
Between the Devil and the Deep Blue Sea: Non-Coding RNAs Associated with Transmissible Cancers in Tasmanian Devil, Domestic Dog and Bivalves


{kind=link}
Abstract
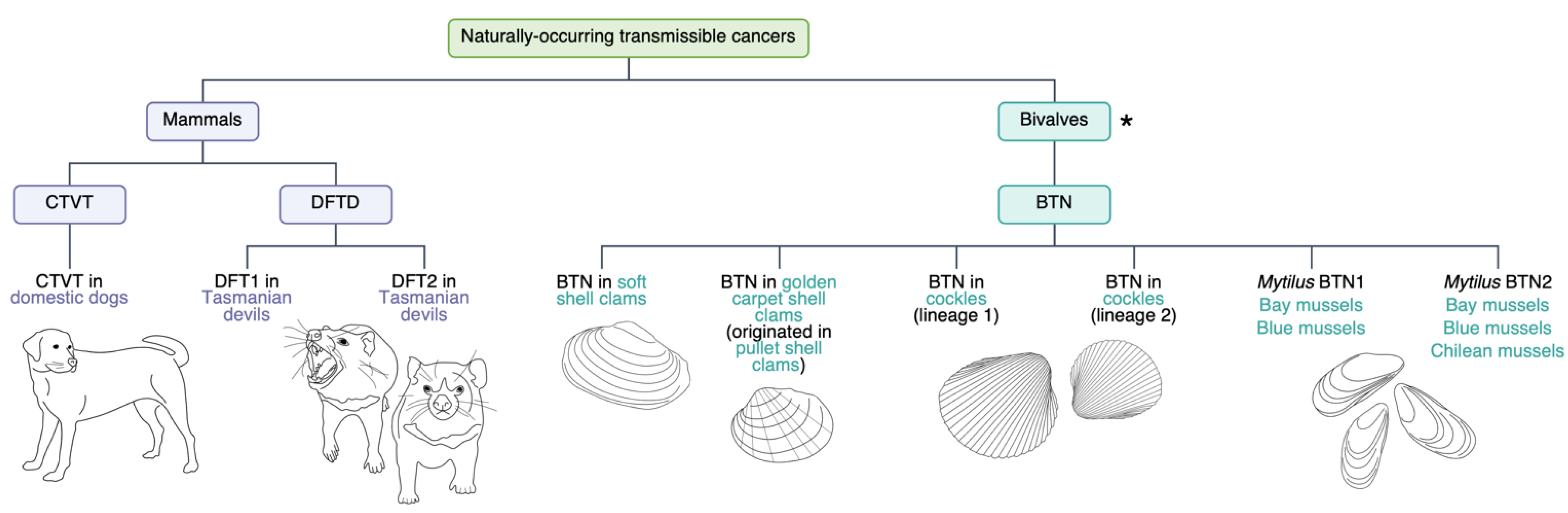
1. Transmissible Cancers
1.1. Canine Transmissible Venereal Tumour (CTVT)
1.2. Devil Facial Tumour Disease (DFTD)
1.3. Bivalve Transmissible Neoplasia (BTN)
2. Non-Coding RNAs
3. CRISPR/Cas
4. The Non-Coding RNA Biology of Transmissible Cancers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Metzger, M.J.; Goff, S.P. A Sixth Modality of Infectious Disease: Contagious Cancer from Devils to Clams and Beyond. PLoS Pathog. 2016, 12, e1005904. [Google Scholar] [CrossRef]
- Dujon, A.; Gatenby, R.A.; Bramwell, G.; MacDonald, N.; Dohrmann, E.; Raven, N.; Schultz, A.; Hamede, R.; Gérard, A.-L.; Giraudeau, M.; et al. Transmissible Cancers in an Evolutionary Perspective. iScience 2020, 23, 101269. [Google Scholar] [CrossRef] [PubMed]
- Ujvari, B.; Gatenby, R.A.; Thomas, F. The evolutionary ecology of transmissible cancers. Infect. Genet. Evol. 2016, 39, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Tolar, J.; Neglia, J. Transplacental and Other Routes of Cancer Transmission Between Individuals. J. Pediatr. Hematol. 2003, 25, 430–434. [Google Scholar] [CrossRef]
- Muehlenbachs, A.; Bhatnagar, J.; Agudelo, C.A.; Hidron, A.; Eberhard, M.L.; Mathison, B.A.; Frace, M.A.; Ito, A.; Metcalfe, M.G.; Rollin, D.C.; et al. Malignant Transformation of Hymenolepis nana in a Human Host. N. Engl. J. Med. 2015, 373, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, H.-V.; Seidl, C.; Luckenbach, C.; Schumm, G.; Seifried, E.; Ritter, H.; Bültmann, B. Genetic Analysis of a Sarcoma Accidentally Transplanted from a Patient to a Surgeon. N. Engl. J. Med. 1996, 335, 1494–1497. [Google Scholar] [CrossRef]
- Scanlon, E.F.; Hawkins, R.A.; Fox, W.W.; Smith, W.S. Fatal homotransplanted melanoma: A case report. Cancer 1965, 18, 782–789. [Google Scholar] [CrossRef]
- Chapman, J.R.; Webster, A.C.; Wong, G. Cancer in the transplant recipient. Cold Spring Harb. Perspect. Med. 2013, 3, a015677. [Google Scholar] [CrossRef]
- Gandhi, M.J.; Strong, D.M. Donor derived malignancy following transplantation: A review. Cell Tissue Bank. 2007, 8, 267–286. [Google Scholar] [CrossRef]
- Engels, E.A.; Pfeiffer, R.M.; Fraumeni, J.F.; Kasiske, B.L.; Israni, A.K.; Snyder, J.J.; Wolfe, R.A.; Goodrich, N.P.; Bayakly, A.R.; Clarke, C.A.; et al. Spectrum of Cancer Risk Among US Solid Organ Transplant Recipients. JAMA 2011, 306, 1891–1901. [Google Scholar] [CrossRef]
- Dujon, A.M.; Bramwell, G.; Roche, B.; Thomas, F.; Ujvari, B. Transmissible cancers in mammals and bivalves: How many examples are there? Predictions indicate widespread occurrence. Bioessays 2021, 43, e2000222. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Das, A.K. Review of Canine Transmissible Venereal Sarcoma. Vet. Res. Commun. 2000, 24, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Wedge, D.C.; Alexandrov, L.B.; Fu, B.; Martincorena, I.; Ning, Z.; Tubio, J.M.C.; Werner, E.I.; Allen, J.; De Nardi, A.B.; et al. Transmissible Dog Cancer Genome Reveals the Origin and History of an Ancient Cell Lineage. Science 2014, 343, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Strakova, A.; Murchison, E.P. The changing global distribution and prevalence of canine transmissible venereal tumour. BMC Vet. Res. 2014, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Baez-Ortega, A.; Gori, K.; Strakova, A.; Allen, J.L.; Allum, K.M.; Bansse-Issa, L.; Bhutia, T.N.; Bisson, J.L.; Briceño, C.; Domracheva, A.C.; et al. Somatic evolution and global expansion of an ancient transmissible cancer lineage. Science 2019, 365. [Google Scholar] [CrossRef]
