
Patterns of Differentially Expressed circRNAs in Human Thymocytes

 ,
,  , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
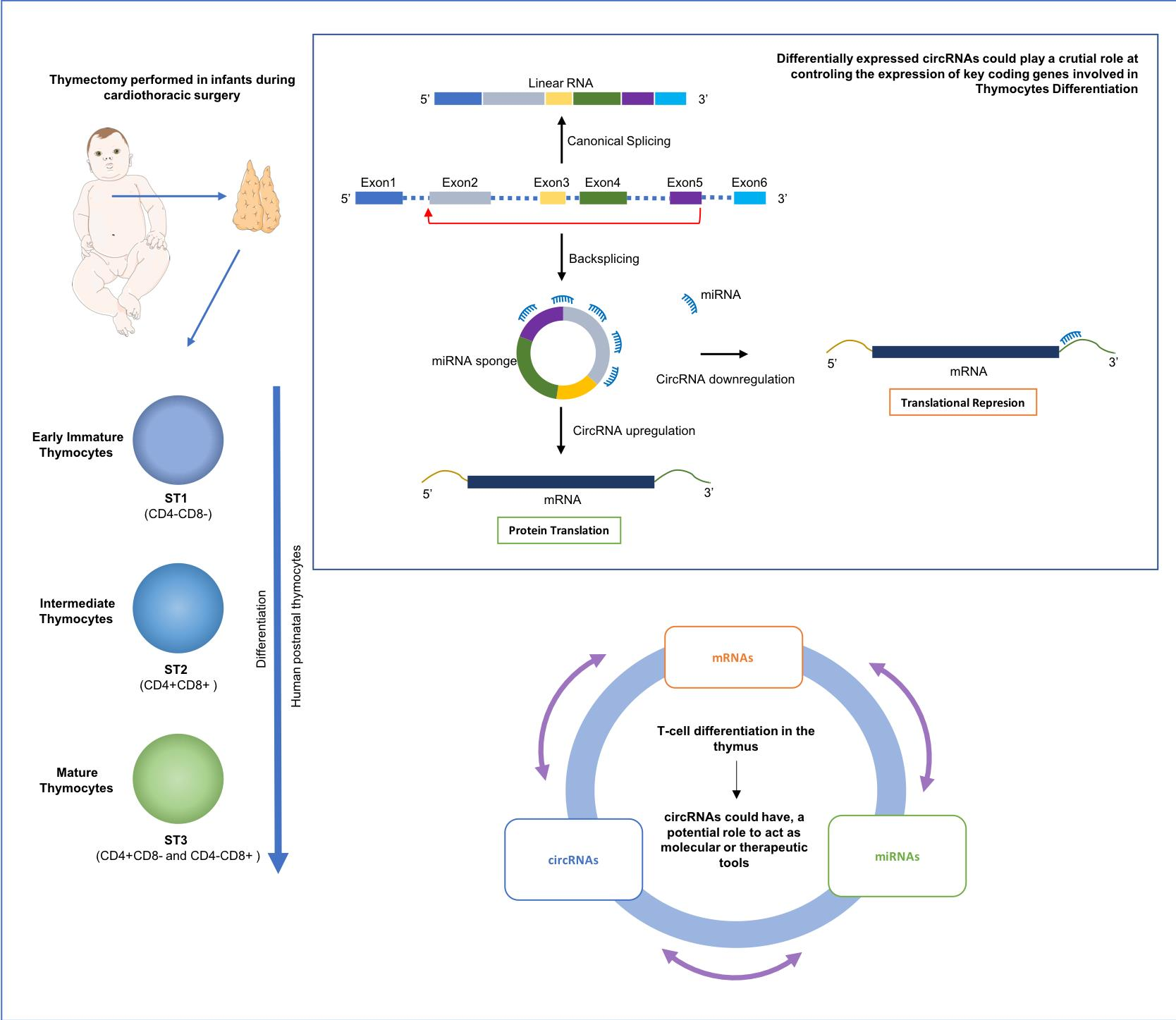
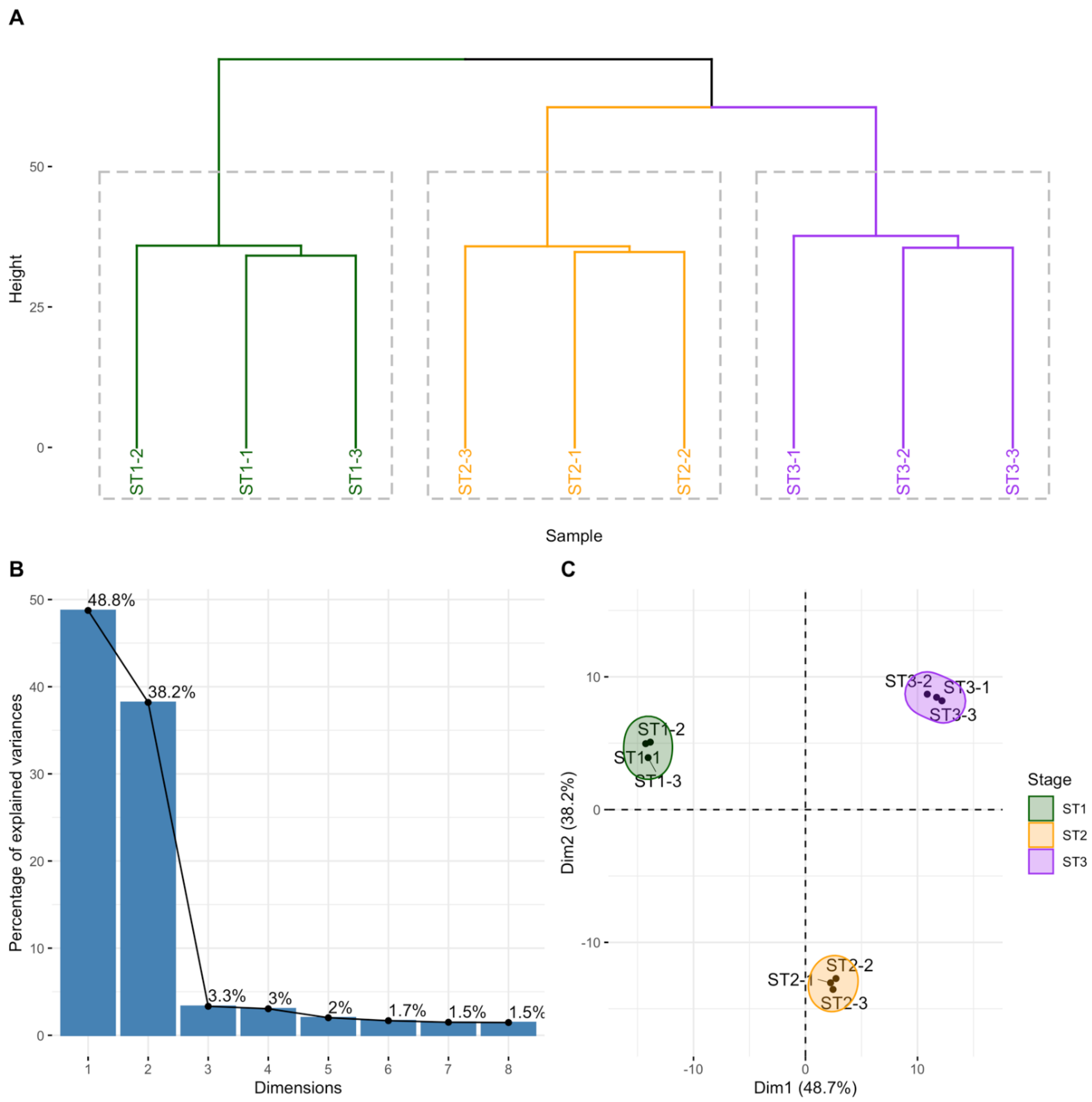
2.1. Isolation of Human Thymocytes at Different Stages during Their Intrathymic Differentiation
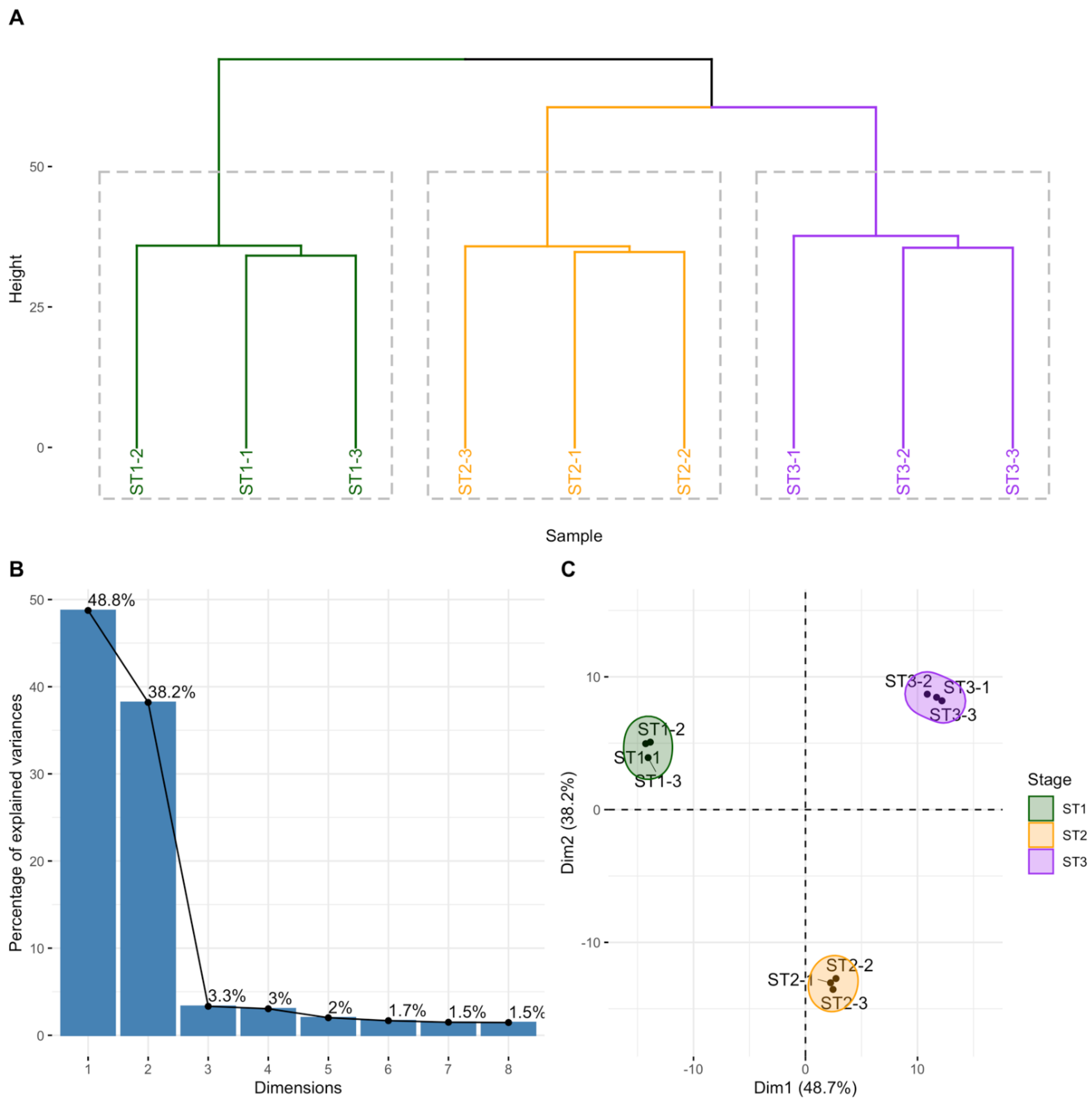
2.2. circRNAs Are Differentially Expressed during Thymocyte Differentiation
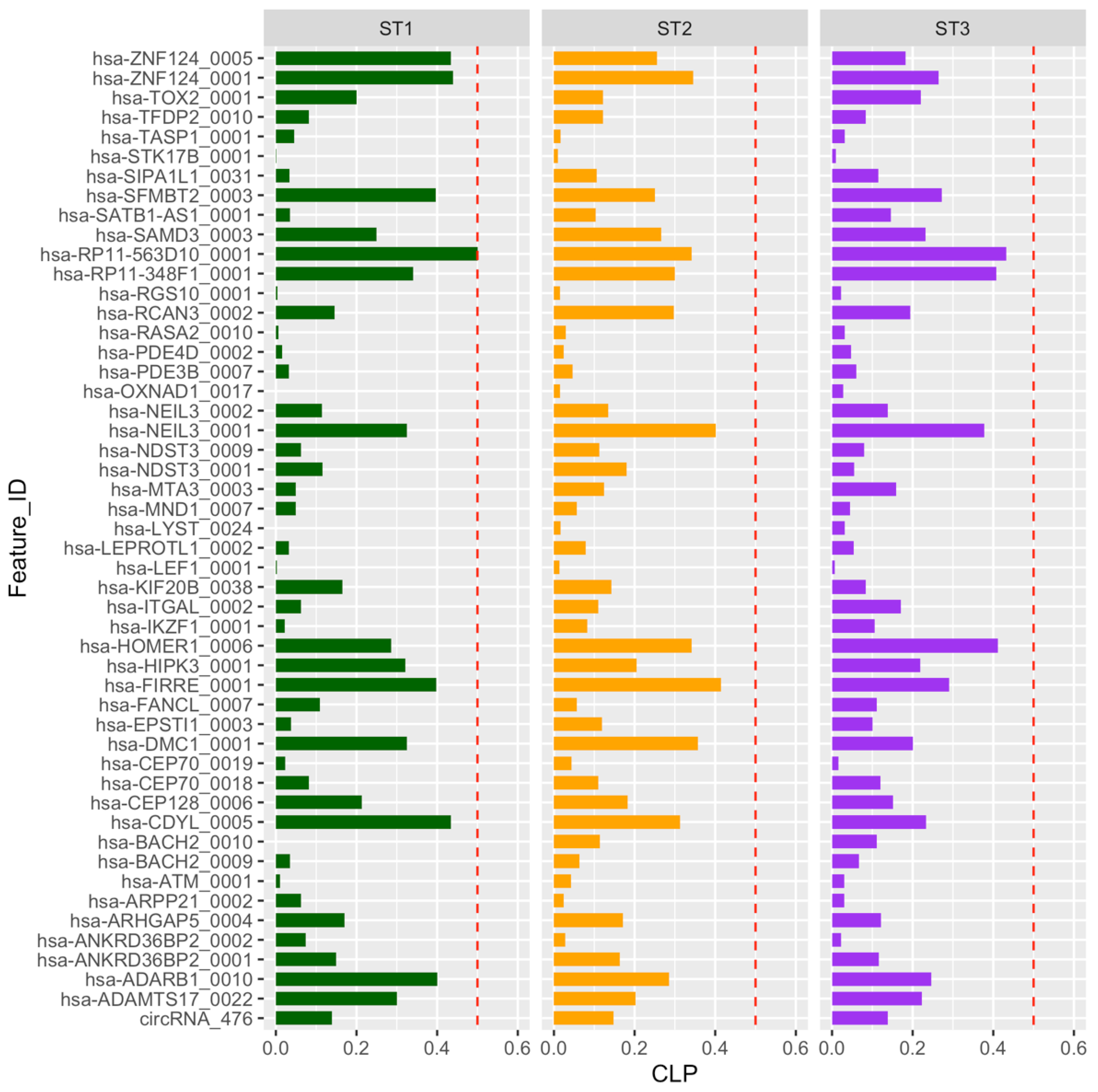
2.3. Circular-to-Linear Expression Proportion
2.4. Validation of Selected circRNAs
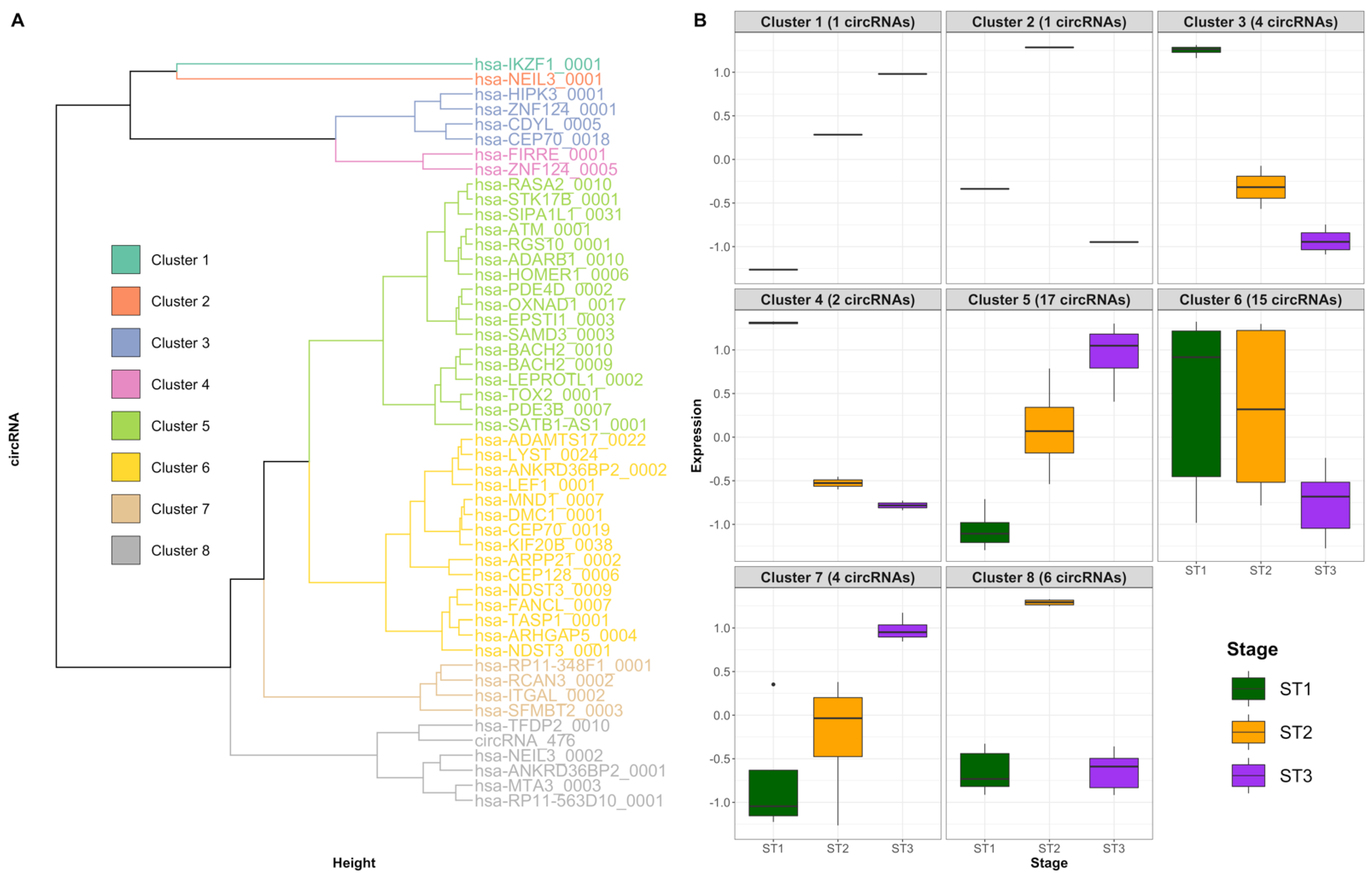
2.5. Patterns of Differentially Expressed circRNAs
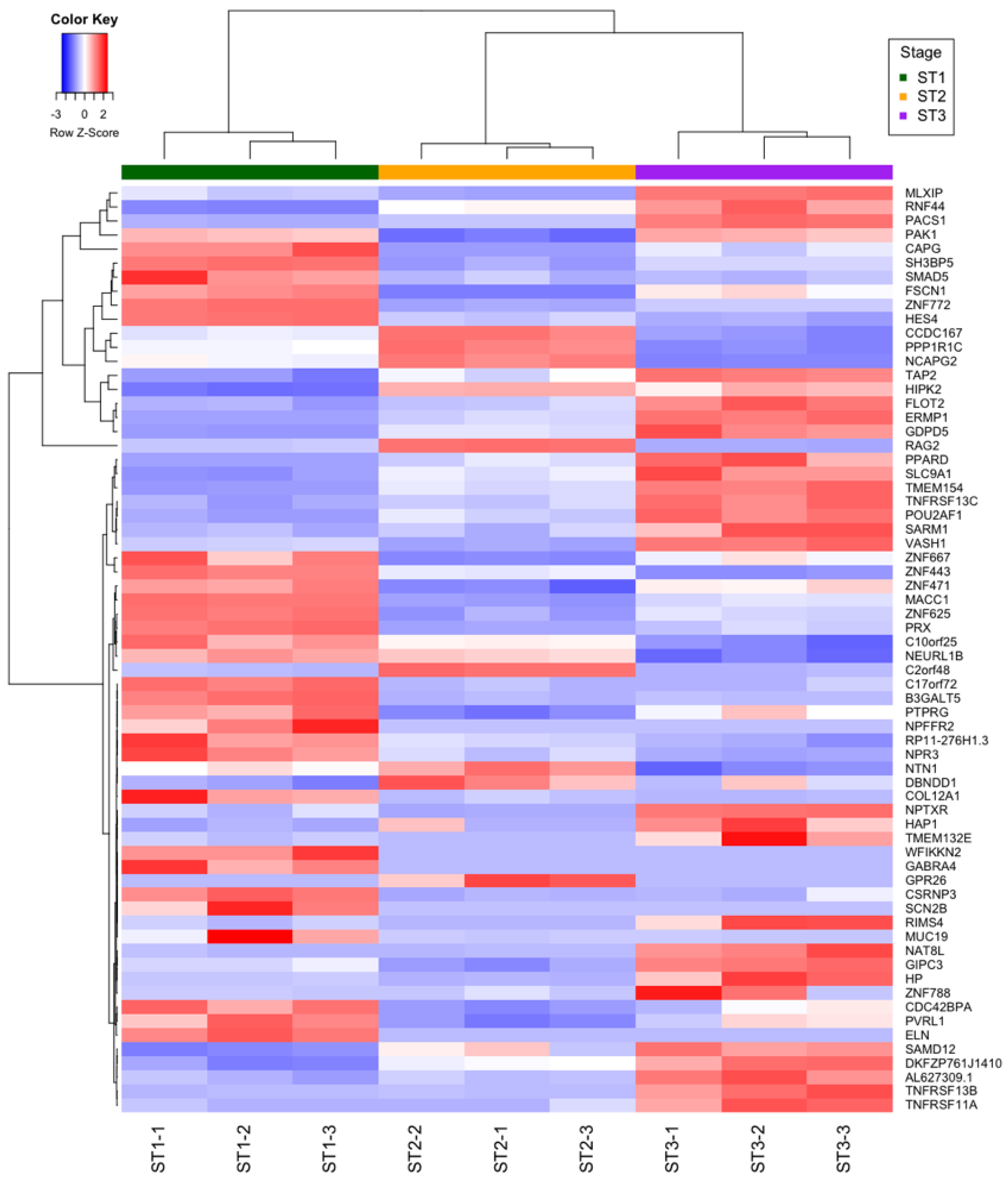
2.6. mRNAs Differentially Expressed during Thymocyte Differentiation
2.7. In Silico Functional Outcome Prediction of Specific circRNAs Differential Expression
3. Materials and Methods
3.1. Patients’ Characteristics
3.2. Isolation of Human Thymocytes at Different Stages of Differentiation
3.3. RNA Isolation
3.4. Quality Control and Trimming
