
FibroDB: Expression Analysis of Protein-Coding and Long Non-Coding RNA Genes in Fibrosis

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
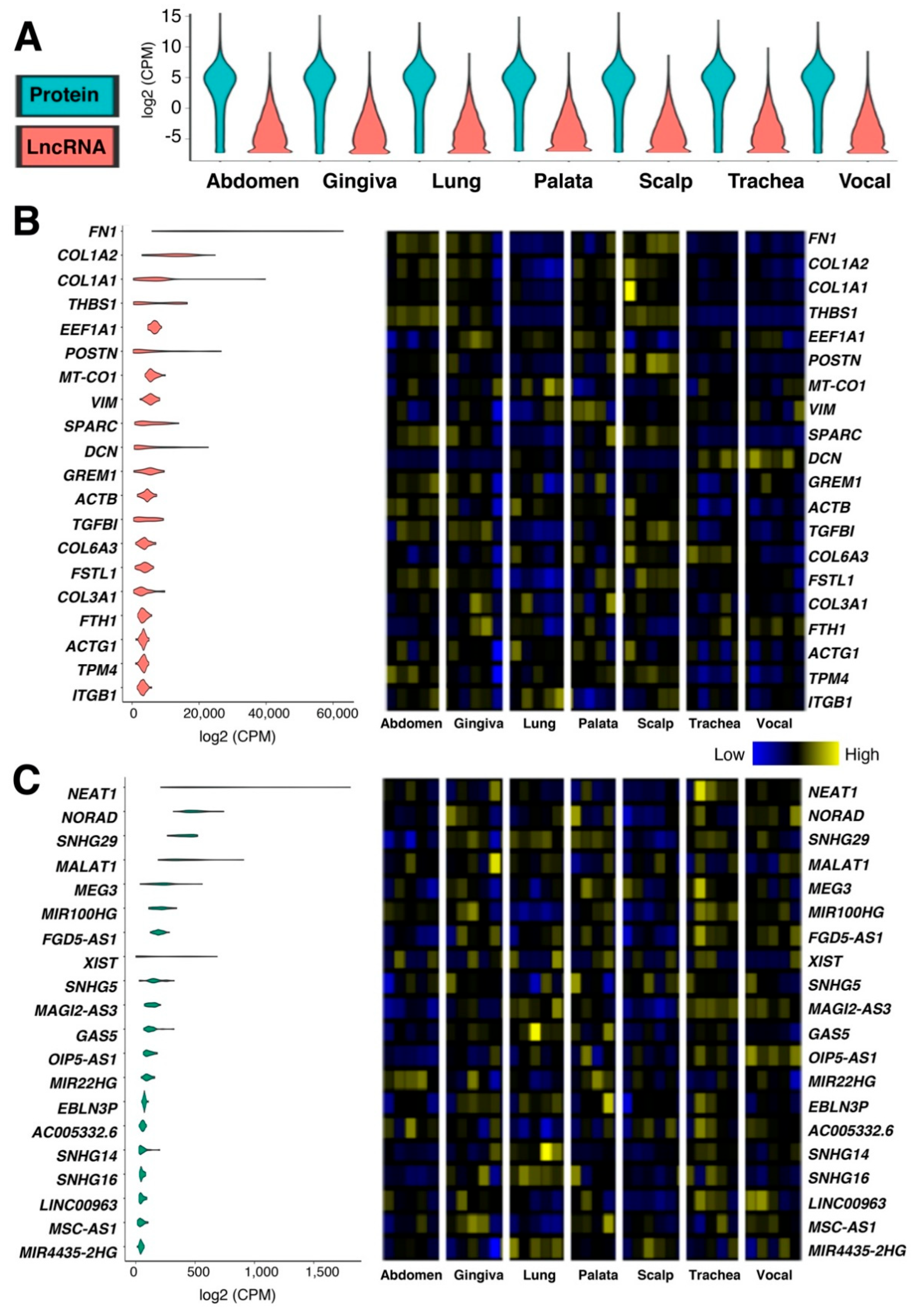
2.1. Tissue-Specific Expressions of Protein-Coding and lncRNA Genes in Human Fibroblasts
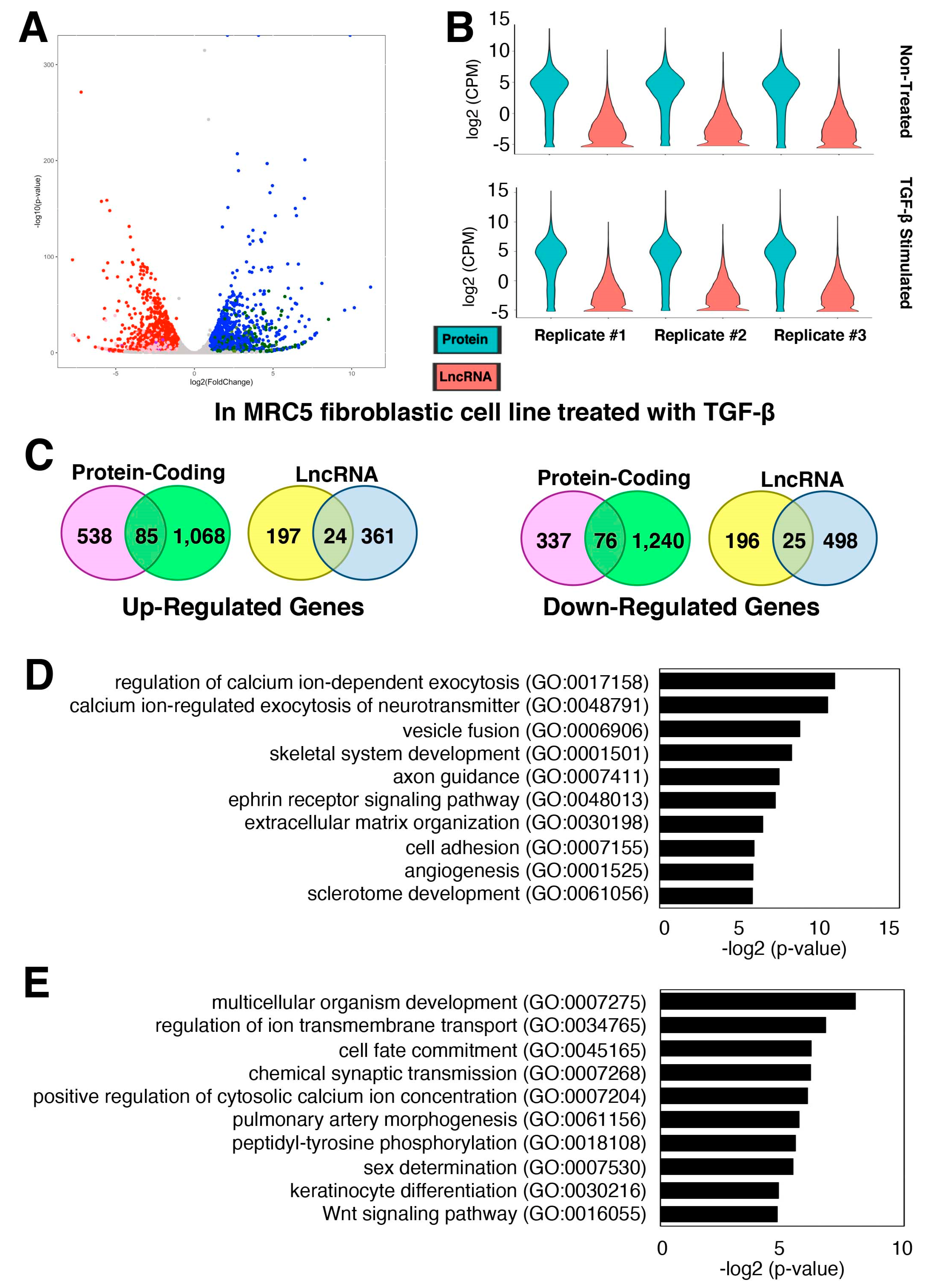
2.2. Gene Expression Changes of Protein-Coding and lncRNA Genes during Pulmonary Fibrosis
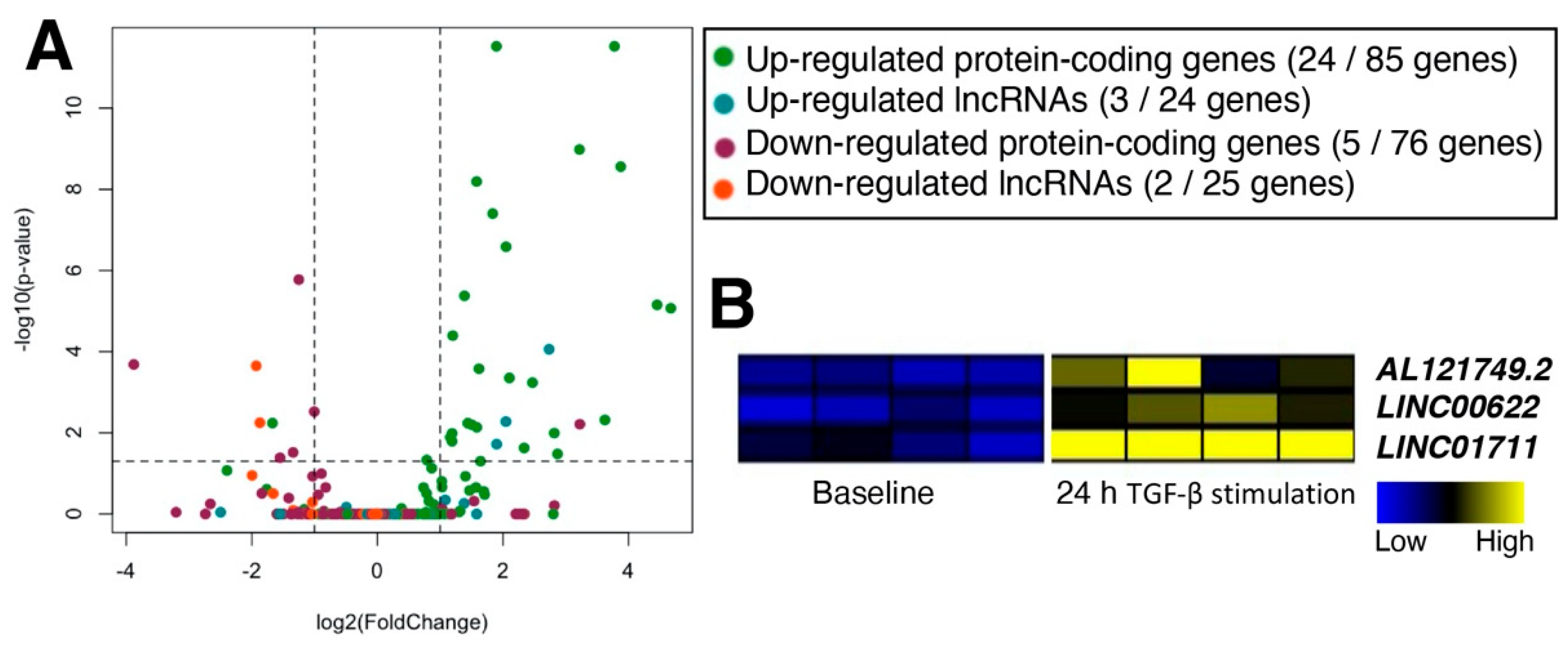
2.3. Characterization of TGF-β-Stimulated lncRNAs in Cardiac Fibroblasts
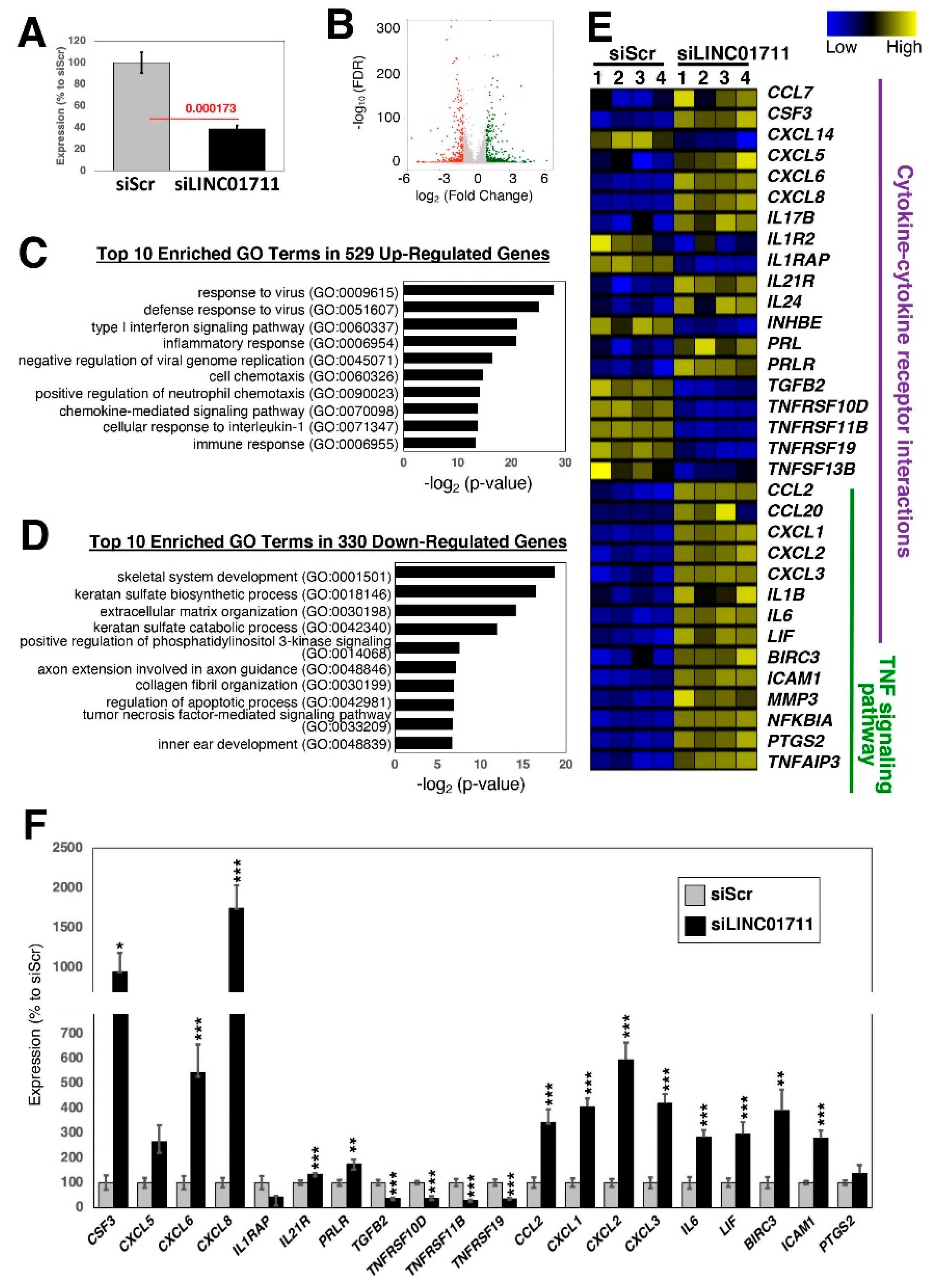
2.4. Loss-of-Function Study of TGF-β-Stimulated lncRNAs in Dermal Fibroblasts
2.5. The Resource for Protein-Coding and lncRNA Genes in Fibrosis: FibroDB
3. Discussion
4. Materials and Methods
4.1. RNA-Seq Data Analysis
4.2. Data Analysis and Visualization
4.3. Cell Culture
4.4. Isolation of Total RNA and RT-PCR
4.5. Immunocytochemistry
4.6. RNA-Seq Experiment
4.7. FibroDB Web Application
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.H. Biology of fibroblasts and myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.K.; Stopak, D.; Wild, P. Fibroblast traction as a mechanism for collagen morphogenesis. Nature 1981, 290, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Heino, J. Connective tissue components in synovial fibroblast cultures exposed to interleukin 1 and prostaglandin E2. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1986, 50, 313–320. [Google Scholar] [CrossRef]
- Pera, F.; Sperling, K. Staining of the spindle apparatus in human lymphocyte and fibroblast cultures. Hum. Genet. 1976, 34, 195–197. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Trejo Bittar, H.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2019, 78, 1379–1387. [Google Scholar] [CrossRef]
