
CRISPR-Based Approaches for the High-Throughput Characterization of Long Non-Coding RNAs


Abstract
:1. Introduction
1.1. Proposed and Proven Functions of lncRNAs
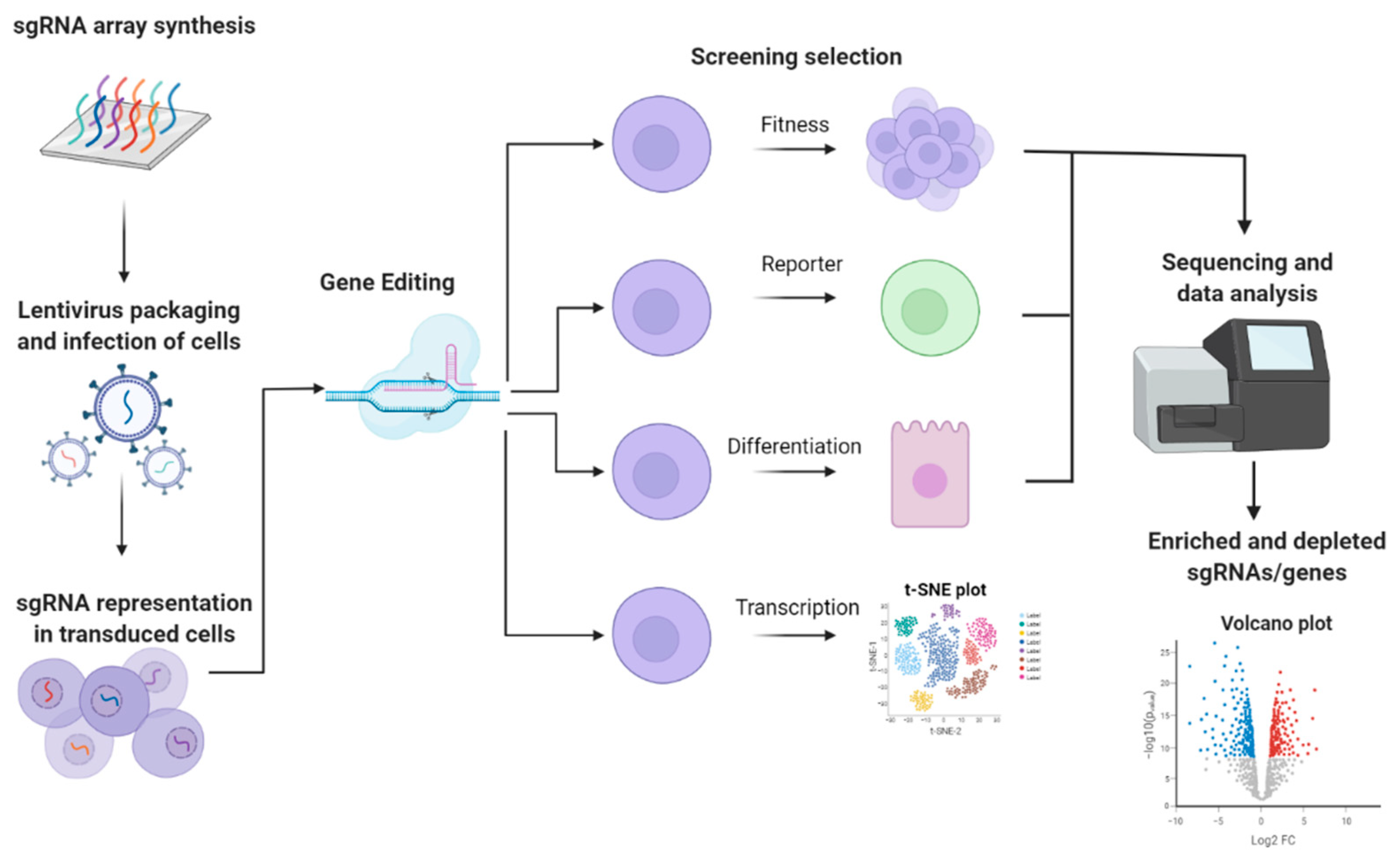
1.2. High-Throughput Reverse Genetic Screening
2. CRISPR-Based Approaches to Study lncRNAs
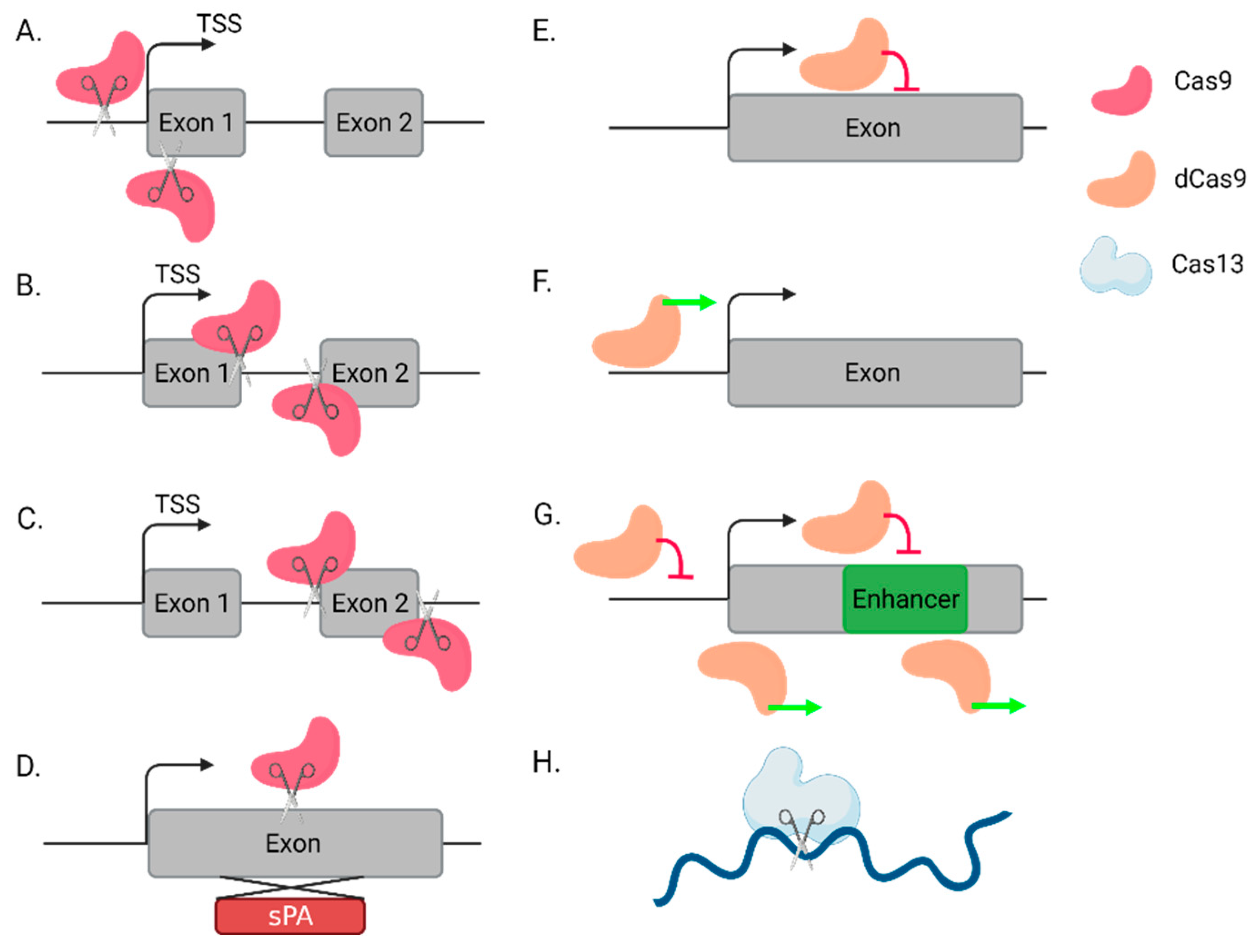
2.1. Using CRISPR-Ko to Directly or Indirectly Knock Out lncRNAs
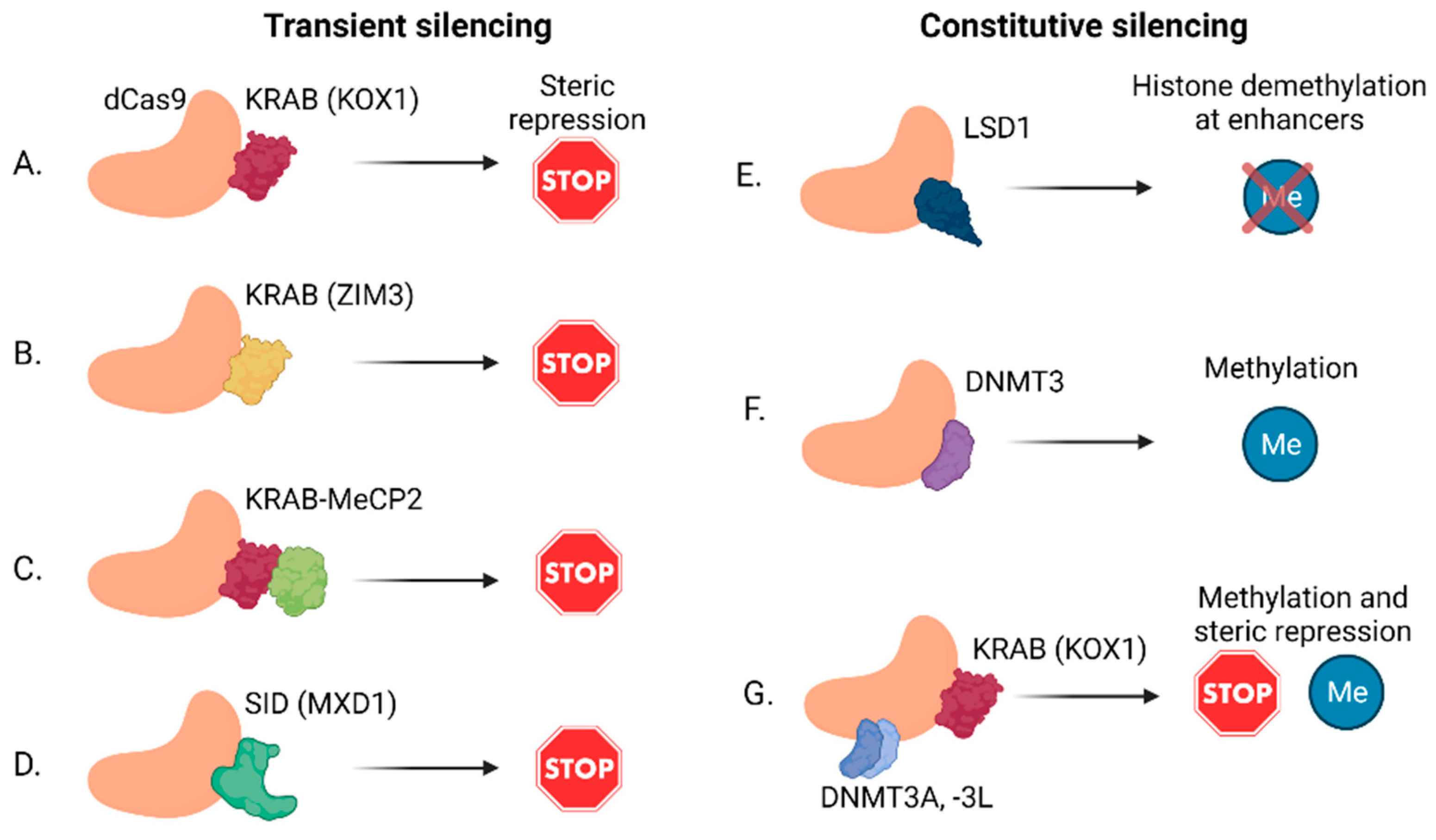
2.2. lncRNA Knockdown by CRISPRi
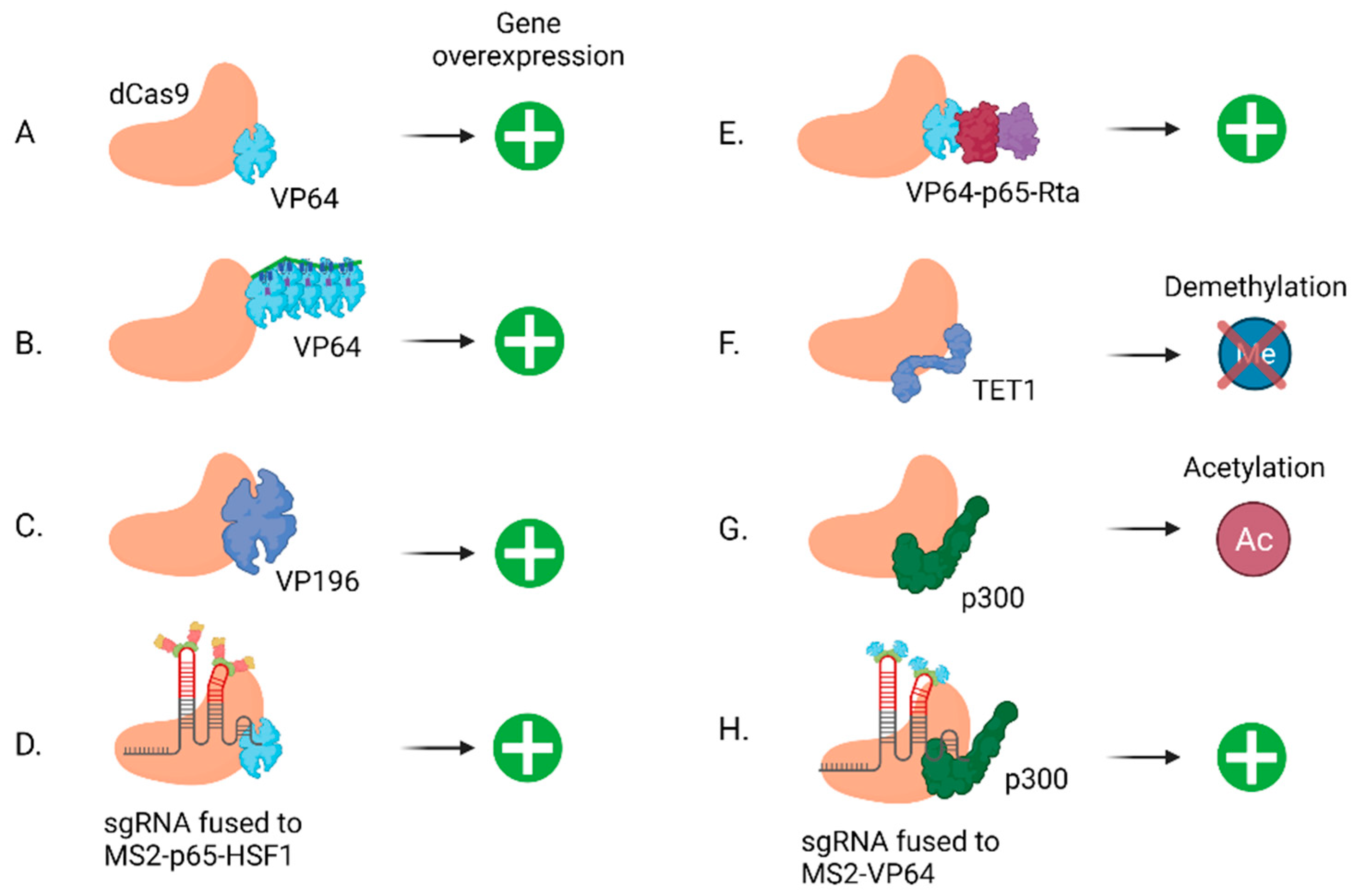
2.3. lncRNA Overexpression by CRISPRa
2.4. Novel Alternative Tools: Cas12 and Cas14
2.5. RNA Targeting by Cas13
3. Approaches Other Than CRISPR
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef]
- Gao, F.; Cai, Y.; Kapranov, P.; Xu, D. Reverse-genetics studies of lncRNAs-what we have learnt and paths forward. Genome Biol. 2020, 21, 93. [Google Scholar] [CrossRef] [Green Version]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of Long Noncoding RNA Evolution Derived from Direct Comparison of Transcriptomes in 17 Species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Szcześniak, M.W.; Wanowska, E.; Mukherjee, N.; Ohler, U.; Makałowska, I. Towards a deeper annotation of human lncRNAs. Biochim. Biophys. Acta-Gene Regul. Mech. 2020, 1863, 194385. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Bowness, J.S.; Wei, G. Progress toward understanding chromosome silencing by Xist RNA. Genes Dev. 2020, 34, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kopp, F.; Chang, T.C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Tichon, A.; Gil, N.; Lubelsky, Y.; Solomon, T.H.; Lemze, D.; Itzkovitz, S.; Stern-Ginossar, N.; Ulitsky, I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 2016, 7, 12209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tichon, A.; Perry, R.B.T.; Stojic, L.; Ulitsky, I. SAM68 is required for regulation of pumilio by the NORAD long noncoding RNA. Genes Dev. 2018, 32, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhu, C.; Jin, Y. The Oncogenic and Tumor Suppressive Functions of the Long Noncoding RNA MALAT1: An Emerging Controversy. Front. Genet. 2020, 11, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onagoruwa, O.T.; Pal, G.; Ochu, C.; Ogunwobi, O.O. Oncogenic Role of PVT1 and Therapeutic Implications. Front. Oncol. 2020, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.B.; Sanjana, N.E. CRISPR Screens to Discover Functional Noncoding Elements. Trends Genet. 2016, 32, 526–529. [Google Scholar] [CrossRef] [Green Version]