3.5. mRNA Pipeline
3.6. circRNA Pipeline
3.7. Quantification and Annotation of circRNAs
3.8. Pairwise Comparations of circRNA and mRNA Expression among the Three Thymocyte Populations
3.9. Functional Annotation of circRNAs
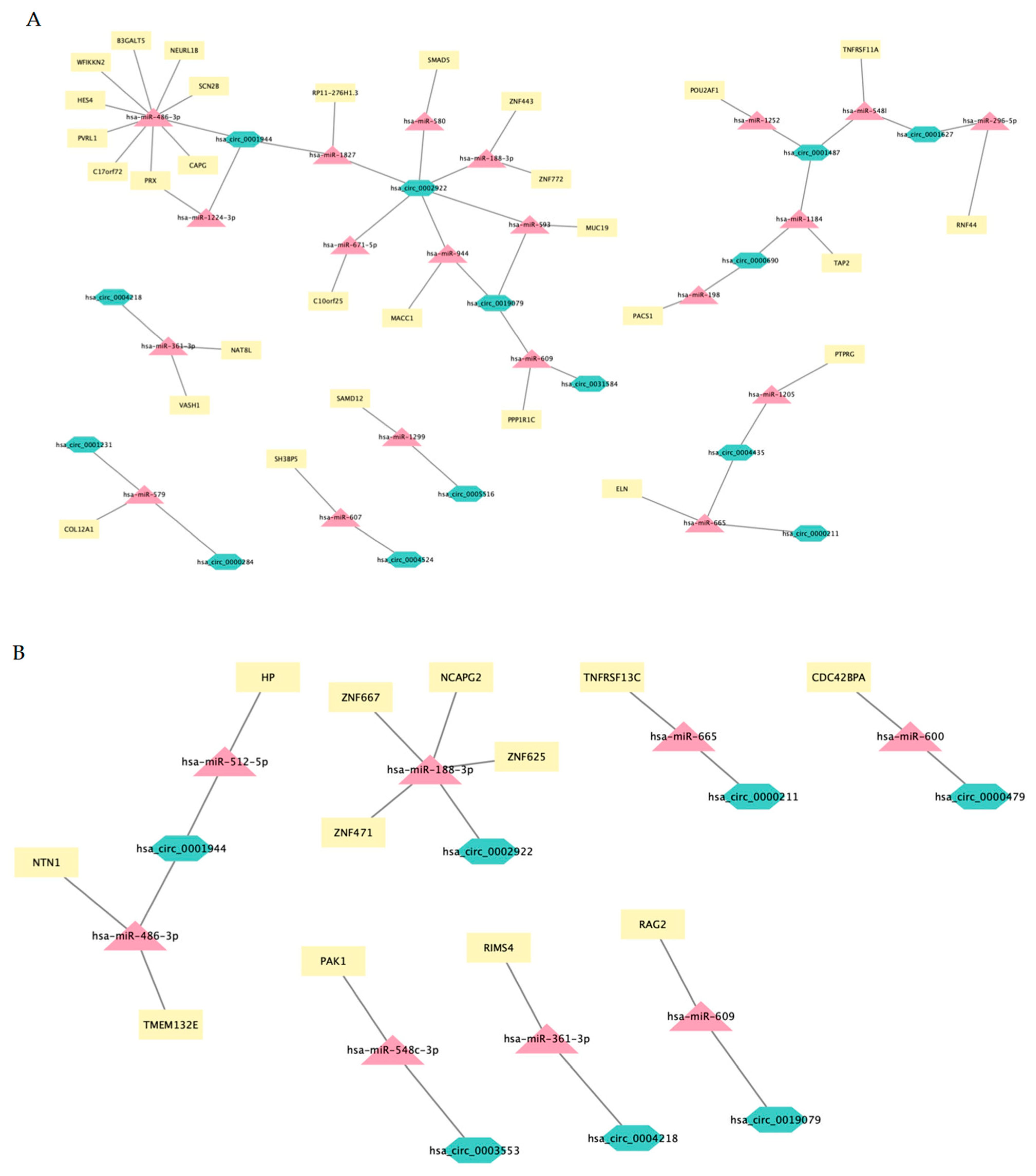
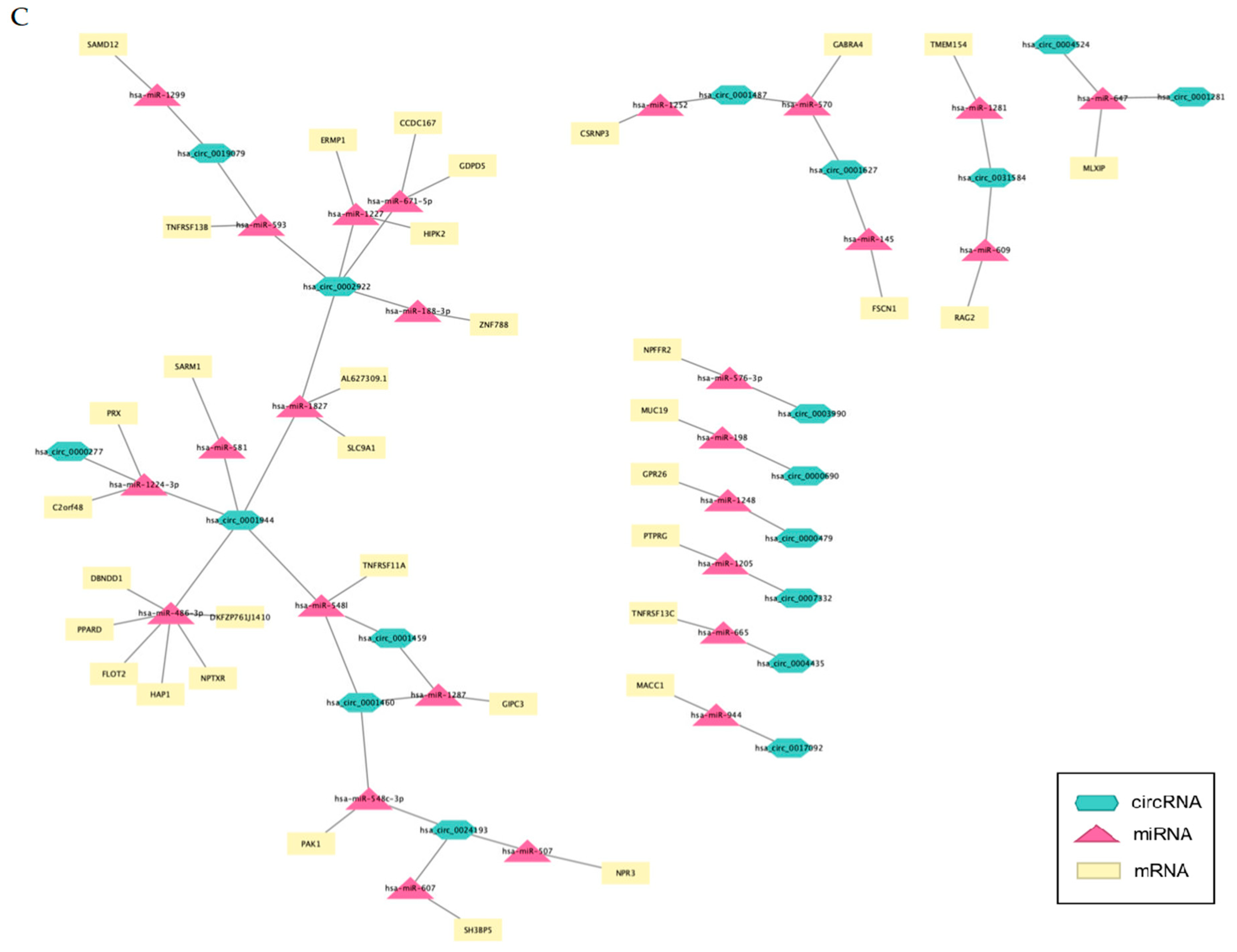
3.10. mRNA-circRNA-miRNA Interaction Network Analysis
3.11. Retrotranscription and Polymerase Chain Reactions
3.12. CircRNA Functional Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhang, X.-O.; Wang, H.-B.; Zhang, Y.; Lu, X.; Chen, L.-L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient MicroRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Meng, X.; Li, X.; Zhang, P.; Wang, J.; Zhou, Y.; Chen, M. Circular RNA: An emerging key player in RNA world. Brief. Bioinform. 2017, 18, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Salzman, J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell. Mol. Life Sci. 2018, 75, 1071–1098. [Google Scholar] [CrossRef] [Green Version]