- Guerrero-Juarez, C.F.; Dedhia, P.H.; Jin, S.; Ruiz-Vega, R.; Ma, D.; Liu, Y.; Yamaga, K.; Shestova, O.; Gay, D.L.; Yang, Z.; et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat. Commun. 2019, 10, 650. [Google Scholar] [CrossRef]
- Xie, T.; Wang, Y.; Deng, N.; Huang, G.; Taghavifar, F.; Geng, Y.; Liu, N.; Kulur, V.; Yao, C.; Chen, P.; et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep. 2018, 22, 3625–3640. [Google Scholar] [CrossRef]
- Muhl, L.; Genove, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 4493. [Google Scholar] [CrossRef]
- Mascharak, S.; Desjardins-Park, H.E.; Longaker, M.T. Fibroblast Heterogeneity in Wound Healing: Hurdles to Clinical Translation. Trends Mol. Med. 2020, 26, 1101–1106. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef]
- Sriram, G.; Bigliardi, P.L.; Bigliardi-Qi, M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur. J. Cell Biol. 2015, 94, 483–512. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, H.; Yue, D.; Blackwell, T.S.; Lv, C.; Song, X. Long non-coding RNAs: Promising new targets in pulmonary fibrosis. J. Gene Med. 2021, 23, e3318. [Google Scholar] [CrossRef]
- Ganguly, N.; Chakrabarti, S. Role of long noncoding RNAs and related epigenetic mechanisms in liver fibrosis (Review). Int. J. Mol. Med. 2021, 47, 1. [Google Scholar] [CrossRef]
- Omote, N.; Sauler, M. Non-coding RNAs as Regulators of Cellular Senescence in Idiopathic Pulmonary Fibrosis and Chronic Obstructive Pulmonary Disease. Front. Med. 2020, 7, 603047. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhang, Y.Y.; Lan, H.Y. LncRNAs in TGF-beta-Driven Tissue Fibrosis. Noncoding RNA 2018, 4, 26. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Long Noncoding RNAs: Molecular Modalities to Organismal Functions. Annu. Rev. Biochem. 2020, 89, 283–308. [Google Scholar] [CrossRef]