- Murgia, C.; Pritchard, J.K.; Kim, S.Y.; Fassati, A.; Weiss, R.A. Clonal Origin and Evolution of a Transmissible Cancer. Cell 2006, 126, 477–487. [Google Scholar] [CrossRef]
- Rebbeck, C.A.; Thomas, R.; Breen, M.; Leroi, A.M.; Burt, A. Origins and evolution of a transmissible cancer. Evolution 2009, 63, 2340–2349. [Google Scholar] [CrossRef]
- Liao, K.-W.; Lin, Z.-Y.; Pao, H.-N.; Kam, S.-Y.; Wang, F.-I.; Chu, R.-M. Identification of canine transmissible venereal tumor cells using in situ polymerase chain reaction and the stable sequence of the long interspersed nuclear element. J. Vet. Diagn. Investig. 2003, 15, 399–406. [Google Scholar] [CrossRef]
- Katzir, N.; Rechavi, G.; Cohen, J.; Unger, T.; Simoni, F.; Segal, S.; Cohen, D.; Givol, D. “Retroposon” insertion into the cellular oncogene c-myc in canine transmissible venereal tumor. Proc. Natl. Acad. Sci. USA 1985, 82, 1054–1058. [Google Scholar] [CrossRef]
- Chu, R.M.; Lin, C.Y.; Liu, C.C.; Yang, S.Y.; Hsiao, Y.W.; Hung, S.W.; Pao, H.N.; Liao, K.W. Proliferation characteristics of canine transmissible venereal tumor. Anticancer Res. 2002, 21, 4017–4024. [Google Scholar]
- Hawkins, C.; Baars, C.; Hesterman, H.; Hocking, G.; Jones, M.; Lazenby, B.; Mann, D.; Mooney, N.; Pemberton, D.; Pyecroft, S.; et al. Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol. Conserv. 2006, 131, 307–324. [Google Scholar] [CrossRef]
- Pearse, A.M.; Swift, K. Allograft theory: Transmission of devil facial-tumour disease. Nature 2006, 439, 549. [Google Scholar] [CrossRef]
- Pyecroft, S.B.; Pearse, A.-M.; Loh, R.; Swift, K.; Belov, K.; Fox, N.; Noonan, E.; Hayes, D.; Hyatt, A.; Wang, L.; et al. Towards a Case Definition for Devil Facial Tumour Disease: What Is It? EcoHealth 2007, 4, 346–351. [Google Scholar] [CrossRef]
- Pye, R.; Pemberton, D.; Tovar, C.; Tubio, J.; Dun, K.; Fox, S.; Darby, J.; Hayes, D.; Knowles, G.W.; Kreiss, A.; et al. A second transmissible cancer in Tasmanian devils. Proc. Natl. Acad. Sci. USA 2015, 113, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Murchison, E.P.; Tovar, C.; Hsu, A.; Bender, H.S.; Kheradpour, P.; Rebbeck, C.A.; Obendorf, D.; Conlan, C.; Bahlo, M.; Blizzard, C.A.; et al. The Tasmanian Devil Transcriptome Reveals Schwann Cell Origins of a Clonally Transmissible Cancer. Science 2010, 327, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Stammnitz, M.; Coorens, T.; Gori, K.; Hayes, D.; Fu, B.; Wang, J.; Martin-Herranz, D.E.; Alexandrov, L.B.; Baez-Ortega, A.; Barthorpe, S.; et al. The Origins and Vulnerabilities of Two Transmissible Cancers in Tasmanian Devils. Cancer Cell 2018, 33, 607–619.e15. [Google Scholar] [CrossRef]
- Murchison, E.P.; Schulz-Trieglaff, O.B.; Ning, Z.; Alexandrov, L.B.; Bauer, M.J.; Fu, B.; Hims, M.; Ding, Z.; Ivakhno, S.; Stewart, C.; et al. Genome Sequencing and Analysis of the Tasmanian Devil and Its Transmissible Cancer. Cell 2012, 148, 780–791. [Google Scholar] [CrossRef]