- Patop, I.L.; Kadener, S. CircRNAs in cancer. Curr. Opin. Genet. Dev. 2018, 48, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Kuo, H.-C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 29. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, B.P.; Engels, S.; Aglialoro, F.; van den Akker, E.; von Lindern, M.; Wolkers, M.C. Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res. 2018, 46, 8168–8180. [Google Scholar] [CrossRef] [PubMed]
- Gaffo, E.; Boldrin, E.; Dal Molin, A.; Bresolin, S.; Bonizzato, A.; Trentin, L.; Frasson, C.; Debatin, K.-M.; Meyer, L.H.; Te Kronnie, G.; et al. Circular RNA differential expression in blood cell populations and exploration of CircRNA deregulation in pediatric acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 14670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratin, A.; Paganin, M.; Gaffo, E.; Dal Molin, A.; Roels, J.; Germano, G.; Siddi, M.T.; Serafin, V.; De Decker, M.; Gachet, S.; et al. Large-scale circular RNA deregulation in T-ALL: Unlocking unique ectopic expression of molecular subtypes. Blood Adv. 2020, 4, 5902–5914. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Taghon, T.; Waegemans, E.; Van de Walle, I. Notch signaling during human T cell development. Notch Regul. Immune Syst. 2012, 360, 75–97. [Google Scholar] [CrossRef] [Green Version]
- Dik, W.A.; Pike-Overzet, K.; Weerkamp, F.; de Ridder, D.; de Haas, E.F.E.; Baert, M.R.M.; van der Spek, P.; Koster, E.E.L.; Reinders, M.J.T.; van Dongen, J.J.M.; et al. New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J. Exp. Med. 2005, 201, 1715–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, R.N. T-cell development and the CD4–CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Blom, B.; Spits, H. Development of human lymphoid cells. Annu. Rev. Immunol. 2006, 24, 287–320. [Google Scholar] [CrossRef]
- Yui, M.A.; Rothenberg, E.V. Developmental gene networks: A triathlon on the course to T cell identity. Nat. Rev. Immunol. 