- DiStefano, J.K. The Emerging Role of Long Noncoding RNAs in Human Disease. Methods Mol. Biol. 2018, 1706, 91–110. [Google Scholar] [CrossRef]
- Pinkney, H.R.; Wright, B.M.; Diermeier, S.D. The lncRNA Toolkit: Databases and In Silico Tools for lncRNA Analysis. Noncoding RNA 2020, 6, 49. [Google Scholar] [CrossRef]
- Neilson, E.G. The Jeremiah Metzger lecture. The origin of fibroblasts and the terminality of epithelial differentiation. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 240–250. [Google Scholar]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef]
- Rinn, J.L.; Bondre, C.; Gladstone, H.B.; Brown, P.O.; Chang, H.Y. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006, 2, e119. [Google Scholar] [CrossRef]
- Higuchi, Y.; Kojima, M.; Ishii, G.; Aoyagi, K.; Sasaki, H.; Ochiai, A. Gastrointestinal Fibroblasts Have Specialized, Diverse Transcriptional Phenotypes: A Comprehensive Gene Expression Analysis of Human Fibroblasts. PLoS ONE 2015, 10, e0129241. [Google Scholar] [CrossRef]
- Foote, A.G.; Wang, Z.; Kendziorski, C.; Thibeault, S.L. Tissue specific human fibroblast differential expression based on RNAsequencing analysis. BMC Genom. 2019, 20, 308. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Zhao, S.; Ye, Z.; Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 2020, 26, 903–909. [Google Scholar] [CrossRef]
- Kim, D.; Chen, Z.; Zhou, L.F.; Huang, S.X. Air pollutants and early origins of respiratory diseases. Chronic. Dis. Transl. Med. 2018, 4, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.W. Atmospheric movement of microorganisms in clouds of desert dust and implications for human health. Clin. Microbiol. Rev. 2007, 20, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Quesnel, C.; Nardelli, L.; Piednoir, P.; Lecon, V.; Marchal-Somme, J.; Lasocki, S.; Bouadma, L.; Philip, I.; Soler, P.; Crestani, B.; et al. Alveolar fibroblasts in acute lung injury: Biological behaviour and clinical relevance. Eur. Respir. J. 2010, 35, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef]