- Deakin, J.E.; Bender, H.; Pearse, A.-M.; Rens, W.; O’Brien, P.C.M.; Ferguson-Smith, M.A.; Cheng, Y.; Morris, K.; Taylor, R.; Stuart, A.; et al. Genomic Restructuring in the Tasmanian Devil Facial Tumour: Chromosome Painting and Gene Mapping Provide Clues to Evolution of a Transmissible Tumour. PLoS Genet. 2012, 8, e1002483. [Google Scholar] [CrossRef]
- McCallum, H.; Tompkins, D.M.; Jones, M.; Lachish, S.; Marvanek, S.; Lazenby, B.; Hocking, G.; Wiersma, J.; Hawkins, C.E. Distribution and Impacts of Tasmanian Devil Facial Tumor Disease. EcoHealth 2007, 4, 318–325. [Google Scholar] [CrossRef]
- Lazenby, B.T.; Tobler, M.W.; Brown, W.E.; Hawkins, C.E.; Hocking, G.J.; Hume, F.; Huxtable, S.; Iles, P.; Jones, M.; Lawrence, C.; et al. Density trends and demographic signals uncover the long-term impact of transmissible cancer in Tasmanian devils. J. Appl. Ecol. 2018, 55, 1368–1379. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.; Guan, K.-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Ujvari, B.; Pearse, A.-M.; Taylor, R.; Pyecroft, S.; Flanagan, C.; Gombert, S.; Papenfuss, A.T.; Madsen, T.; Belov, K. Telomere Dynamics and Homeostasis in a Transmissible Cancer. PLoS ONE 2012, 7, e44085. [Google Scholar] [CrossRef]
- Siddle, H.V.; Kreiss, A.; Tovar, C.; Yuen, C.K.; Cheng, Y.; Belov, K.; Swift, K.; Pearse, A.-M.; Hamede, R.; Jones, M.; et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 5103–5108. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.E.B.; Lyons, A.B.; Woods, G.M.; Flies, A.S. Inducible IFN-γ Expression for MHC-I Upregulation in Devil Facial Tumor Cells. Front. Immunol. 2019, 9, 3117. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, A.; Coleby, R.; Tovar, C.; Stammnitz, M.R.; Kwon, Y.M.; Owen, R.S.; Tringides, M.; Murchison, E.P.; Skjødt, K.; Thomas, G.J.; et al. The newly-arisen Devil facial tumour disease 2 (DFT2) reveals a mechanism for the emergence of a contagious cancer. eLife 2018, 7, e35314. [Google Scholar] [CrossRef] [PubMed]
- Epstein, B.; Jones, M.; Hamede, R.; Hendricks, S.; McCallum, H.; Murchison, E.P.; Schönfeld, B.; Wiench, C.; Hohenlohe, P.; Storfer, A. Rapid evolutionary response to a transmissible cancer in Tasmanian devils. Nat. Commun. 2016, 7, 12684. [Google Scholar] [CrossRef] [PubMed]
- Wright, B.; Willet, C.E.; Hamede, R.; Jones, M.; Belov, K.; Wade, C.M. Variants in the host genome may inhibit tumour growth in devil facial tumours: Evidence from genome-wide association. Sci. Rep. 2017, 7, 423. [Google Scholar] [CrossRef] [PubMed]
- Ujvari, B.; Hamede, R.; Peck, S.; Pemberton, D.; Jones, M.; Belov, K.; Madsen, T. Immunoglubolin dynamics and cancer prevalence in Tasmanian devils (Sarcophilus harrisii). Sci. Rep. 2016, 6, 25093. [Google Scholar] [CrossRef]
- Farley, C.A. Sarcomatoid Proliferative Disease in a Wild Population of Blue Mussels (Mytilus edulis). J. Natl. Cancer Inst. 1969, 43, 509–516. [Google Scholar] [CrossRef]
- Farley, C.A. Probable neoplastic disease of the hematopoietic system in oysters, Crassostrea virginica and Crassostrea gigas. Natl. Cancer Inst. Monogr. Ser. 1969, 31, 541–555. [Google Scholar]
- Carballal, M.J.; Barber, B.J.; Iglesias, D.; Villalba, A. Neoplastic diseases of marine bivalves. J. Invertebr. Pathol. 2015, 131, 83–106. [Google Scholar] [CrossRef] [PubMed]