- Esposito, R.; Bosch, N.; Lanzós, A.; Polidori, T.; Pulido-Quetglas, C.; Johnson, R. Hacking the Cancer Genome: Profiling Therapeutically Actionable Long Non-coding RNAs Using CRISPR-Cas9 Screening. Cancer Cell 2019, 35, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Szczelkun, M.D.; Tikhomirova, M.S.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, A.; Huangfu, D. Decoding the noncoding genome via large-scale CRISPR screens. Curr. Opin. Genet. Dev. 2018, 52, 70–76. [Google Scholar] [CrossRef]
- Engreitz, J.; Abudayyeh, O.; Gootenberg, J.; Zhang, F. CRISPR tools for systematic studies of RNA regulation. Cold Spring Harb. Perspect. Biol. 2019, 11, a035386. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Myacheva, K.; Groß, M.; Klingenberg, M.; Duran Arqué, B.; Diederichs, S. Challenges of CRISPR/Cas9 applications for long non-coding RNA genes. Nucleic Acids Res. 2017, 45, e12. [Google Scholar] [CrossRef] [Green Version]
- Phelan, J.D.; Staudt, L.M. CRISPR-based technology to silence the expression of IncRNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 8225–8227. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Rahman, J.A.; Wessels, H.-H.; Méndez-Mancilla, A.; Haro, D.; Chen, X.; Sanjana, N.E. Transcriptome-wide Cas13 guide RNA design for model organisms and viral RNA pathogens. Cell Genom. 2021, 1, 100001. [Google Scholar] [CrossRef]
- Nötzold, L.; Frank, L.; Gandhi, M.; Polycarpou-Schwarz, M.; Groß, M.; Gunkel, M.; Beil, N.; Erfle, H.; Harder, N.; Rohr, K.; et al. The long non-coding RNA LINC00152 is essential for cell cycle progression through mitosis in HeLa cells. Sci. Rep. 2017, 7, 2265. [Google Scholar] [CrossRef] [Green Version]
- Ramilowski, J.A.; Yip, C.W.; Agrawal, S.; Chang, J.C.; Ciani, Y.; Kulakovskiy, I.V.; Mendez, M.; Ooi, J.L.C.; Ouyang, J.F.; Parkinson, N.; et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res. 2020, 30, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016, 44, 863–877. [Google Scholar] [CrossRef] [Green Version]
- Stojic, L.; Lun, A.T.L.; Mangei, J.; Mascalchi, P.; Quarantotti, V.; Barr, A.R.; Bakal, C.; Marioni, J.C.; Gergely, F.; Odom, D.T. Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res. 2018, 46, 5950–5966. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, G.; Lopes, R.; Ugalde, A.P.; Nevedomskaya, E.; Han, R.; Myacheva, K.; Zwart, W.; Elkon, R.; Agami, R. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Mu, Q.; Zhang, Y.; Xie, Z. Construction of a CRISPR-based paired-sgRNA library for chromosomal deletion of long non-coding RNAs. Quant. Biol. 2020, 8, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, Z.; Wang, Y.; Guo, Y.; Xu, P.; Yuan, P.; Liu, Z.; He, Y.; Wei, W. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 2018, 36, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, M.; Findlay, G.M.; McKenna, A.; Milbank, J.H.; Lee, C.; Zhang, M.D.; Cusanovich, D.A.; Shendure, J. CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet. 2017, 101, 192–205. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Harmston, N.; Glaser, T.L.; Wong, Y.; Zhong, Z.; Madan, B.; Virshup, D.M.; Petretto, E. Wnt-regulated lncRNA discovery enhanced by in vivo identification and CRISPRi functional validation. Genome Med. 2020, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S.; et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019, 176, 377–390.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355, eaah7111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020, 21. [Google Scholar] [CrossRef]
- Haswell, J.R.; Mattioli, K.; Gerhardinger, C.; Maass, P.G.; Foster, D.J.; Fernandez, P.P.; Wang, X.; Rinn, J.L.; Slack, F.J. Genome-Wide CRISPR Interference Screen Identifies Long Non-Coding RNA Loci Required for Differentiation and Pluripotency. PLoS ONE 2021, 16, e0252848. [Google Scholar] [CrossRef]
- Fulco, C.P.; Munschauer, M.; Anyoha, R.; Munson, G.; Grossman, S.R.; Perez, E.M.; Kane, M.; Cleary, B.; Lander, E.S.; Engreitz, J.M. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 2016, 354, 769–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef]
- Xie, S.; Duan, J.; Li, B.; Zhou, P.; Hon, G.C. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 2017, 66, 285–299.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef] [PubMed]
- Shariati, S.A.; Dominguez, A.; Xie, S.; Wernig, M.; Qi, L.S.; Skotheim, J.M. Reversible Disruption of Specific Transcription Factor-DNA Interactions Using CRISPR/Cas9. Mol. Cell 2019, 74, 622–633.e4. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An Integrated Genome-wide CRISPRa Approach to Functionalize lncRNAs in Drug Resistance. Cell 2018, 173, 649–664.e20. [Google Scholar] [CrossRef]
- Koirala, P.; Huang, J.; Ho, T.T.; Wu, F.; Ding, X.; Mo, Y.Y. LncRNA AK023948 is a positive regulator of AKT. Nat. Commun. 2017, 8, 14422. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, D.R.; Gowen, B.G.; Boontanrart, M.; Roth, T.L.; Gagnon, J.D.; Mumbach, M.R.; Satpathy, A.T.; Lee, Y.; Bray, N.L.; Chan, A.Y.; et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 2017, 549, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Wessels, H.H.; Méndez-Mancilla, A.; Guo, X.; Legut, M.; Daniloski, Z.; Sanjana, N.E. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 2020, 38, 722–727. [Google Scholar] [CrossRef]