- Kim, N.H.; Delcroix, M.; Jais, X.; Madani, M.M.; Matsubara, H.; Mayer, E.; Ogo, T.; Tapson, V.F.; Ghofrani, H.A.; Jenkins, D.P. Chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Bochenek, M.L.; Rosinus, N.S.; Lankeit, M.; Hobohm, L.; Bremmer, F.; Schutz, E.; Klok, F.A.; Horke, S.; Wiedenroth, C.B.; Munzel, T.; et al. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2017, 117, 769–783. [Google Scholar] [CrossRef]
- Sharma, S.; Hofbauer, T.M.; Ondracek, A.S.; Chausheva, S.; Alimohammadi, A.; Artner, T.; Panzenboeck, A.; Rinderer, J.; Shafran, I.; Mangold, A.; et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood 2021, 137, 1104–1116. [Google Scholar] [CrossRef]
- Clark, R.A.; McCoy, G.A.; Folkvord, J.M.; McPherson, J.M. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: A fibronectin matrix-dependent event. J. Cell Physiol. 1997, 170, 69–80. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-beta pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56. [Google Scholar] [CrossRef]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-beta: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6. [Google Scholar] [CrossRef]
- Savary, G.; Dewaeles, E.; Diazzi, S.; Buscot, M.; Nottet, N.; Fassy, J.; Courcot, E.; Henaoui, I.S.; Lemaire, J.; Martis, N.; et al. The Long Noncoding RNA DNM3OS Is a Reservoir of FibromiRs with Major Functions in Lung Fibroblast Response to TGF-beta and Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 184–198. [Google Scholar] [CrossRef]
- De Langhe, E.; Aznar-Lopez, C.; De Vooght, V.; Vanoirbeek, J.A.; Luyten, F.P.; Lories, R.J. Secreted frizzled related proteins inhibit fibrosis in vitro but appear redundant in vivo. Fibrogenes. Tissue Repair 2014, 7, 14. [Google Scholar] [CrossRef]