- da Silva, P.M.; Farias, N.D.; Queiroga, F.R.; Hégaret, H.; Soudant, P. Disseminated neoplasia in cultured Crassostrea gasar oysters from northeast Brazil. J. Invertebr. Pathol. 2018, 159, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.J.; Reinisch, C.; Sherry, J.; Goff, S.P. Horizontal Transmission of Clonal Cancer Cells Causes Leukemia in Soft-Shell Clams. Cell 2015, 161, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.J.; Villalba, A.; Carballal, M.J.; Iglesias, D.; Sherry, J.; Reinisch, C.; Muttray, A.F.; Baldwin, S.A.; Goff, S.P. Widespread transmission of independent cancer lineages within multiple bivalve species. Nature 2016, 534, 705–709. [Google Scholar] [CrossRef]
- Yonemitsu, A.M.; Giersch, R.M.; Polo-Prieto, M.; Hammel, M.; Simon, A.; Cremonte, F.; Avilés, F.T.; Merino-Véliz, N.; Burioli, E.A.; Muttray, A.F.; et al. A single clonal lineage of transmissible cancer identified in two marine mussel species in South America and Europe. eLife 2019, 8, e47788. [Google Scholar] [CrossRef]
- Murchison, E.P. Cancer: Transmissible tumours under the sea. Nature 2016, 534, 628–629. [Google Scholar] [CrossRef][Green Version]
- Skazina, M.; Odintsova, N.; Maiorova, M.; Ivanova, A.; Väinölä, R.; Strelkov, P. First description of a widespread Mytilus trossulus-derived bivalve transmissible cancer lineage in M. trossulus itself. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Aguilera, F. Neoplasia in Mollusks: What Does it Tell us about Cancer in Humans?—A Review. J. Genet. Disord. 2017, 1, 7. [Google Scholar]
- Walker, C.W.; Van Beneden, R.J.; Muttray, A.F.; Böttger, S.A.; Kelley, M.L.; Tucker, A.E.; Thomas, W.K. p53 Superfamily Proteins in Marine Bivalve Cancer and Stress Biology. Adv. Mar. Biol. 2011, 59, 1–36. [Google Scholar] [CrossRef]
- Sunila, I.; Farley, C. Environmental limits for survival of sarcoma cells from the soft-shell clam Mya arenaria. Dis. Aquat. Org. 1989, 7, 111–115. [Google Scholar] [CrossRef]
- Green, T.; Jones, B.J.; Adlard, R.D.; Barnes, A. Parasites, pathological conditions and mortality in QX-resistant and wild-caught Sydney rock oysters, Saccostrea glomerata. Aquaculture 2008, 280, 35–38. [Google Scholar] [CrossRef]
- Alderman, D.; Van Banning, P.; Perez-Colomer, A. Two European oyster (Ostrea edulis) mortalities associated with an abnormal haemocytic condition. Aquaculture 1977, 10, 335–340. [Google Scholar] [CrossRef]
- Bramwell, G.; Schultz, A.G.; Sherman, C.D.; Giraudeau, M.; Thomas, F.; Ujvari, B.; Dujon, A.M. A review of the potential effects of climate change on disseminated neoplasia with an emphasis on efficient detection in marine bivalve populations. Sci. Total Environ. 2021, 775, 145134. [Google Scholar] [CrossRef]
- Olivier, A.V.D.S.; Jones, L.; Le Vay, L.; Christie, M.; Wilson, J.; Malham, S.K. A global review of the ecosystem services provided by bivalve aquaculture. Rev. Aquac. 2018, 12, 3–25. [Google Scholar] [CrossRef]
- Ohno, S. So much “junk” DNA in our genome. Brookhaven Symp. Boil. 1972, 23, 366–370. [Google Scholar]
- Ohno, S.; Yomo, T. The grammatical rule for all DNA: Junk and coding sequences. Electrophoresis 1991, 12, 103–108. [Google Scholar] [CrossRef]
- Mouse Genome Sequencing Consortium; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar]