- Zhang, Y.; Nguyen, T.M.; Zhang, X.O.; Wang, L.; Phan, T.; Clohessy, J.G.; Pandolfi, P.P. Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol. 2021, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1794. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, X.; Xue, W.; Zhang, L.; Yang, L.Z.; Cao, S.M.; Lei, Y.N.; Liu, C.X.; Guo, S.K.; Shan, L.; et al. Screening for functional circular RNAs using the CRISPR–Cas13 system. Nat. Methods 2021, 18, 51–59. [Google Scholar] [CrossRef]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Clemson, C.M.; McNeil, J.A.; Willard, H.F.; Lawrence, J.B. XIST RNA paints the inactive X chromosome at interphase: Evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol. 1996, 132, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T.; Jaenisch, R. Long-range cis effects of ectopic X-inactivation centres on a mouse autosome. Nature 1997, 386, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Clerc, P.; Avner, P. X-chromosome inactivation in mammals. Annu. Rev. Genet. 1997, 31, 571–610. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-b-induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Nakajima, K.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 2015, 75, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Luan, W.; Li, L.; Shi, Y.; Bu, X.; Xia, Y.; Wang, J.; Djangmah, H.S.; Liu, X.; You, Y.; Xu, B. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget 2016, 7, 63901–63912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, Z.H.; Roche, V.; Chew, X.H.; Fadieieva, A.; Tay, Y. A non-canonical tumor suppressive role for the long non-coding RNA MALAT1 in colon and breast cancers. Int. J. Cancer 2018, 143, 668–678. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef]
- Guan, Y.; Kuo, W.L.; Stilwell, J.L.; Takano, H.; Lapuk, A.V.; Fridlyand, J.; Mao, J.H.; Yu, M.; Miller, M.A.; Santos, J.L.; et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 2007, 13, 5745–5755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef]
- Li, T.; Meng, X.; Yang, W. Long Noncoding RNA PVT1 Acts as a “Sponge” to Inhibit microRNA-152 in Gastric Cancer Cells. Dig. Dis. Sci. 2017, 62, 3021–3028. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Li, S.; Sun, D.; Yang, S. LncRNA PVT1 accelerates progression of non-small cell lung cancer via targeting miRNA-526b/EZH2 regulatory loop. Oncol. Lett. 2020, 19, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Xu, J.; Sun, R.; Mumbach, M.R.; Carter, A.C.; Chen, Y.G.; Yost, K.E.; Kim, J.; He, J.; Nevins, S.A.; et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 2018, 173, 1398–1412.