- Chothani, S.; Schafer, S.; Adami, E.; Viswanathan, S.; Widjaja, A.A.; Langley, S.R.; Tan, J.; Wang, M.; Quaife, N.M.; Jian Pua, C.; et al. Widespread Translational Control of Fibrosis in the Human Heart by RNA-Binding Proteins. Circulation 2019, 140, 937–951. [Google Scholar] [CrossRef]
- Huang, H.C.; Klein, P.S. The Frizzled family: Receptors for multiple signal transduction pathways. Genome Biol. 2004, 5, 234. [Google Scholar] [CrossRef]
- Spanjer, A.I.; Baarsma, H.A.; Oostenbrink, L.M.; Jansen, S.R.; Kuipers, C.C.; Lindner, M.; Postma, D.S.; Meurs, H.; Heijink, I.H.; Gosens, R.; et al. TGF-beta-induced profibrotic signaling is regulated in part by the WNT receptor Frizzled-8. FASEB J. 2016, 30, 1823–1835. [Google Scholar] [CrossRef]
- Guo, M.; Li, D.; Feng, Y.; Li, M.; Yang, B. Adipose-derived stem cell-derived extracellular vesicles inhibit neuroblastoma growth by regulating GABBR1 activity through LINC00622-mediated transcription factor AR. J. Leukoc. Biol. 2021, 111, 19–32. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhao, H. Identification of a nomogram based on long non-coding RNA to improve prognosis prediction of esophageal squamous cell carcinoma. Aging 2020, 12, 1512–1526. [Google Scholar] [CrossRef]
- Xu, M.L.; Liu, T.C.; Dong, F.X.; Meng, L.X.; Ling, A.X.; Liu, S. Exosomal lncRNA LINC01711 facilitates metastasis of esophageal squamous cell carcinoma via the miR-326/FSCN1 axis. Aging 2021, 13, 19776–19788. [Google Scholar] [CrossRef]
- Gee, H.Y.; Tang, B.L.; Kim, K.H.; Lee, M.G. Syntaxin 16 binds to cystic fibrosis transmembrane conductance regulator and regulates its membrane trafficking in epithelial cells. J. Biol. Chem. 2010, 285, 35519–35527. [Google Scholar] [CrossRef]