- Goodstadt, L.; Ponting, C.P. Phylogenetic Reconstruction of Orthology, Paralogy, and Conserved Synteny for Dog and Human. PLoS Comput. Biol. 2006, 2, e133. [Google Scholar] [CrossRef] [PubMed]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, A.B.; McCue, K.; Schaeffer, L.; Wold, B.J. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Palade, G.E. A small particulate component of the cytoplasm. J. Cell Biol. 1955, 1, 59–68. [Google Scholar] [CrossRef]
- Hoagland, M.B.; Stephenson, M.L.; Scott, J.F.; Hecht, L.I.; Zamecnik, P.C. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 1958, 231, 241–257. [Google Scholar] [CrossRef]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Brown, C.; Ballabio, A.; Rupert, J.L.; LaFreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef]
- Brannan, C.I.; Dees, E.C.; Ingram, R.S.; Tilghman, S.M. The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 1990, 10, 28–36. [Google Scholar] [CrossRef]
- Zhang, Y.; Tycko, B. Monoallelic expression of the human H19 gene. Nat. Genet. 1992, 1, 40–44. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.; Mattick, J. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional Maps of 10 Human Chromosomes at 5-Nucleotide Resolution. Science 2005, 308, 1149–1154. [Google Scholar] [CrossRef]
- Frith, M.; Pheasant, M.; Mattick, J. Genomics: The amazing complexity of the human transcriptome. Eur. J. Hum. Genet. 2005, 13, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Rahman, L.; Bliskovski, V.; Kaye, F.J.; Zajac-Kaye, M. Evolutionary conservation of a 2-kb intronic sequence flanking a tissue-specific alternative exon in the PTBP2 gene. Genomics 2003, 83, 76–84. [Google Scholar] [CrossRef]
- Kim, T.-K.; Hemberg, M.; Gray, J.M. Enhancer RNAs: A Class of Long Noncoding RNAs Synthesized at Enhancers: Figure 1. Cold Spring Harb. Perspect. Biol. 2015, 7, a018622. [Google Scholar] [CrossRef]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol. 2012, 13, 1–26. [Google Scholar] [CrossRef]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Prensner, J.; Chinnaiyan, A.M. The Emergence of lncRNAs in Cancer Biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.; Ackley, A.; Turner, A.-M.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K. An HIV-Encoded Antisense Long Noncoding RNA Epigenetically Regulates Viral Transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol. 2012, 19, 1068–1075. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Lee, J.; Davidow, L.S.; Warshawsky, D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 1999, 21, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Smilinich, N.J.; Day, C.D.; Fitzpatrick, G.V.; Caldwell, G.M.; Lossie, A.C.; Cooper, P.; Smallwood, A.C.; Joyce, J.A.; Schofield, P.N.; Reik, W.; et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 8064–8069. [Google Scholar] [CrossRef]
- Wutz, A.; Smrzka, O.W.; Schweifer, N.; Schellander, K.; Wagner, E.F.; Barlow, D.P. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nat. Cell Biol. 1997, 389, 745–749. [Google Scholar] [CrossRef]
