
The Interplay of NEAT1 and miR-339-5p Influences on Mesangial Gene Expression and Function in Various Diabetic-Associated Injury Models

 ,
,  and
and
Abstract
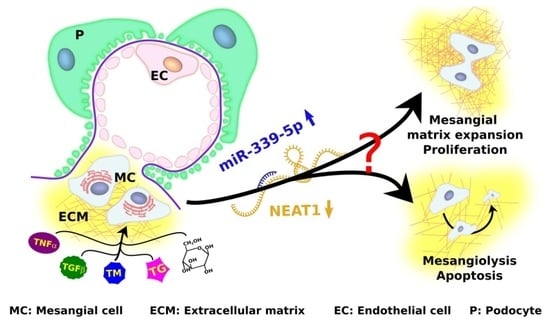
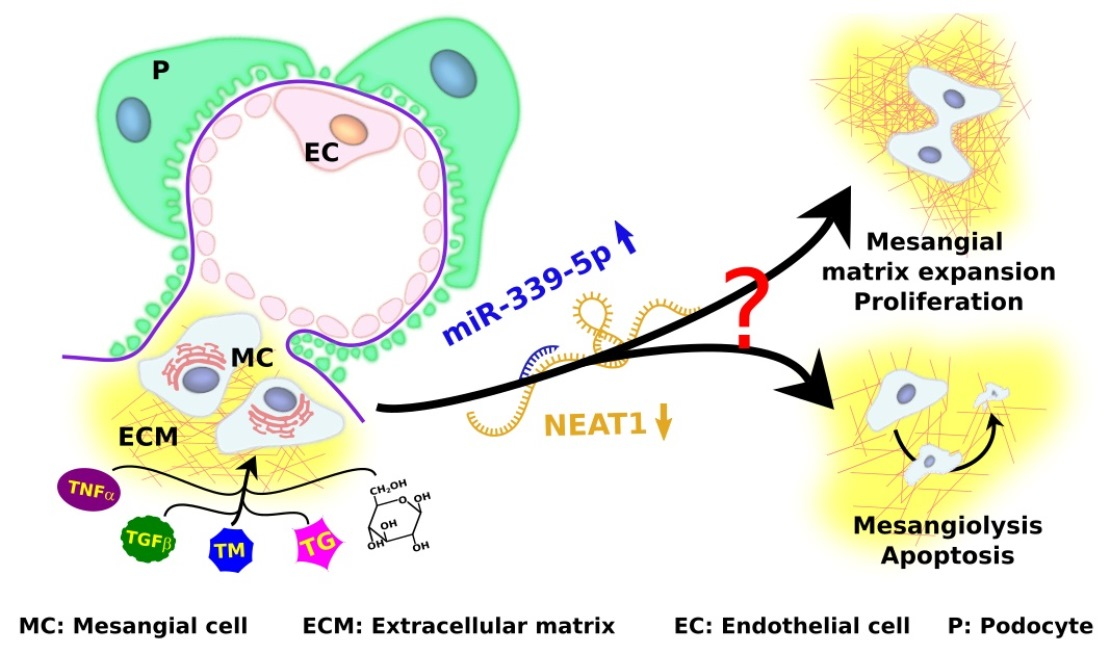
1. Introduction
2. Results
2.1. NEAT1 Expression in Human Mesangial Cells
2.2. NEAT1 Knockdown in Human Mesangial Cells Affects Proliferation, Cell Size, and Migration
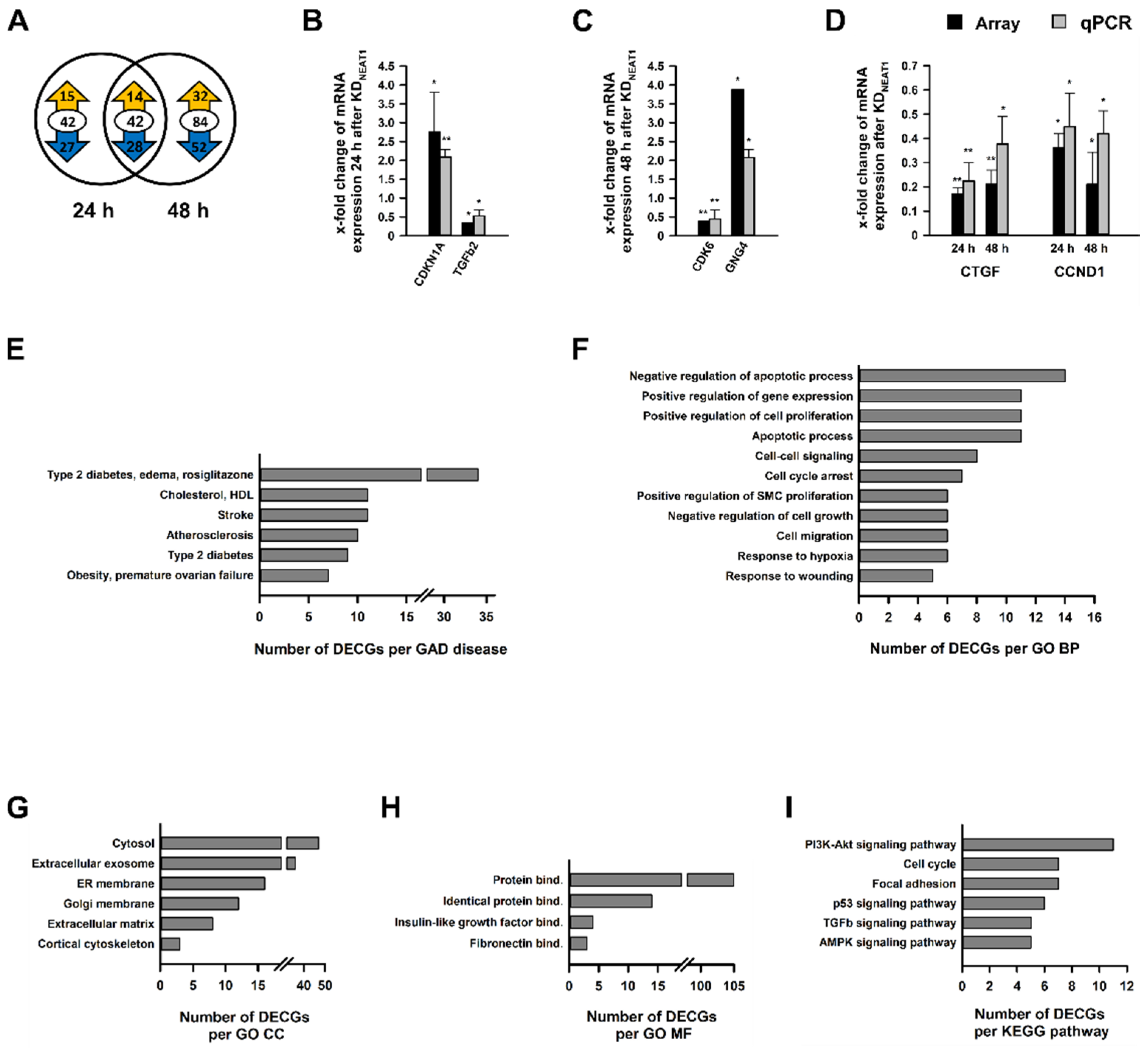
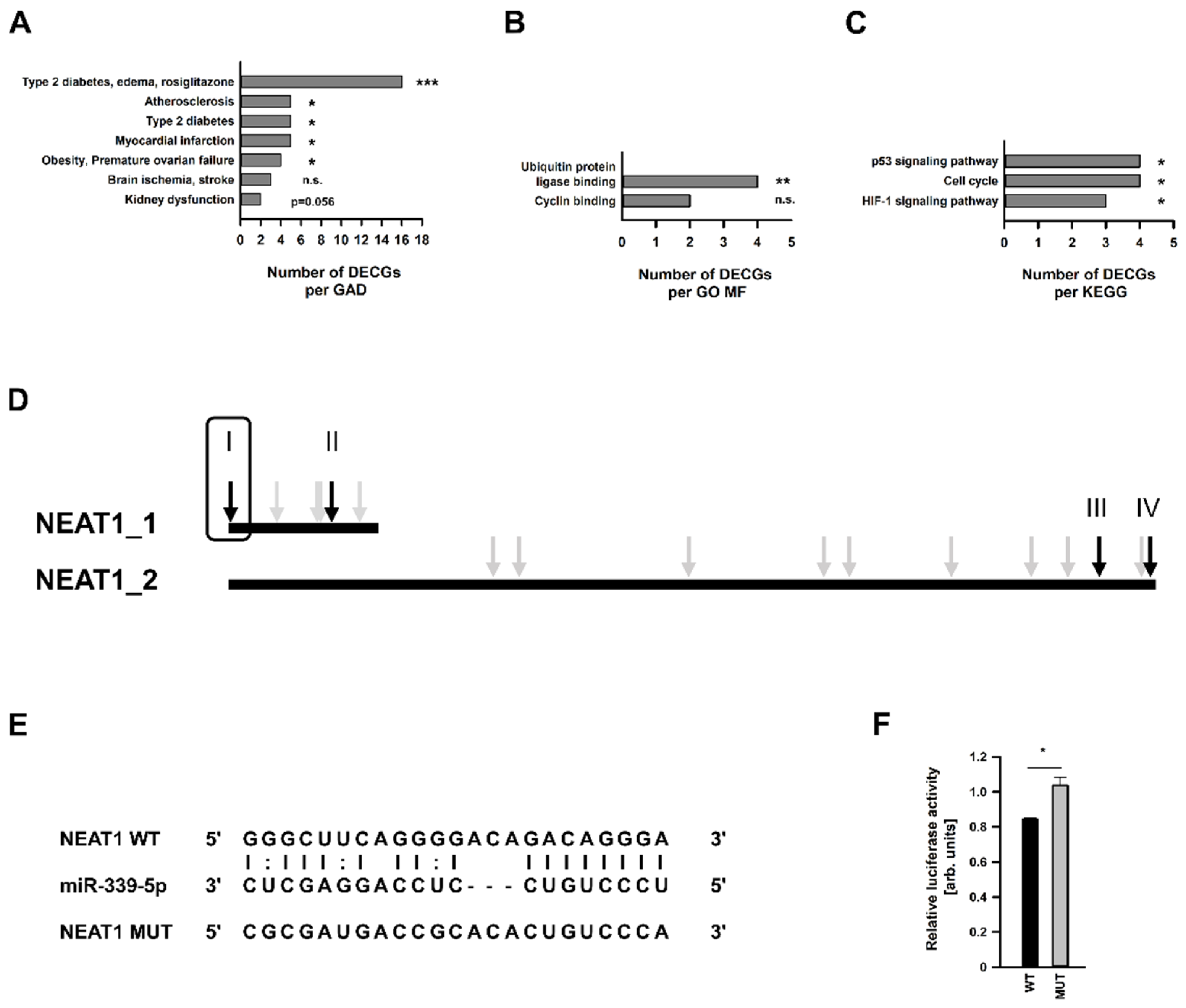
2.3. Analysis of Differentially Expressed Coding Genes after NEAT1 Knockdown
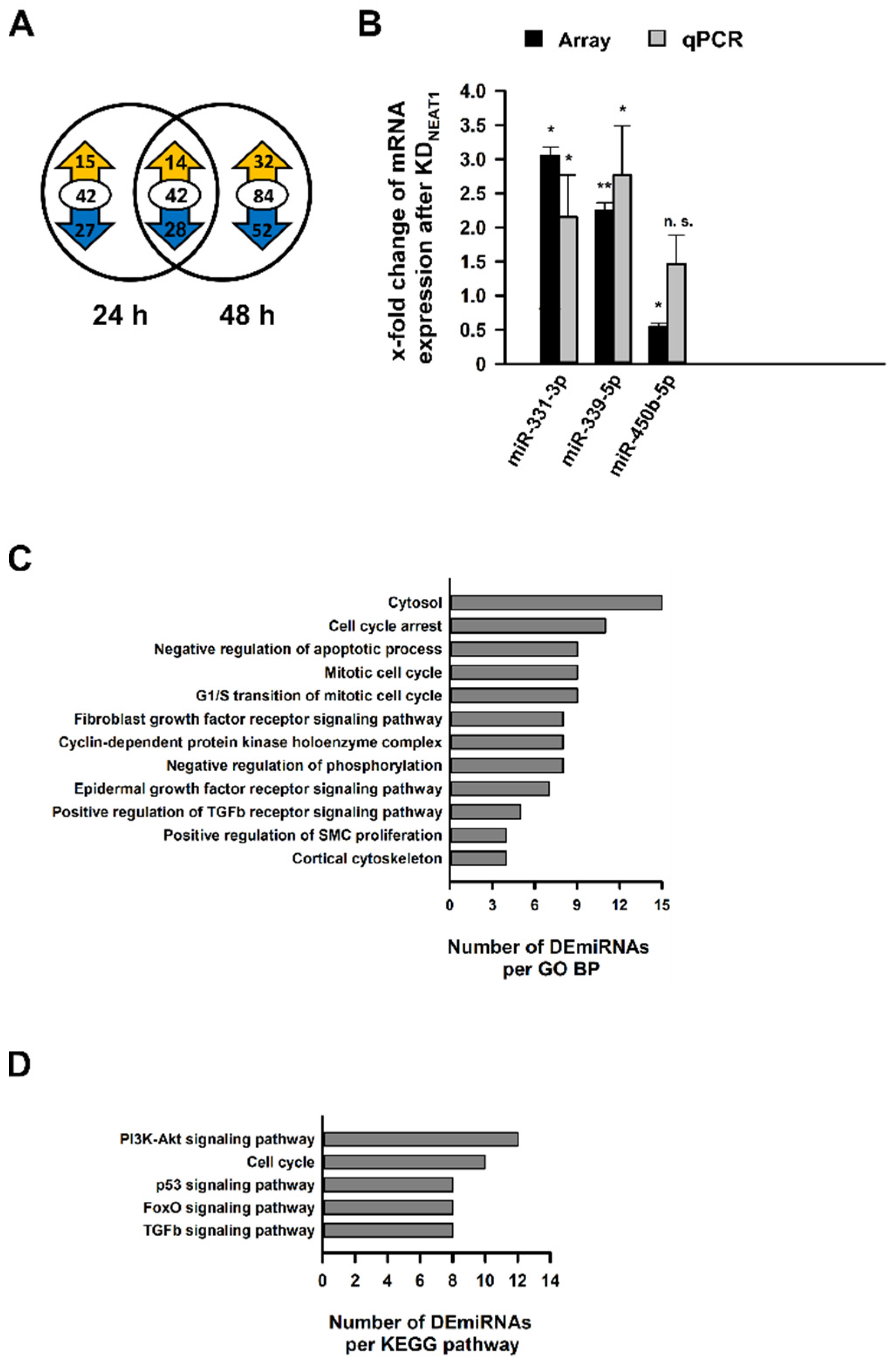
2.4. Analysis of Differentially Expressed miRNAs after NEAT1 Knockdown
2.5. Identification and In Silico-Characterization of the NEAT1 Binding miRNA miR-339-5p
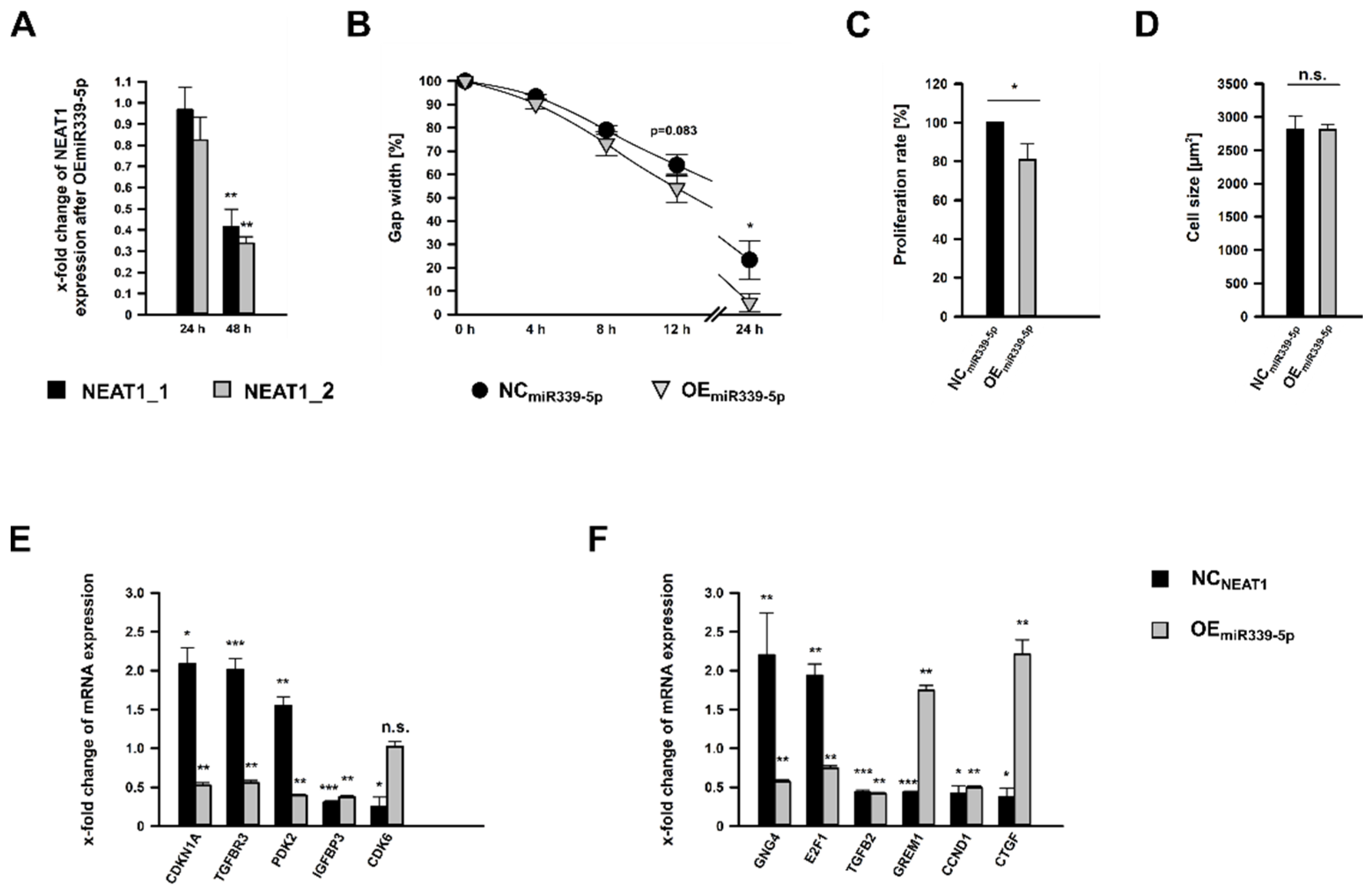
2.6. Treatment of hMCs with miR-339-5p Mimics
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Cell Culture Maintenance
4.3. ISH Localization in hMCs
4.4. In-Vitro Stimulation Experiments
4.5. NEAT1 Knockdown
4.6. Overexpression of miR-339-5p
4.7. Proliferation Assay
4.8. Wound Healing Assay
4.9. Cell Size Measurement
4.10. Cloning of Luciferase Reporter Gene Vectors and Luciferase Assay
4.11. Expression Analysis of DECGs and DEmiRNAs
4.12. Clariom™ S Assay Human and GeneChip™ miRNA 4.0 Array
4.13. Functional Enrichment Analyses
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. CJASN 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. JASN 2015, 26, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.W.; Moon, J.-Y. The role of inflammation in diabetic kidney disease. Korean J. Intern. Med. 2021, 36, 753–766. [Google Scholar] [CrossRef]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef]
- White, K.E.; Bilous, R.W. Structural alterations to the podocyte are related to proteinuria in type 2 diabetic patients. Nephrol. Dial. Transplant. 2004, 19, 1437–1440. [Google Scholar] [CrossRef]
- Fineberg, D.; Jandeleit-Dahm, K.A.M.; Cooper, M.E. Diabetic nephropathy: Diagnosis and treatment. Nat. Rev. Endocrinol. 2013, 9, 713–723. [Google Scholar] [CrossRef]
