CrossRef]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040–1041, 45–53. [Google Scholar] [CrossRef]
- de Wergifosse, M.; Seibert, J.; Grimme, S. Simplified time-dependent density functional theory (sTD-DFT) for molecular optical rotation. J. Chem. Phys. 2020, 153, 084116. [Google Scholar] [CrossRef] [PubMed]
- Clar, E. Polycyclic Hydrocarbons; Academic Press: Cambridge, MA, USA; Springer: London, UK, 1964; pp. 86–104. [Google Scholar]
- Platt, J.R. Classification of Spectra of Cata-Condensed Hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Clar, E. The Aromatic Sextet; John Wiley & Sons: London, UK; New York, NY, USA; Sydney, Australia; Toronto, ON, Canada, 1972; pp. 1–128. [Google Scholar]
- Sola, M. Forty years of Clar’s aromatic π-sextet rule. Front. Chem. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed]
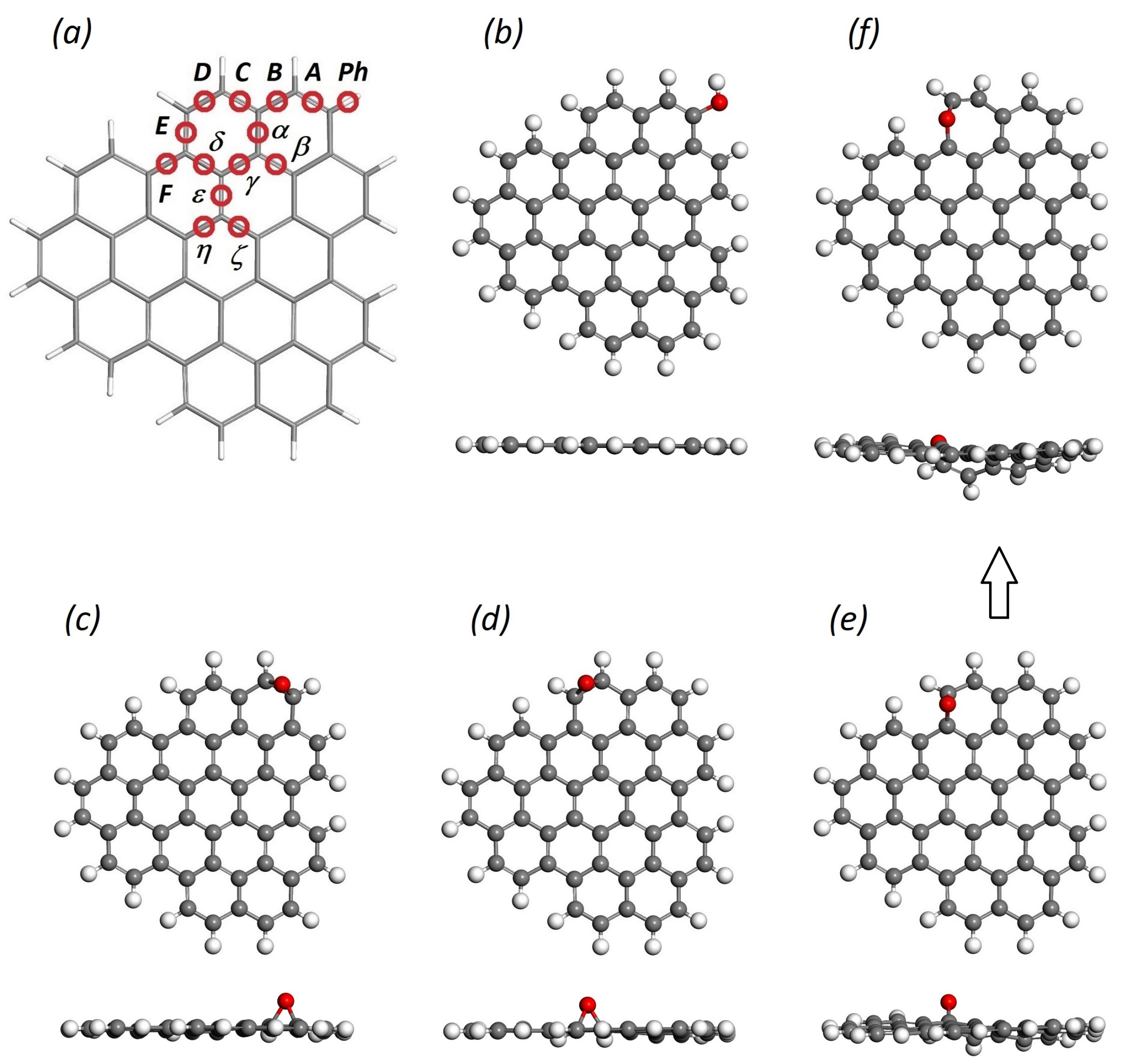
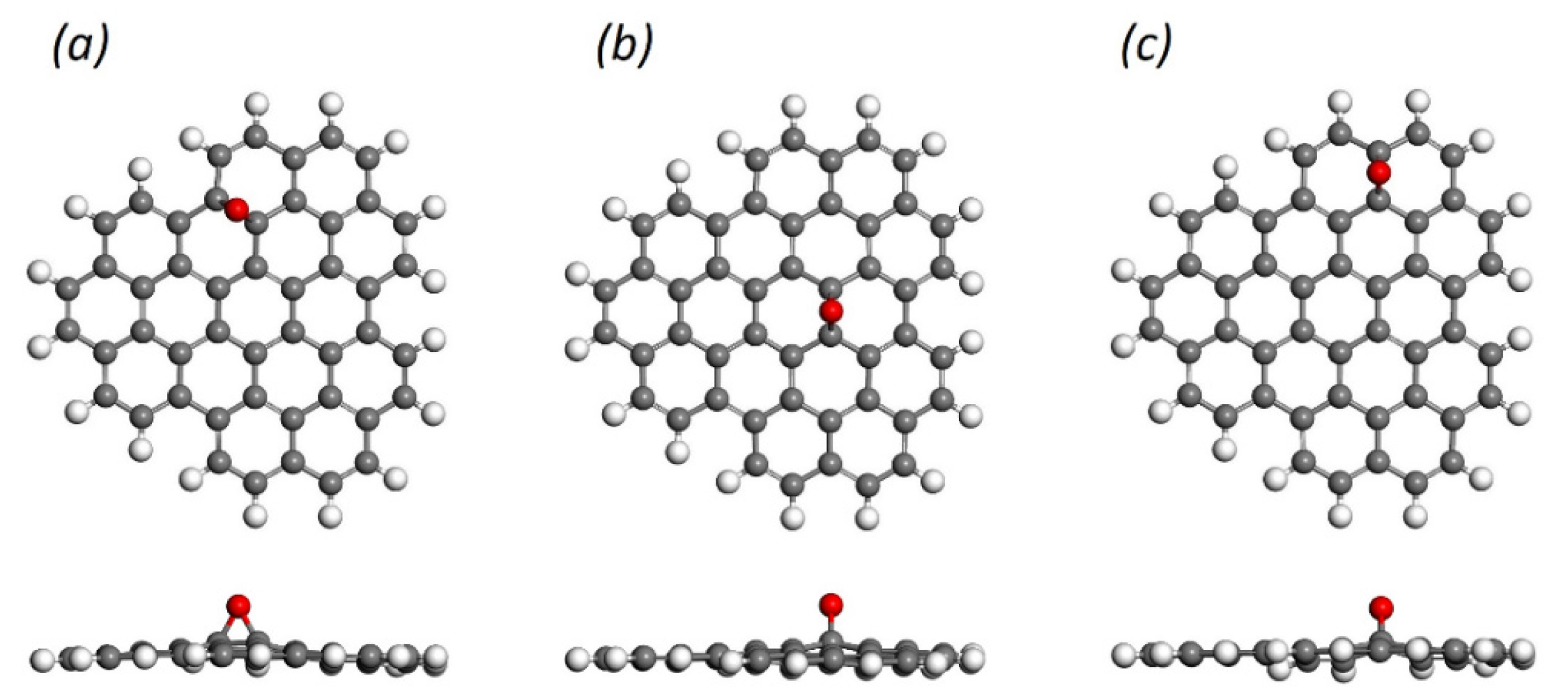
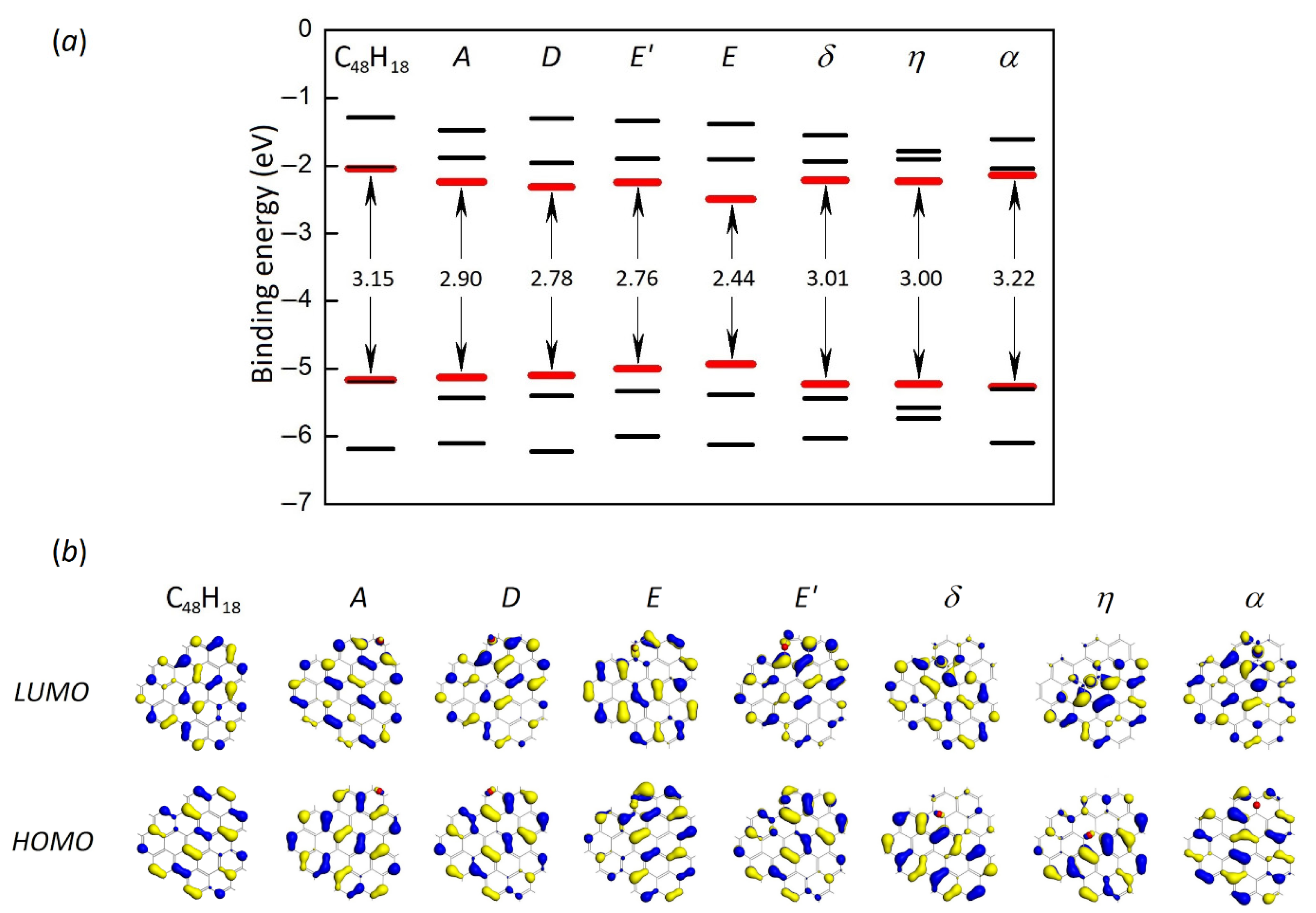
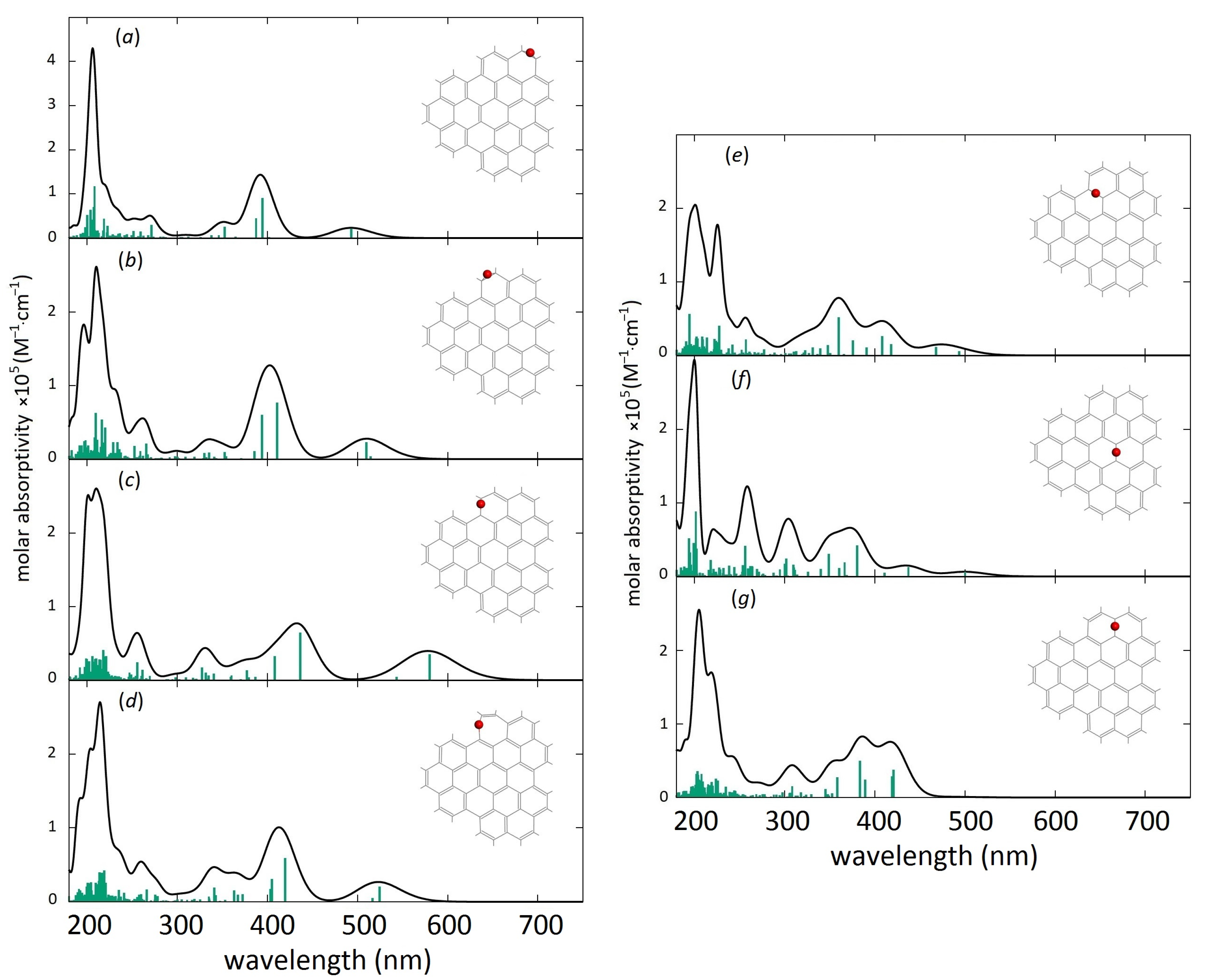



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
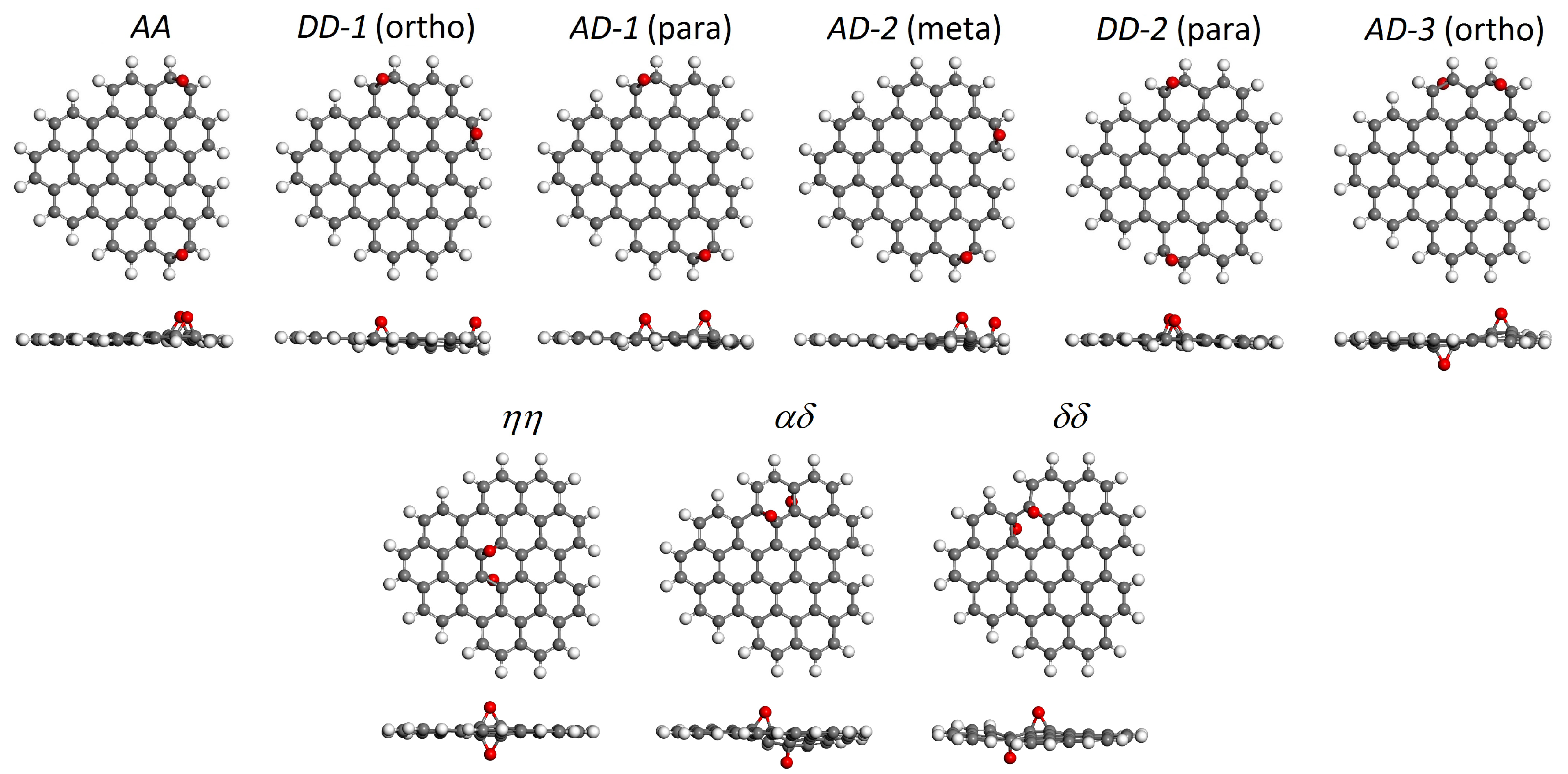