- Chen, J.; Sun, M.; Kent, W.J.; Huang, X.; Xie, H.; Wang, W.; Zhou, G.; Shi, R.Z.; Rowley, J.D. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res. 2004, 32, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Zhang, M.; Huang, J.; Modarresi, F.; Van der Brug, M.P.; Nalls, M.A.; Cookson, M.R.; St-Laurent, G., 3rd; Wahlestedt, C. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 2010, 11, R56. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Henriksson, S.; Corcoran, M.; Méndez-Vidal, C.; Wiman, K.; Farnebo, M. Wrap53, a Natural p53 Antisense Transcript Required for p53 Induction upon DNA Damage. Mol. Cell 2009, 33, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Tufarelli, C.; Sloane-Stanley, J.A.; Garrick, D.; Sharpe, J.A.; Ayyub, H.; Wood, W.G.; Higgs, D.R. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat. Genet. 2003, 34, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.; Santoso, S.; Turner, A.-M.; Pastori, C.; Hawkins, P.G. Bidirectional Transcription Directs Both Transcriptional Gene Activation and Suppression in Human Cells. PLoS Genet. 2008, 4, e1000258. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Alvarez, A.B.; Peña, R.; Bonilla, F.; de Herreros, A.G. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef]
- Choi, P.; Jordan, C.D.; Mendez, E.; Houck, J.; Yueh, B.; Farwell, D.G.; Futran, N.; Chen, C. Examination of Oral Cancer Biomarkers by Tissue Microarray Analysis. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 539–546. [Google Scholar] [CrossRef]
- Taniue, K.; Akimitsu, N. The Functions and Unique Features of LncRNAs in Cancer Development and Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 632. [Google Scholar] [CrossRef]
- Rinn, J.; Kertesz, M.; Wang, J.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Wang, W.-T.; Han, C.; Sun, Y.-M.; Chen, T.-Q.; Chen, Y.-Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Lister, N.; Shevchenko, G.; Walshe, J.L.; Groen, J.; Johnsson, P.; Vidarsdóttir, L.; Grander, D.; Ataide, S.F.; Morris, K.V. The molecular dynamics of long noncoding RNA control of transcription in PTEN and its pseudogene. Proc. Natl. Acad. Sci. USA 2017, 114, 9942–9947. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.-O.; Corcoran, M.; Grandér, D.; Morris, K. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Karvelis, T.; Gasiunas, G.; Miksys, A.; Barrangou, R.; Horvath, P.; Siksnys, V. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013, 10, 841–851. [Google Scholar] [CrossRef]
- Hsu, P.; Scott, D.A.; Weinstein, J.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.-L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.; Doudna, J.A.; Weissman, J.S.; Arkin, A.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Polstein, L.R.; Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Bledsoe, P.; Song, L.; Safi, A.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Genome-wide specificity of DNA binding, gene regulation, and chromatin remodeling by TALE-and CRISPR/Cas9-based transcriptional activators. Genome Res. 2015, 25, 1158–1169. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 654–658. [Google Scholar] [CrossRef]
- Sánchez-Rivera, F.J.; Jacks, T. Applications of the CRISPR–Cas9 system in cancer biology. Nat. Rev. Cancer 2015, 15, 387–393. [Google Scholar] [CrossRef]
- Paralkar, V.R.; Taborda, C.C.; Huang, P.; Yao, Y.; Kossenkov, A.V.; Prasad, R.; Luan, J.; Davies, J.; Hughes, J.R.; Hardison, R.; et al. Unlinking An lncRNA from Its Associated cis Element. Mol. Cell 2016, 62, 104–110. [Google Scholar] [CrossRef]