- National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2012, 60, 850–886. [Google Scholar] [CrossRef]
- Schlöndorff, D.; Banas, B. The mesangial cell revisited: No cell is an island. JASN 2009, 20, 1179–1187. [Google Scholar] [CrossRef]
- Tonneijck, L.; Muskiet, M.; Smits, M.; Van Bommel, E.J.; Heerspink, H.J.L.; Van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. JASN 2017, 28, 1023–1039. [Google Scholar] [CrossRef]
- Kong, Y.; Shen, Y.; Ni, J.; Shao, D.; Miao, N.; Xu, J.; Zhou, L.; Xue, H.; Zhang, W.; Wang, X.; et al. Insulin deficiency induces rat renal mesangial cell dysfunction via activation of IGF-1/IGF-1R pathway. Acta Pharm. Sin. 2016, 37, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Li, Z.; Ehara, T.; Yang, L.; Wang, D.; Feng, L.; Zhang, Y.; Wang, K.; Shi, Y.; Duan, H.; et al. Fatty Acid-Binding Protein 4 mediates apoptosis via endoplasmic reticulum stress in mesangial cells of diabetic nephropathy. Mol. Cell Endocrinol. 2015, 411, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, M.; Zoja, C.; Zanchi, C.; Corna, D.; Villa, S.; Bolognini, S.; Novelli, R.; Perico, L.; Remuzzi, G.; Benigni, A.; et al. Manipulating Sirtuin 3 pathway ameliorates renal damage in experimental diabetes. Sci. Rep. 2020, 10, 8418. [Google Scholar] [CrossRef] [PubMed]
- Hudkins, K.L.; Pinchaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Mühlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2010, 21, 1533–1542. [Google Scholar] [CrossRef]
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.G.; McCann, O.T.; Collins, J.E.; Oliver, K.; Willey, D.; Gribble, S.M.; Yang, F.; McLaren, K.; Rogers, J.; Ning, Z.; et al. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]
- Höck, J.; Meister, G. The Argonaute protein family. Genome Biol. 2008, 9, 210. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Angrand, P.-O.; Vennin, C.; Le Bourhis, X.; Adriaenssens, E. The role of long non-coding RNAs in genome formatting and expression. Front. Genet. 2015, 6, 165. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Chittka, D.; Banas, B.; Lennartz, L.; Putz, F.J.; Eidenschink, K.; Beck, S.; Stempfl, T.; Moehle, C.; Reichelt-Wurm, S.; Banas, M.C. Long-term expression of glomerular genes in diabetic nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2018, 33, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Reichelt-Wurm, S.; Wirtz, T.; Chittka, D.; Lindenmeyer, M.; Reichelt, R.M.; Beck, S.; Politis, P.; Charonis, A.; Kretz, M.; Huber, T.B.; et al. Glomerular expression pattern of long non-coding RNAs in the type 2 diabetes mellitus BTBR mouse model. Sci. Rep. 2019, 9, 9765. [Google Scholar] [CrossRef] [PubMed]
- Arvaniti, E.; Moulos, P.; Vakrakou, A.; Chatziantoniou, C.; Chadjichristos, C.; Kavvadas, P.; Charonis, A.; Politis, P.K. Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci. Rep. 2016, 6, 26235. [Google Scholar] [CrossRef]
- Sunwoo, H.; Dinger, M.E.; Wilusz, J.E.; Amaral, P.P.; Mattick, J.S.; Spector, D.L. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009, 19, 347–359. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakagawa, S.; Tanigawa, A.; Sasaki, Y.; Goshima, N.; Hirose, T. Alternative 3’-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 2012, 31, 4020–4034. [Google Scholar] [CrossRef]
- Sasaki, Y.T.F.; Ideue, T.; Sano, M.; Mituyama, T.; Hirose, T. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 2009, 106, 2525–2530. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, Y.; Wang, N.; Yu, N. NEAT1 negatively regulates miR-218 expression and promotes breast cancer progression. CBM 2017, 20, 247–254. [Google Scholar] [CrossRef]
- Pang, Y.; Wu, J.; Li, X.; Wang, C.; Wang, M.; Liu, J.; Yang, G. NEAT1/miR-124/STAT3 feedback loop promotes breast cancer progression. Int. J. Oncol. 2019, 55, 745–754. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, X.-Q.; Zhou, X.-Q.; Liu, Q.-B.; Chen, L.; Cai, F. NEAT1 induces osteosarcoma development by modulating the miR-339-5p/TGF-β1 pathway. J. Cell Physiol. 2019, 234, 5097–5105. [Google Scholar] [CrossRef]