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverri, C.J.; Perrimon, N. High-throughput RNAi screening in cultured cells: A user’s guide. Nat. Rev. Genet. 2006, 7, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S. The art and design of genetic screens: Mammalian culture cells. Nat. Rev. Genet. 2004, 5, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Roux, B.T.; Lindsay, M.A.; Heward, J.A. Knockdown of nuclear-located enhancer RNAs and long ncRNAs using locked nucleic acid gapmers. Methods Mol. Biol. 2017, 1468, 11–18. [Google Scholar] [CrossRef]
- Raffeiner, P.; Hart, J.R.; García-Caballero, D.; Bar-Peled, L.; Weinberg, M.S.; Vogt, P.K. An MXD1-derived repressor peptide identifies noncoding mediators of MYC-driven cell proliferation. Proc. Natl. Acad. Sci. USA 2020, 117, 6571–6579. [Google Scholar] [CrossRef] [Green Version]
- Horlbeck, M.A.; Liu, S.J.; Chang, H.Y.; Lim, D.A.; Weissman, J.S. Fitness effects of CRISPR/Cas9-targeting of long noncoding RNA genes. Nat. Biotechnol. 2020, 38, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Cao, Z.; Wei, W. Reply to: Fitness effects of CRISPR/Cas9-targeting of long noncoding RNA genes. Nat. Biotechnol. 2020, 38, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Souquere, S.; Chujo, T.; Kobelke, S.; Chong, Y.S.; Fox, A.H.; Bond, C.S.; Nakagawa, S.; Pierron, G.; Hirose, T. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 2018, 70, 1038–1053.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaertner, B.; van Heesch, S.; Schneider-Lunitz, V.; Schulz, J.F.; Witte, F.; Blachut, S.; Nguyen, S.; Wong, R.; Matta, I.; Hübner, N.; et al. A human esc-based screen identifies a role for the translated lncrna linc00261 in pancreatic endocrine differentiation. ELife 2020, 9, e58659. [Google Scholar] [CrossRef]
- Aparicio-Prat, E.; Arnan, C.; Sala, I.; Bosch, N.; Guigó, R.; Johnson, R. DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genom. 2015, 16, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Pulido, T.H.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. Rna 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-Ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.M.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef] [Green Version]
- Dang, Y.; Jia, G.; Choi, J.; Ma, H.; Anaya, E.; Ye, C.; Shankar, P.; Wu, H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015, 16, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radzisheuskaya, A.; Shlyueva, D.; Müller, I.; Helin, K. Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 2016, 44, e141. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Xiao, T.; Chen, C.H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Horlbeck, M.A.; Witkowsky, L.B.; Guglielmi, B.; Replogle, J.M.; Gilbert, L.A.; Villalta, J.E.; Torigoe, S.E.; Tjian, R.; Weissman, J.S. Nucleosomes impede cas9 access to DNA in vivo and in vitro. ELife 2016, 5, e12677. [Google Scholar] [CrossRef]