- Geng, X.Q.; Ma, A.; He, J.Z.; Wang, L.; Jia, Y.L.; Shao, G.Y.; Li, M.; Zhou, H.; Lin, S.Q.; Ran, J.H.; et al. Ganoderic acid hinders renal fibrosis via suppressing the TGF-beta/Smad and MAPK signaling pathways. Acta Pharmacol. Sin. 2020, 41, 670–677. [Google Scholar] [CrossRef]
- Lee, J.; An, J.N.; Hwang, J.H.; Lee, H.; Lee, J.P.; Kim, S.G. p38 MAPK activity is associated with the histological degree of interstitial fibrosis in IgA nephropathy patients. PLoS ONE 2019, 14, e0213981. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Madala, S.K.; Schmidt, S.; Davidson, C.; Ikegami, M.; Wert, S.; Hardie, W.D. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 380–388. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1 30 31–31 30 33. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Abbasi, S.; Sinha, S.; Labit, E.; Rosin, N.L.; Yoon, G.; Rahmani, W.; Jaffer, A.; Sharma, N.; Hagner, A.; Shah, P.; et al. Distinct Regulatory Programs Control the Latent Regenerative Potential of Dermal Fibroblasts during Wound Healing. Cell Stem Cell 2020, 27, 396–412.e396. [Google Scholar] [CrossRef] [PubMed]
- DeSisto, J.; O’Rourke, R.; Jones, H.E.; Pawlikowski, B.; Malek, A.D.; Bonney, S.; Guimiot, F.; Jones, K.L.; Siegenthaler, J.A. Single-Cell Transcriptomic Analyses of the Developing Meninges Reveal Meningeal Fibroblast Diversity and Function. Dev. Cell 2020, 54, 43–59.e44. [Google Scholar] [CrossRef] [PubMed]
- Waise, S.; Parker, R.; Rose-Zerilli, M.J.J.; Layfield, D.M.; Wood, O.; West, J.; Ottensmeier, C.H.; Thomas, G.J.; Hanley, C.J. An optimised tissue disaggregation and data processing pipeline for characterising fibroblast phenotypes using single-cell RNA sequencing. Sci. Rep. 2019, 9, 9580. [Google Scholar] [CrossRef] [PubMed]
- Molder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable data analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef]