- Yang, D.-Y.; Zhou, X.; Liu, Z.-W.; Xu, X.-Q.; Liu, C. LncRNA NEAT1 accelerates renal tubular epithelial cell damage by modulating mitophagy via miR-150-5p-DRP1 axis in diabetic nephropathy. Exp. Physiol. 2021, 106, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, Y.; Sun, Y.; Li, S.; Chen, L.; Jin, X.; Hou, X.; Liu, X.; Chen, Q.; Li, J.; et al. Knockdown of NEAT1 induces tolerogenic phenotype in dendritic cells by inhibiting activation of NLRP3 inflammasome. Theranostics 2019, 9, 3425–3442. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Shi, L.; Wang, X.; Luo, L.; Ling, L.; Yin, J.; Song, Y.; Zhang, Z.; Qiu, N.; Liu, H.; et al. KLF5 regulated lncRNA RP1 promotes the growth and metastasis of breast cancer via repressing p27kip1 translation. Cell Death Dis. 2019, 10, 373. [Google Scholar] [CrossRef]
- Slyne, J.; Slattery, C.; McMorrow, T.; Ryan, M.P. New developments concerning the proximal tubule in diabetic nephropathy: In vitro models and mechanisms. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2015, 30 (Suppl. 4), iv60–iv67. [Google Scholar]
- Campos-Melo, D.; Droppelmann, C.A.; Volkening, K.; Strong, M.J. Comprehensive luciferase-based reporter gene assay reveals previously masked up-regulatory effects of miRNAs. Int. J. Mol. Sci. 2014, 15, 15592–15602. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Prasanth, K.V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating gene expression through RNA nuclear retention. Cell 2005, 123, 249–263. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, X.-H.; Wu, Y.; Cao, G.-K.; Han, D. LncRNA NEAT1 regulates pulmonary fibrosis through miR-9-5p and TGF-β signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8483–8492. [Google Scholar]
- Zhang, P.; Cao, L.; Zhou, R.; Yang, X.; Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019, 10, 1495. [Google Scholar] [CrossRef]
- Castanotto, D.; Zhang, X.; Alluin, J.; Zhang, X.; Rüger, J.; Armstrong, B.; Rossi, J.; Riggs, A.; Stein, C.A. A stress-induced response complex (SIRC) shuttles miRNAs, siRNAs, and oligonucleotides to the nucleus. Proc. Natl. Acad. Sci. USA 2018, 115, E5756–E5765. [Google Scholar] [CrossRef]