- Liu, G.; Yin, K.; Zhang, Q.; Gao, C.; Qiu, J.L. Modulating chromatin accessibility by transactivation and targeting proximal dsgRNAs enhances Cas9 editing efficiency in vivo. Genome Biol. 2019, 20, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Uusi-Mäkelä, M.I.E.; Barker, H.R.; Bäuerlein, C.A.; Häkkinen, T.; Nykter, M.; Rämet, M. Chromatin accessibility is associated with CRISPR-Cas9 efficiency in the zebrafish (Danio rerio). PLoS ONE 2018, 13, e0196238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alerasool, N.; Segal, D.; Lee, H.; Taipale, M. An efficient KRAB domain for CRISPRi applications in human cells. Nat. Methods 2020, 17, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [Green Version]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [Green Version]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Replogle, J.M.; Norman, T.M.; Xu, A.; Hussmann, J.A.; Chen, J.; Cogan, J.Z.; Meer, E.J.; Terry, J.M.; Riordan, D.P.; Srinivas, N.; et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 2020, 38, 954–961. [Google Scholar] [CrossRef]
- Song, Q.; Song, Q.; Ni, K.; Ni, K.; Liu, M.; Liu, M.; Li, Y.; Li, Y.; Wang, L.; Wang, L.; et al. Direct-seq: Programmed gRNA scaffold for streamlined scRNA-seq in CRISPR screen. Genome Biol. 2020, 21, 136. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016, 167, 1883–1896.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866.e17. [Google Scholar] [CrossRef] [Green Version]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, D.; Weltner, J.; Eurola, S.; Trokovic, R.; Wartiovaara, K.; Otonkoski, T. Conditionally Stabilized dCas9 Activator for Controlling Gene Expression in Human Cell Reprogramming and Differentiation. Stem Cell Rep. 2015, 5, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Liu, J.; Zhou, C.; Gao, N.; Rao, Z.; Li, H.; Hu, X.; Li, C.; Yao, X.; Shen, X.; et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 2018, 21, 440–446. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Liu, Y.; Cao, H.; Zhang, Y.; Gu, Z.; Liu, X.; Yu, A.; Kaphle, P.; Dickerson, K.E.; Ni, M.; et al. Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat. Commun. 2020, 11, 485. [Google Scholar] [CrossRef]
- Braun, C.J.; Bruno, P.M.; Horlbeck, M.A.; Gilbert, L.A.; Weissman, J.S.; Hemann, M.T. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc. Natl. Acad. Sci. USA 2016, 113, E3892–E3900. [Google Scholar] [CrossRef] [Green Version]