- Weirick, T.; John, D.; Dimmeler, S.; Uchida, S. C-It-Loci: A knowledge database for tissue-enriched loci. Bioinformatics 2015, 31, 3537–3543. [Google Scholar] [CrossRef][Green Version]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, 2699. [Google Scholar] [CrossRef]
- Park, C.; Yu, N.; Choi, I.; Kim, W.; Lee, S. lncRNAtor: A comprehensive resource for functional investigation of long non-coding RNAs. Bioinformatics 2014, 30, 2480–2485. [Google Scholar] [CrossRef]
- Li, Z.; Liu, L.; Jiang, S.; Li, Q.; Feng, C.; Du, Q.; Zou, D.; Xiao, J.; Zhang, Z.; Ma, L. LncExpDB: An expression database of human long non-coding RNAs. Nucleic Acids Res. 2021, 49, D962–D968. [Google Scholar] [CrossRef]
- Jiang, S.; Cheng, S.J.; Ren, L.C.; Wang, Q.; Kang, Y.J.; Ding, Y.; Hou, M.; Yang, X.X.; Lin, Y.; Liang, N.; et al. An expanded landscape of human long noncoding RNA. Nucleic Acids Res. 2019, 47, 7842–7856. [Google Scholar] [CrossRef]
- Muller, R.; Weirick, T.; John, D.; Militello, G.; Chen, W.; Dimmeler, S.; Uchida, S. ANGIOGENES: Knowledge database for protein-coding and noncoding RNA genes in endothelial cells. Sci. Rep. 2016, 6, 32475. [Google Scholar] [CrossRef]
- Weirick, T.; Militello, G.; Ponomareva, Y.; John, D.; Doring, C.; Dimmeler, S.; Uchida, S. Logic programming to infer complex RNA expression patterns from RNA-seq data. Brief. Bioinform. 2018, 19, 199–209. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. In Use R! 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Bedre, R. Reneshbedre/Bioinfokit: Bioinformatics Data Analysis and Visualization Toolkit; Version v0.9; Zenodo, 2020. [Google Scholar] [CrossRef]
- Howe, E.A.; Sinha, R.; Schlauch, D.; Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Chang, W.; Cheng, J.; Allaire, J.; Xie, Y.; McPherson, J. Shiny: Web application framework for R. R Package Version 2017, 1, 2017. [Google Scholar]