| Oxygen Positions | Binding Energy, eV | Energy Difference, eV | HOMO-LUMO Gap, eV |
|---|---|---|---|
| C48H18O edge-oxidized | |||
| A | –2.95 | 0.00 | 2.90 |
| D | –2.86 | +0.09 | 2.78 |
| E | –1.95 | +1.00 | 2.44 |
| E’ (ether) | –2.77 | +0.18 | 2.76 |
| B’ (ether) | –2.78 | +0.17 | 2.85 |
| Ph | –4.54 | –1.59 | 3.07 |
| C48H18O basal-oxidized | |||
| δ | –1.88 | +1.07 | 3.01 |
| η | –1.76 | +1.19 | 3.00 |
| α | –1.72 | +1.23 | 3.22 |
| C48H18O2 edge-oxidized | |||
| AA | –2.96 | 0.00 | 3.04 |
| DD-1 | –2.93 | +0.03 | 2.85 |
| AD-1 | –2.89 | +0.07 | 2.51 |
| AD-2 | –2.87 | +0.09 | 2.66 |
| DD-2 | –2.87 | +0.09 | 3.00 |
| AD-3 | –2.77 | +0.19 | 2.78 |
| C48H18O2 basal-oxidized | |||
| ηη | –2.12 | +0.84 | 3.37 |
| αδ | –2.11 | +0.85 | 2.94 |
| δδ | –2.10 | +0.86 | 3.29 |
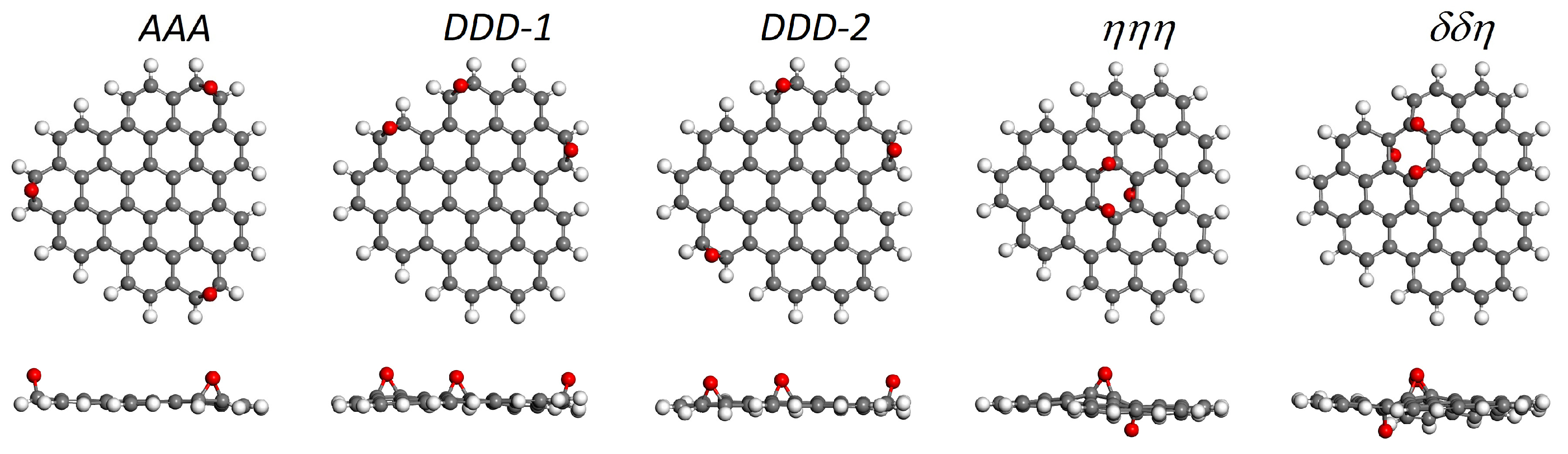
| C48H18O3 edge-oxidized | |||
| AAA | –2.97 | 0.00 | 3.43 |
| DDD-1 | –2.93 | +0.04 | 2.70 |
| DDD-2 | –2.92 | +0.05 | 3.01 |
| C48H18O3 basal-oxidized | |||
| ηηη | –2.32 | +0.65 | 3.50 |
| δδη | –2.26 | +0.71 | 2.95 |
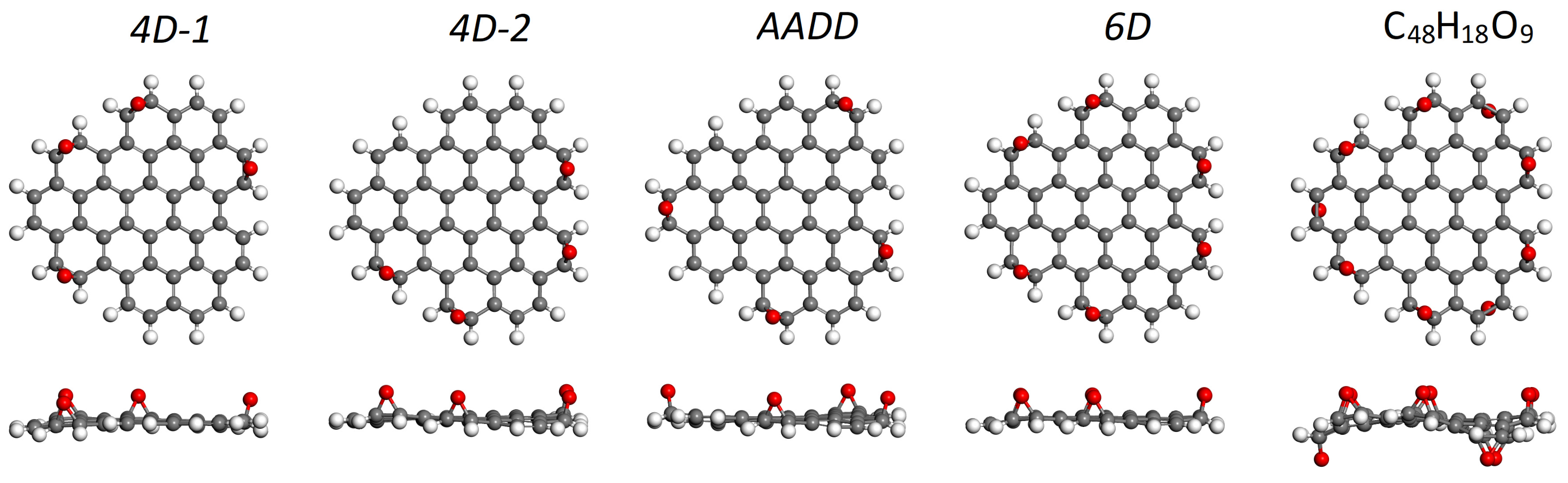