- Colic, M.; Wang, G.; Zimmermann, M.; Mascall, K.; McLaughlin, M.; Bertolet, L.; Lenoir, W.F.; Moffat, J.; Angers, S.; Durocher, D.; et al. Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med. 2019, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 2017, 68, 210–223.e6. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Hunnewell, P.; Alfonse, L.E.; Carte, J.M.; Keston-Smith, E.; Sothiselvam, S.; Garrity, A.J.; Chong, S.; Makarova, K.S.; Koonin, E.V.; et al. Functionally diverse type V CRISPR-Cas systems. Science 2019, 363, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [Green Version]
- Bandaru, S.; Tsuji, M.H.; Shimizu, Y.; Usami, K.; Lee, S.; Takei, N.K.; Yoshitome, K.; Nishimura, Y.; Otsuki, T.; Ito, T. Structure-based design of gRNA for Cas13. Sci. Rep. 2020, 10, 11610. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Shi, X.; Tjian, R.; Lionnet, T.; Singer, R.H. CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11870–11875. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-mediated live imaging of genome editing and transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Wu, X.; Mao, S.; Ying, Y.; Krueger, C.J.; Chen, A.K. Progress and Challenges for Live-cell Imaging of Genomic Loci Using CRISPR-based Platforms. Genom. Proteom. Bioinforma 2019, 17, 119–128. [Google Scholar] [CrossRef]
- Yang, L.Z.; Wang, Y.; Li, S.Q.; Yao, R.W.; Luan, P.F.; Wu, H.; Carmichael, G.G.; Chen, L.L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997.e7. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhao, B.S.; Myers, S.A.; Carr, S.A.; He, C.; Ting, A.Y. RNA-protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc. Natl. Acad. Sci. USA 2020, 117, 22068–22079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, W.; Shi, T.; Lu, P.; Zhuang, M.; Liu, J.L. Capturing RNA-protein interaction via CRUIS. Nucleic Acids Res. 2020, 48, e52. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Ketting, R.F. The Many Faces of RNAi. Dev. Cell 2011, 20, 148–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, M.; Ahringer, J. The art and design of genetic screens: RNA interference. Nat. Rev. Genet. 2008, 9, 554–566. [Google Scholar] [CrossRef]
- Stojic, L.; Lun, A.T.L.; Mascalchi, P.; Ernst, C.; Redmond, A.M.; Mangei, J.; Barr, A.R.; Bousgouni, V.; Bakal, C.; Marioni, J.C.; et al. A high-content RNAi screen reveals multiple roles for long noncoding RNAs in cell division. Nat. Commun. 2020, 11, 1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Singh, S.K.; Nielsen, P.; Koshkin, A.A.; Wengel, J. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 4, 455–456. [Google Scholar] [CrossRef]