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; Chapman and Hall: London, UK; CRC: Boca Raton, FL, USA, 2020. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, W. enrichR: Provides an R Interface to ‘Enrichr’; Version 3.0; 2021. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.A.J.; Grolemund, G. R Markdown: The Definitive Guide; Chapman and Hall: London, UK; CRC: Boca Raton, FL, USA, 2018. [Google Scholar]
- Fritah, S.; Niclou, S.P.; Azuaje, F. Databases for lncRNAs: A comparative evaluation of emerging tools. RNA 2014, 20, 1655–1665. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilieva, M.; Miller, H.E.; Agarwal, A.; Paulus, G.K.; Madsen, J.H.; Bishop, A.J.R.; Kauppinen, S.; Uchida, S. FibroDB: Expression Analysis of Protein-Coding and Long Non-Coding RNA Genes in Fibrosis. Non-Coding RNA 2022, 8, 13. https://doi.org/10.3390/ncrna8010013
Ilieva M, Miller HE, Agarwal A, Paulus GK, Madsen JH, Bishop AJR, Kauppinen S, Uchida S. FibroDB: Expression Analysis of Protein-Coding and Long Non-Coding RNA Genes in Fibrosis. Non-Coding RNA. 2022; 8(1):13. https://doi.org/10.3390/ncrna8010013
Chicago/Turabian StyleIlieva, Mirolyuba, Henry E. Miller, Arav Agarwal, Gabriela K. Paulus, Jens Hedelund Madsen, Alexander J. R. Bishop, Sakari Kauppinen, and Shizuka Uchida. 2022. "FibroDB: Expression Analysis of Protein-Coding and Long Non-Coding RNA Genes in Fibrosis" Non-Coding RNA 8, no. 1: 13. https://doi.org/10.3390/ncrna8010013
APA StyleIlieva, M., Miller, H. E., Agarwal, A., Paulus, G. K., Madsen, J. H., Bishop, A. J. R., Kauppinen, S., & Uchida, S. (2022). FibroDB: Expression Analysis of Protein-Coding and Long Non-Coding RNA Genes in Fibrosis. Non-Coding RNA, 8(1), 13. https://doi.org/10.3390/ncrna8010013