- Yi, Z.; Yan, P.; Lu, J.; Song, G.; Zhu, Y.; Li, Z.; Zhao, Y.; Shen, B.; Huang, X.; Zhu, H.; et al. Opposing Roles for the lncRNA Haunt and Its Genomic Locus in Regulating HOXA Gene Activation during Embryonic Stem Cell Differentiation. Cell Stem. Cell 2015, 16, 504–516. [Google Scholar] [CrossRef]
- Rankin, C.R.; Treger, J.; Faure-Kumar, E.; Benhammou, J.; Anisman-Posner, D.; Bollinger, A.E.; Pothoulakis, C.; Padua, D.M. Overexpressing Long Noncoding RNAs Using Gene-activating CRISPR. J. Vis. Exp. 2019, 145, e59233. [Google Scholar] [CrossRef]
- Ghosh, S.; Tibbit, C.; Liu, J.L. Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic Acids Res. 2016, 44, e84. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Liu, J.; Chen, C.-H.; Liao, Q.; Xu, P.; Xu, H.; Xiao, T.; Cao, Z.; Peng, J.; et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat. Biotechnol. 2016, 34, 1279–1286. [Google Scholar] [CrossRef]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355, eaah7111. [Google Scholar] [CrossRef] [PubMed]
- Zayas, Y.R.; Molina, M.A.F.; Guerra, R.T.; Padilla, C.R. Evaluation of a canine transmissible venereal tumour cell line with tumour immunity capacity but without tumorigenic property. J. Vet. Res. 2019, 63, 225–233. [Google Scholar] [CrossRef]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef]
- Oliva-Rico, D.; Herrera, L.A. Regulated expression of the lncRNA TERRA and its impact on telomere biology. Mech. Ageing Dev. 2017, 167, 16–23. [Google Scholar] [CrossRef]
- Nelson, A.D.L.; Shippen, D.E. Evolution of TERT-interacting lncRNAs: Expanding the regulatory landscape of telomerase. Front. Genet. 2015, 6, 277. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Magiorkinis, G.; Lim, A.G.; Gupta, S.; Belshaw, R.; Gifford, R. Larger Mammalian Body Size Leads to Lower Retroviral Activity. PLoS Pathog. 2014, 10, e1004214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Qi, F.; Wu, F.; Nie, H.; Song, Y.; Shao, L.; Han, J.; Wu, Z.; Saiyin, H.; Wei, G.; et al. Endogenous Retrovirus-Derived Long Noncoding RNA Enhances Innate Immune Responses via Derepressing RELA Expression. mBio 2019, 10, e00937-19. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.A. The devil is in the details: Transposable element analysis of the Tasmanian devil genome. Mob. Genet. Elem. 2015, 6, e1119926. [Google Scholar] [CrossRef][Green Version]
- Gallus, S.; Hallström, B.M.; Kumar, V.; Dodt, W.G.; Janke, A.; Schumann, G.G.; Nilsson, M.A. Evolutionary Histories of Transposable Elements in the Genome of the Largest Living Marsupial Carnivore, the Tasmanian Devil. Mol. Biol. Evol. 2015, 32, 1268–1283. [Google Scholar] [CrossRef][Green Version]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Wakefield, M.J.; Aken, B.; Amemiya, C.T.; Chang, J.L.; Duke, S.; Garber, M.; Gentles, A.J.; Goodstadt, L.; Heger, A.; et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 2007, 447, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Calegari-Silva, T.C.; Vivarini, A.C.; Pereira, R.M.S.; Dias-Teixeira, K.L.; Rath, C.T.; Pacheco, A.S.S.; Silva, G.B.L.; Pinto, C.A.S.; Dos Santos, J.V.; Saliba, A.M.; et al. Leishmania amazonensis downregulates macrophage iNOS expression via Histone Deacetylase 1 (HDAC1): A novel parasite evasion mechanism. Eur. J. Immunol. 2018, 48, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Piertney, S.B.; Oliver, M.K. The evolutionary ecology of the major histocompatibility complex. Heredity 2005, 96, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, N.S.; Shieh, M.; Monos, D. Regulatory noncoding RNAs and the major histocompatibility complex. Hum. Immunol. 2020, 82, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kryczek, I.; Nam, J.; Li, X.; Li, S.; Li, J.; Wei, S.; Grove, S.; Vatan, L.; Zhou, J.; et al. LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nature 2021, 23, 526–537. [Google Scholar] [CrossRef]