- Sarma, K.; Levasseur, P.; Aristarkhov, A.; Lee, J.T. Locked nucleic acids (LNAs) reveal sequence requirements and kinetics of Xist RNA localization to the X chromosome. Proc. Natl. Acad. Sci USA 2010, 107, 22196–22201. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; Mac Leod, A.R.; Wheeler, T.M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamola, P.J.; Kitson, J.D.A.; Turner, G.; Maratou, K.; Eriksson, S.; Panjwani, A.; Warnock, L.C.; Douillard Guilloux, G.A.; Moores, K.; Koppe, E.L.; et al. In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. 2015, 43, 8638–8650. [Google Scholar] [CrossRef] [PubMed] [Green Version]

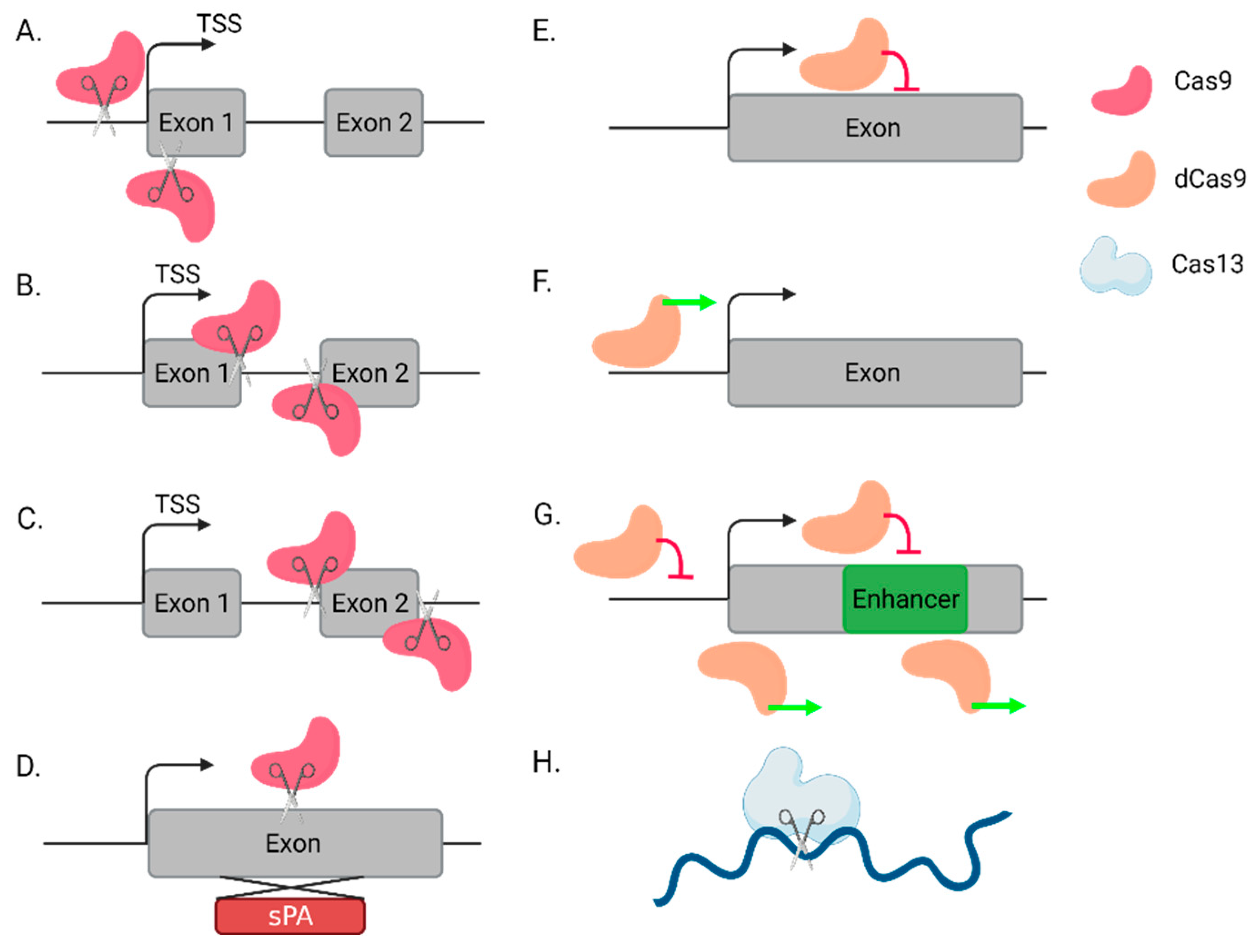{kind=link}
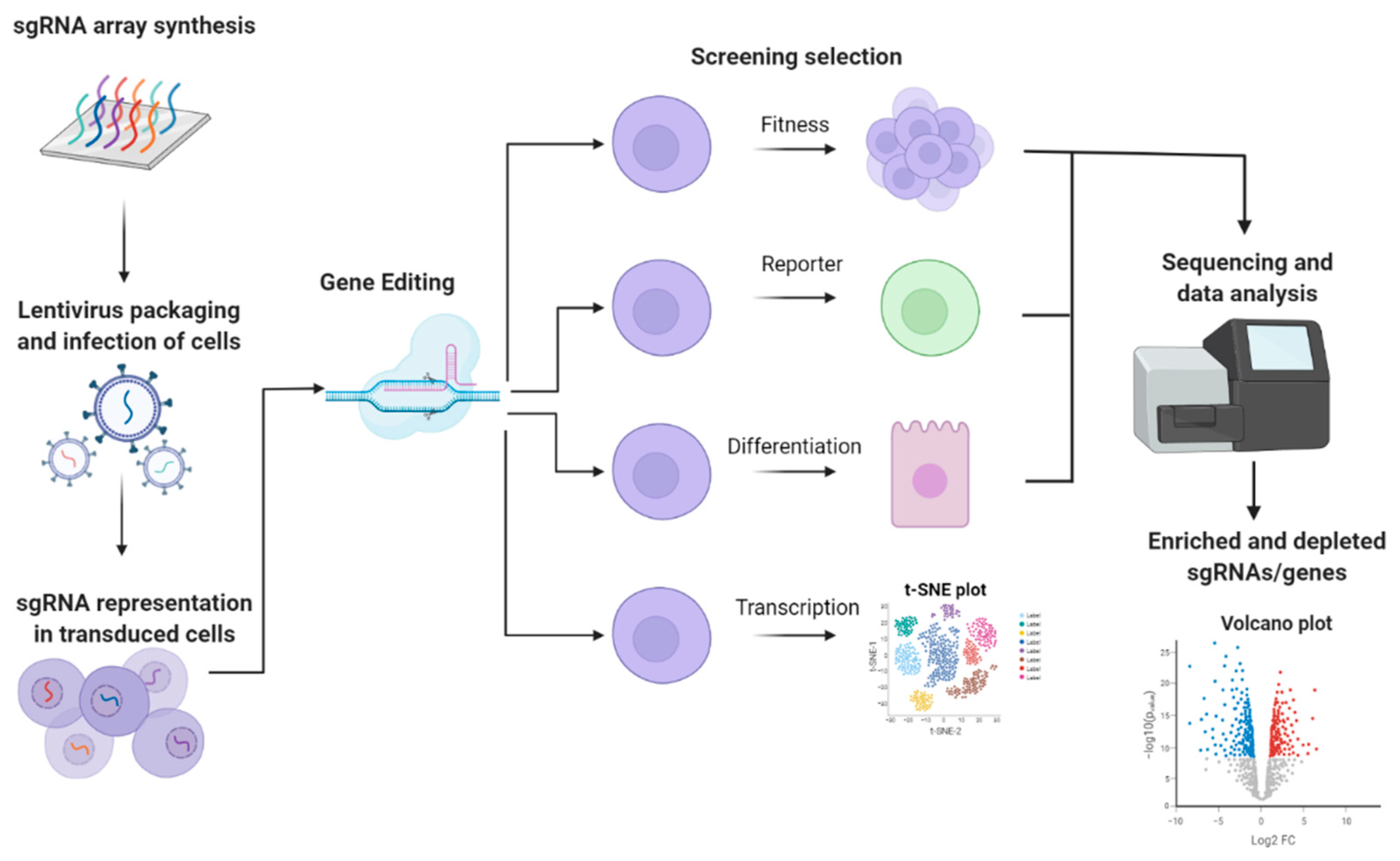{kind=link}
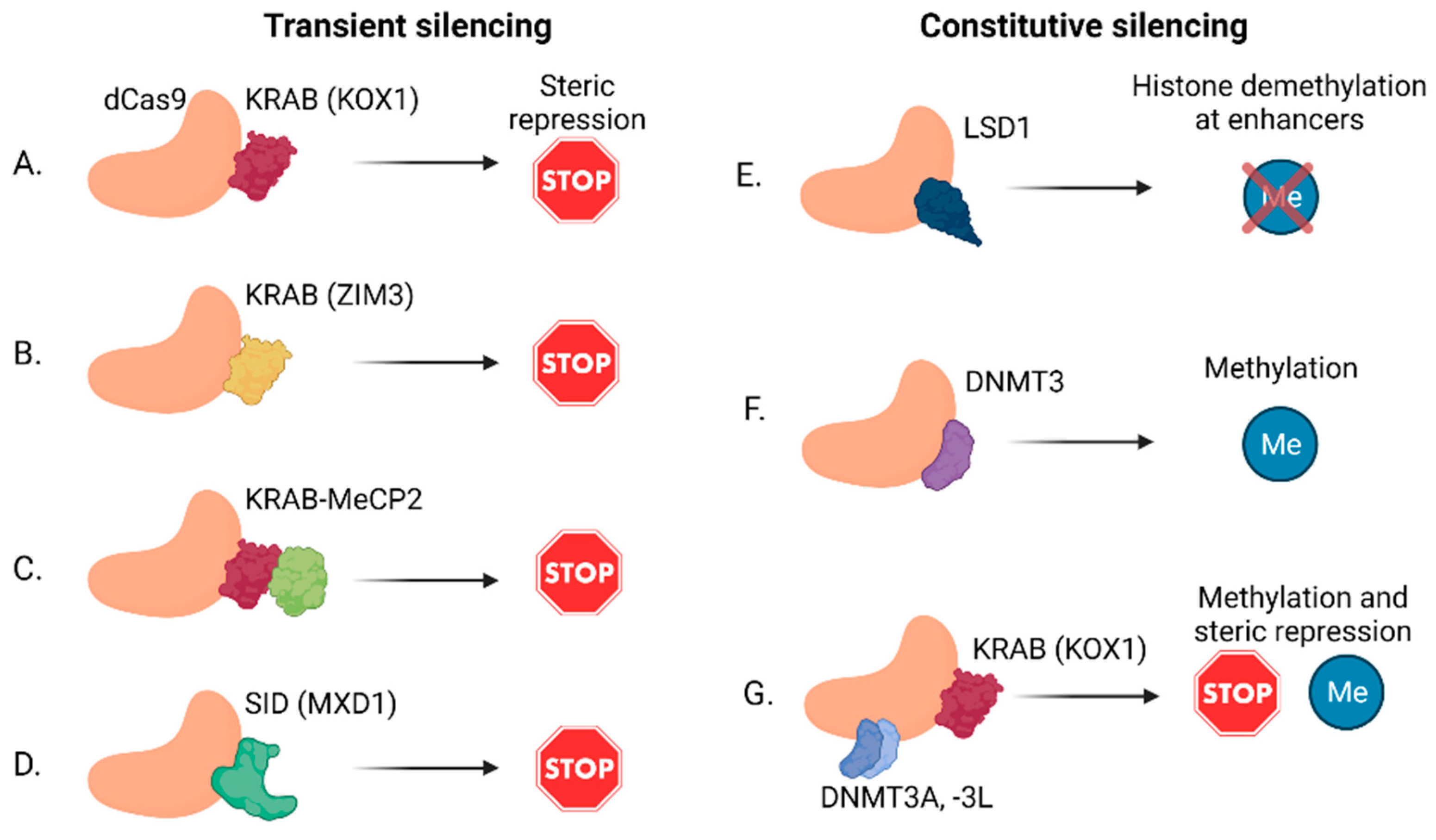{kind=link}
{kind=link}
| Perturbation Method | Effect | Element(s) Targeted | Reference(s) |
|---|---|---|---|
| CRISPR-ko | Mutagenesis | Enhancers, protein-coding genes (PCG) | [37,38,39] |
| CRISPR-ko | Transcription start site/whole sequence deletion | lncRNAs | [40,41] |
| CRISPR-ko | Splice site mutation | lncRNAs | [42] |
| CRISPR-ko | Tiling deletion | lncRNAs | [43] |
| CRISPR-ki | Synthetic polyadenylation signal (spA) insertion | lncRNAs | [44] |
| CRISPRi | Transcription inhibition | lncRNAs, enhancers, PCG | [45,46,47,48,49,50,51,52] |
| CRISPRi | Epigenetic silencing | lncRNAs, enhancers, PCG | [53] |
| CRISPRi | Binding site interactions | Enhancers | [50,51,52,54] |
| CRISPRa | Transcription activation | lncRNAs, enhancers, PCG | [51,55,56,57,58] |
| CRISPRa | Epigenetic activation | lncRNAs, enhancers, PCG | [53] |
| Cas13 | RNA targeting | lncRNAs, circRNA, PCG | [59,60,61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hazan, J.; Bester, A.C. CRISPR-Based Approaches for the High-Throughput Characterization of Long Non-Coding RNAs. Non-Coding RNA 2021, 7, 79. https://doi.org/10.3390/ncrna7040079
Hazan J, Bester AC. CRISPR-Based Approaches for the High-Throughput Characterization of Long Non-Coding RNAs. Non-Coding RNA. 2021; 7(4):79. https://doi.org/10.3390/ncrna7040079
Chicago/Turabian StyleHazan, Joshua, and Assaf Chanan Bester. 2021. "CRISPR-Based Approaches for the High-Throughput Characterization of Long Non-Coding RNAs" Non-Coding RNA 7, no. 4: 79. https://doi.org/10.3390/ncrna7040079
APA StyleHazan, J., & Bester, A. C. (2021). CRISPR-Based Approaches for the High-Throughput Characterization of Long Non-Coding RNAs. Non-Coding RNA, 7(4), 79. https://doi.org/10.3390/ncrna7040079