- Jiang, L.; Shao, C.; Wu, Q.-J.; Chen, G.; Zhou, J.; Yang, B.; Li, H.; Gou, L.-T.; Zhang, Y.; Wang, Y.; et al. NEAT1 scaffolds RNA-binding proteins and the Microprocessor to globally enhance pri-miRNA processing. Nat. Struct. Mol. Biol. 2017, 24, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Petrica, L.; Hogea, E.; Gadalean, F.; Vlad, A.; Vlad, M.; Dumitrascu, V.; Velciov, S.; Gluhovschi, C.; Bob, F.; Ursoniu, S.; et al. Long noncoding RNAs may impact podocytes and proximal tubule function through modulating miRNAs expression in Early Diabetic Kidney Disease of Type 2 Diabetes Mellitus patients. Int. J. Med. Sci. 2021, 18, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Chen, J.; Zhang, C.; Guo, Y.; Liu, W.; Liu, W.; Duan, L.; Liu, Z.; Hu, J.; Lu, J. LncRNA NEAT1 Promotes High Glucose-Induced Mesangial Cell Hypertrophy by Targeting miR-222-3p/CDKN1B Axis. Front. Mol. Biosci. 2020, 7, 627827. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhao, N.; Du, L.; Wang, Y. Downregulation of lncRNA NEAT1 inhibits mouse mesangial cell proliferation, fibrosis, and inflammation but promotes apoptosis in diabetic nephropathy. Int. J. Clin. Exp. Pathol. 2019, 12, 1174–1183. [Google Scholar]
- Li, C.; Liu, Y.-F.; Huang, C.; Chen, Y.-X.; Xu, C.-Y.; Chen, Y. Long noncoding RNA NEAT1 sponges miR-129 to modulate renal fibrosis by regulation of collagen type I. Am. J. Physiol. -Ren. Physiol. 2020, 319, F93–F105. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.-F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Okado, T.; Terada, Y.; Tanaka, H.; Inoshita, S.; Nakao, A.; Sasaki, S. Smad7 mediates transforming growth factor-beta-induced apoptosis in mesangial cells. Kidney Int. 2002, 62, 1178–1186. [Google Scholar] [CrossRef]
- Jardim, D.P.; Poço, P.C.E.; Campos, A.H. Dact1, a Wnt-Pathway Inhibitor, Mediates Human Mesangial Cell TGF-β1-Induced Apoptosis. J. Cell Physiol. 2017, 232, 2104–2111. [Google Scholar] [CrossRef]
- Lampropoulou, I.T.; Stangou, M.; Sarafidis, P.; Gouliovaki, A.; Giamalis, P.; Tsouchnikas, I.; Didangelos, T.; Papagianni, A. TNF-α pathway and T-cell immunity are activated early during the development of diabetic nephropathy in Type II Diabetes Mellitus. Clin. Immunol. Orlando Fla. 2020, 215, 108423. [Google Scholar] [CrossRef]