- Cheng, Y.; Makara, M.; Peel, E.; Fox, S.; Papenfuss, A.T.; Belov, K. Tasmanian devils with contagious cancer exhibit a constricted T-cell repertoire diversity. Commun. Biol. 2019, 2, 99. [Google Scholar] [CrossRef]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef]
- Wang, P.; Xue, Y.; Han, Y.; Lin, L.; Wu, C.; Xu, S.; Jiang, Z.; Xu, J.; Liu, Q.; Cao, X. The STAT3-Binding Long Noncoding RNA lnc-DC Controls Human Dendritic Cell Differentiation. Science 2014, 344, 310–313. [Google Scholar] [CrossRef]
- Gomez, J.A.; Wapinski, O.L.; Yang, Y.W.; Bureau, J.F.; Gopinath, S.; Monack, D.M.; Chang, H.Y.; Brahic, M.; Kirkegaard, K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell 2013, 152, 743–754. [Google Scholar] [CrossRef]
- Yang, C.; Feng, T.; Lin, F.; Gong, T.; Yang, S.; Tao, Y.; Li, H. Long noncoding RNA TANCR promotes γδ T cells activation by regulating TRAIL expression in cis. Cell Biosci. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Allam, B.; Raftos, D. Immune responses to infectious diseases in bivalves. J. Invertebr. Pathol. 2015, 131, 121–136. [Google Scholar] [CrossRef]
- Abo-Al-Ela, H.G.; Faggio, C. MicroRNA-mediated stress response in bivalve species. Ecotoxicol. Environ. Saf. 2021, 208, 111442. [Google Scholar] [CrossRef] [PubMed]
- Rosani, U.; Bortoletto, E.; Bai, C.-M.; Novoa, B.; Figueras, A.; Venier, P.; Fromm, B. Digging into bivalve miRNAomes: Between conservation and innovation. Philos. Trans. R. Soc. B Biol. Sci. 2021, 376, 20200165. [Google Scholar] [CrossRef] [PubMed]
- Botta, R.; Asche, F.; Borsum, J.S.; Camp, E.V. A review of global oyster aquaculture production and consumption. Mar. Policy 2020, 117, 103952. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lister, N.C.; Milton, A.M.; Hanrahan, B.J.; Waters, P.D. Between the Devil and the Deep Blue Sea: Non-Coding RNAs Associated with Transmissible Cancers in Tasmanian Devil, Domestic Dog and Bivalves. Non-Coding RNA 2021, 7, 72. https://doi.org/10.3390/ncrna7040072
Lister NC, Milton AM, Hanrahan BJ, Waters PD. Between the Devil and the Deep Blue Sea: Non-Coding RNAs Associated with Transmissible Cancers in Tasmanian Devil, Domestic Dog and Bivalves. Non-Coding RNA. 2021; 7(4):72. https://doi.org/10.3390/ncrna7040072
Chicago/Turabian StyleLister, Nicholas C., Ashley M. Milton, Benjamin J. Hanrahan, and Paul D. Waters. 2021. "Between the Devil and the Deep Blue Sea: Non-Coding RNAs Associated with Transmissible Cancers in Tasmanian Devil, Domestic Dog and Bivalves" Non-Coding RNA 7, no. 4: 72. https://doi.org/10.3390/ncrna7040072
APA StyleLister, N. C., Milton, A. M., Hanrahan, B. J., & Waters, P. D. (2021). Between the Devil and the Deep Blue Sea: Non-Coding RNAs Associated with Transmissible Cancers in Tasmanian Devil, Domestic Dog and Bivalves. Non-Coding RNA, 7(4), 72. https://doi.org/10.3390/ncrna7040072