2014, 14, 529–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, E.V.; Ungerbäck, J.; Champhekar, A. Forging T-lymphocyte identity: Intersecting networks of transcriptional control. Adv. Immunol. 2016, 129, 109–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallaert, A.; Durinck, K.; Taghon, T.; Van Vlierberghe, P.; Speleman, F. T-all and thymocytes: A message of noncoding RNAs. J. Hematol. Oncol. 2017, 10, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant CircHIPK3 that regulates cell growth by sponging multiple MiRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Poggi, A.; Costa, P.; Morelli, L.; Cantoni, C.; Pella, N.; Spada, F.; Biassoni, R.; Nanni, L.; Revello, V.; Tomasello, E.; et al. Expression of human NKRP1A by CD34+ immature thymocytes: NKRP1A-mediated regulation of proliferation and cytolytic activity. Eur. J. Immunol. 1996, 26, 1266–1272. [Google Scholar] [CrossRef]
- Le, J.; Park, J.E.; Ha, V.L.; Luong, A.; Branciamore, S.; Rodin, A.S.; Gogoshin, G.; Li, F.; Loh, Y.-H.E.; Camacho, V.; et al. Single-cell RNA-seq mapping of human thymopoiesis reveals lineage specification trajectories and a commitment spectrum in T cell development. Immunity 2020, 52, 1105–1118.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhu, W.; Wang, Q.; Gu, J.; Huang, L.F.; Sun, X. Differential regulatory network-based quantification and prioritization of key genes underlying cancer drug resistance based on time-course RNA-Seq data. PLoS Comput. Biol. 2019, 15, e1007435. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.R.; Damen, F.; Aspiras, A.; Tabin, J.A.; McGaugh, S.; Tabin, C.J. Evolution of gastrointestinal tract morphology and plasticity in cave-adapted mexican tetra, astyanax mexicanus. bioRxiv 2020, 852814. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, T.; Hu, W.; Chen, B.; Dai, M.; Xu, G. Genome-wide methylation patterns in androgen-independent prostate cancer cells: A comprehensive analysis combining MeDIP-bisulfite, RNA, and MicroRNA sequencing data. Genes 2018, 9, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, R.D.; Majety, M.; Chen, S. The aging transcriptome and cellular landscape of the human lung in relation to SARS-CoV-2. Nat. Commun. 2021, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-Seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Jakobi, T.; Uvarovskii, A.; Dieterich, C. Circtools-a one-stop software solution for circular RNA research. Bioinformatics 2019, 35, 2326–2328. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Metge, F.; Dieterich, C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 2016, 32, 1094–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019, baz003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and MicroRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of MicroRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Nieva, P.; Fernández-Navarro, P.; Cobos-Fernández, M.Á.; González-Vasconcellos, I.; Sánchez Pérez, R.; Aroca, Á.; Fernández-Piqueras, J.; Santos, J. Patterns of Differentially Expressed circRNAs in Human Thymocytes. Non-Coding RNA 2022, 8, 26. https://doi.org/10.3390/ncrna8020026
López-Nieva P, Fernández-Navarro P, Cobos-Fernández MÁ, González-Vasconcellos I, Sánchez Pérez R, Aroca Á, Fernández-Piqueras J, Santos J. Patterns of Differentially Expressed circRNAs in Human Thymocytes. Non-Coding RNA. 2022; 8(2):26. https://doi.org/10.3390/ncrna8020026
Chicago/Turabian StyleLópez-Nieva, Pilar, Pablo Fernández-Navarro, María Ángeles Cobos-Fernández, Iria González-Vasconcellos, Raúl Sánchez Pérez, Ángel Aroca, José Fernández-Piqueras, and Javier Santos. 2022. "Patterns of Differentially Expressed circRNAs in Human Thymocytes" Non-Coding RNA 8, no. 2: 26. https://doi.org/10.3390/ncrna8020026
APA StyleLópez-Nieva, P., Fernández-Navarro, P., Cobos-Fernández, M. Á., González-Vasconcellos, I., Sánchez Pérez, R., Aroca, Á., Fernández-Piqueras, J., & Santos, J. (2022). Patterns of Differentially Expressed circRNAs in Human Thymocytes. Non-Coding RNA, 8(2), 26. https://doi.org/10.3390/ncrna8020026