- Navarro, J.F.; Mora, C.; Rivero, A.; Gallego, E.; Chahin, J.; Macía, M.; Méndez, M.L.; García, J. Urinary protein excretion and serum tumor necrosis factor in diabetic patients with advanced renal failure: Effects of pentoxifylline administration. Am. J. Kidney Dis. 1999, 33, 458–463. [Google Scholar] [CrossRef]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Huang, J.; Shu, H.-B.; Baichwal, V.; Goeddel, D.V. TNF-Dependent Recruitment of the Protein Kinase RIP to the TNF Receptor-1 Signaling Complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Xu, Y.; He, N.; Leng, L.; Li, Z. Tumor necrosis factor-α regulates matrix metalloproteinase-2 expression and cell migration via ERK pathway in rat glomerular mesangial cells. Cell Biol. Int. 2014, 38, 1060–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, X.; Hu, Q.; Chen, X.; Chen, Y.; Hunag, L. Galectin-3 activates TLR4/NF-κB signaling to promote lung adenocarcinoma cell proliferation through activating lncRNA-NEAT1 expression. BMC Cancer 2018, 18, 580. [Google Scholar] [CrossRef]
- Zhou, Y.; Sha, Z.; Yang, Y.; Wu, S.; Chen, H. lncRNA NEAT1 regulates gastric carcinoma cell proliferation, invasion and apoptosis via the miR-500a-3p/XBP-1 axis. Mol. Med. Rep. 2021, 24, 503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Wang, X.; Wu, R.; Lin, M.; Laddha, S.V.; Yang, Q.; Chan, C.S.; Feng, Z. MicroRNA-339-5p inhibits colorectal tumorigenesis through regulation of the MDM2/p53 signaling. Oncotarget 2014, 5, 9106–9117. [Google Scholar] [CrossRef]
- Toda, N.; Mori, K.; Kasahara, M.; Ishii, A.; Koga, K.; Ohno, S.; Mori, K.P.; Kato, Y.; Osaki, K.; Kuwabara, T.; et al. Crucial Role of Mesangial Cell-derived Connective Tissue Growth Factor in a Mouse Model of Anti-Glomerular Basement Membrane Glomerulonephritis. Sci. Rep. 2017, 7, 42114. [Google Scholar] [CrossRef]
- Marine, J.-C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef]