| C48H18O4 edge-oxidized | |||
| 4D-1 | –2.95 | 0.00 | 2.93 |
| 4D-2 | –2.93 | +0.02 | 2.80 |
| AADD | –2.91 | +0.04 | 2.69 |
| C48H18O6 edge-oxidized | |||
| 6D | –2.97 | – | 3.57 |
| C48H18O9 edge-oxidized | |||
| all A and D | –2.77 | – | 3.15 |
| A | D | E | E’ | δ | η | α | ||
|---|---|---|---|---|---|---|---|---|
| Average C-C bond length (Å) | Oxidized | 1.478 | 1.477 | 1.563 | 2.329 | 1.524 | 1.514 | 1.554 |
| Initial | 1.371 | 1.372 | 1.425 | 1.425 | 1.422 | 1.418 | 1.424 | |
| C-C bond stretching (%) | 7.8 | 7.7 | 9.7 | 63 | 7.2 | 6.8 | 9.1 | |
| Average C-O bond length (Å) | 1.430 | 1.432 | 1.418 | 1.374 | 1.438 | 1.438 | 1.428 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ershov, I.V.; Lavrentyev, A.A.; Romanov, D.L.; Holodova, O.M. Tuning Optical Excitations of Graphene Quantum Dots Through Selective Oxidation: Effect of Epoxy Groups. C 2025, 11, 51. https://doi.org/10.3390/c11030051
Ershov IV, Lavrentyev AA, Romanov DL, Holodova OM. Tuning Optical Excitations of Graphene Quantum Dots Through Selective Oxidation: Effect of Epoxy Groups. C. 2025; 11(3):51. https://doi.org/10.3390/c11030051
Chicago/Turabian StyleErshov, Igor V., Anatoly A. Lavrentyev, Dmitry L. Romanov, and Olga M. Holodova. 2025. "Tuning Optical Excitations of Graphene Quantum Dots Through Selective Oxidation: Effect of Epoxy Groups" C 11, no. 3: 51. https://doi.org/10.3390/c11030051
APA StyleErshov, I. V., Lavrentyev, A. A., Romanov, D. L., & Holodova, O. M. (2025). Tuning Optical Excitations of Graphene Quantum Dots Through Selective Oxidation: Effect of Epoxy Groups. C, 11(3), 51. https://doi.org/10.3390/c11030051