- Jansson, M.D.; Damas, N.D.; Lees, M.; Jacobsen, A.; Lund, A.H. miR-339-5p regulates the p53 tumor-suppressor pathway by targeting MDM2. Oncogene 2015, 34, 1908–1918. [Google Scholar] [CrossRef]
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics 2015, 5, 1122–1143. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, L.; Li, L.; Fan, M.; Hou, L. MiR-339 attenuates LPS-induced intestinal epithelial cells inflammatory responses and apoptosis by targeting TLR4. Genes Genom 2020, 42, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Afgar, A.; Fard-Esfahani, P.; Mehrtash, A.; Azadmanesh, K.; Khodarahmi, F.; Ghadir, M.; Teimoori-Toolabi, L. MiR-339 and especially miR-766 reactivate the expression of tumor suppressor genes in colorectal cancer cell lines through DNA methyltransferase 3B gene inhibition. Cancer Biol. Ther. 2016, 17, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yao, T.; Zhang, Y.; Li, W.; Wang, Z. NEAT1 as a competing endogenous RNA in tumorigenesis of various cancers: Role, mechanism and therapeutic potential. Int. J. Biol. Sci. 2021, 17, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, L.; Yin, L. lncRNA NEAT1 Binds to MiR-339-5p to Increase HOXA1 and Alleviate Ischemic Brain Damage in Neonatal Mice. Mol. Ther.-Nucleic Acids 2020, 20, 117–127. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, G.; Mi, J.; Wang, G.; Yu, M.; Jin, D.; Tong, X.; Wang, X. The Impact of lncRNA on Diabetic Kidney Disease: Systematic Review and In Silico Analyses. Comput. Intell. Neurosci. 2022, 2022, 8400106. [Google Scholar] [CrossRef]
- Hiyama, H.; Iavarone, A.; Reeves, S.A. Regulation of the cdk inhibitor p21 gene during cell cycle progression is under the control of the transcription factor E2F. Oncogene 1998, 16, 1513–1523. [Google Scholar] [CrossRef]
- Banas, B.; Luckow, B.; Möller, M.; Klier, C.; Nelson, P.J.; Schadde, E.; Brigl, M.; Halevy, D.; Holthöfer, H.; Reinhart, B.; et al. Chemokine and chemokine receptor expression in a novel human mesangial cell line. J. Am. Soc. Nephrol. JASN 1999, 10, 2314–2322. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated DECGs | Downregulated DECGs | ||
|---|---|---|---|
| Name | xFC | Name | xFC |
| SERPINE1 | −11.82 | EPB41 | 2.03 |
| EDN1 | −11.13 | CBFA2T2 | 2.07 |
| RGS4 | −4.77 | RAB15 | 2.08 |
| IGFBP3 | −4.19 | PDK2 | 2.17 |
| SKP2 | −3.55 | CUX1 | 2.39 |
| NT5DC3 | −3.3 | MXD4 | 2.54 |
| DDAH1 | −3.17 | ITPK1 | 2.55 |
| VGLL3 | −3.09 | SMC1A | 2.65 |
| FLNA | −3.01 | CDKN1A | 2.75 |
| ENTPD7 | −2.97 | TGFBR3 | 2.89 |
| LBH | −2.95 | BTBD2 | 2.92 |
| DCP2 | −2.93 | MAN2A2 | 3.19 |
| SRGN | −2.91 | OAS3 | 7.21 |
| HMGCR | −2.66 | ||
| CDK6 | −2.57 | ||
| NAV3 | −2.53 | ||
| LBR | −2.45 | ||
| GSKIP | −2.4 | ||
| RUFY2 | −2.29 | ||
| ATXN1 | −2.2 | ||
| TACC1 | −2.2 | ||
| PDE4D | −2.19 | ||
| COPZ1 | −2.19 | ||
| CCDC50 | −2.17 | ||
| DUSP5 | −2.16 | ||
| TMEM2 | −2.08 | ||
| ENC1 | −2.03 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reichelt-Wurm, S.; Pregler, M.; Wirtz, T.; Kretz, M.; Holler, K.; Banas, B.; Banas, M.C. The Interplay of NEAT1 and miR-339-5p Influences on Mesangial Gene Expression and Function in Various Diabetic-Associated Injury Models. Non-Coding RNA 2022, 8, 52. https://doi.org/10.3390/ncrna8040052
Reichelt-Wurm S, Pregler M, Wirtz T, Kretz M, Holler K, Banas B, Banas MC. The Interplay of NEAT1 and miR-339-5p Influences on Mesangial Gene Expression and Function in Various Diabetic-Associated Injury Models. Non-Coding RNA. 2022; 8(4):52. https://doi.org/10.3390/ncrna8040052
Chicago/Turabian StyleReichelt-Wurm, Simone, Matthias Pregler, Tobias Wirtz, Markus Kretz, Kathrin Holler, Bernhard Banas, and Miriam C. Banas. 2022. "The Interplay of NEAT1 and miR-339-5p Influences on Mesangial Gene Expression and Function in Various Diabetic-Associated Injury Models" Non-Coding RNA 8, no. 4: 52. https://doi.org/10.3390/ncrna8040052
APA StyleReichelt-Wurm, S., Pregler, M., Wirtz, T., Kretz, M., Holler, K., Banas, B., & Banas, M. C. (2022). The Interplay of NEAT1 and miR-339-5p Influences on Mesangial Gene Expression and Function in Various Diabetic-Associated Injury Models. Non-Coding RNA, 8(4), 52. https://doi.org/10.3390/ncrna8040052