
Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine


Abstract
1. Introduction
2. Genetic Variability in the Cancer Genome
2.1. Structural Classification of Mutations in Cancer
2.2. Functional Classification of Mutations in Cancer
3. Non-Transcribed Regulatory Variants
3.1. Genetic Variability in Promoters
3.2. Genetic Variability in Enhancers
3.3. Genetic Variability in Silencer Elements
3.4. Genetic Variability in Insulator Elements
4. Transcribed Non-Coding Variants
4.1. Non-Coding Variants Affecting miRNA Targeting and Biogenesis
4.2. Non-Coding Variants Affecting lncRNA Function
5. Methodologies to Functionally Characterize Non-Coding Variants
5.1. Scanning for Regulatory Sequences Based on Open-Chromatin State
5.2. Scanning for Regulatory Variants in Trans Regulatory Elements
5.3. Scanning for Regulatory Variants Based on Chromatin Interactions
5.4. Scanning for Regulatory Variants Based on RNA-Chromatin Interactions
5.5. Methodologies to Functionally Dissect Transcribed Non-Coding Variants
5.6. Validating Regulatory Variants with CRISPR-Based Approaches
6. Utilizing Non-Coding Variants in Clinomics
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Della Chiara, G.; Gervasoni, F.; Fakiola, M.; Godano, C.; D’Oria, C.; Azzolin, L.; Bonnal, R.J.P.; Moreni, G.; Drufuca, L.; Rossetti, G.; et al. Epigenomic landscape of human colorectal cancer unveils an aberrant core of pan-cancer enhancers orchestrated by YAP/TAZ. Nat. Commun. 2021, 12, 2340. [Google Scholar] [CrossRef]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat. Commun. 2021, 12, 2242. [Google Scholar] [CrossRef]
- Xiong, L.; Wu, F.; Wu, Q.; Xu, L.; Cheung, O.K.; Kang, W.; Mok, M.T.; Szeto, L.L.M.; Lun, C.Y.; Lung, R.W.; et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat. Commun. 2019, 10, 335. [Google Scholar] [CrossRef]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- See, Y.X.; Wang, B.Z.; Fullwood, M.J. Chromatin Interactions and Regulatory Elements in Cancer: From Bench to Bedside. Trends Genet. 2019, 35, 145–158. [Google Scholar] [CrossRef]
- Savarese, F.; Grosschedl, R. Blurring cis and trans in gene regulation. Cell 2006, 126, 248–250. [Google Scholar] [CrossRef]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet 2020, 21, 71–87. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Furlan-Magaril, M.; Mifsud, B.; Tavares-Cadete, F.; Sugar, R.; Javierre, B.M.; Nagano, T.; Katsman, Y.; Sakthidevi, M.; Wingett, S.W.; et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015, 25, 582–597. [Google Scholar] [CrossRef]
- Furlong, E.E.M.; Levine, M. Developmental enhancers and chromosome topology. Science 2018, 361, 1341–1345. [Google Scholar] [CrossRef]
- Maass, P.G.; Barutcu, A.R.; Rinn, J.L. Interchromosomal interactions: A genomic love story of kissing chromosomes. J. Cell Biol. 2019, 218, 27–38. [Google Scholar] [CrossRef]
- Gasperini, M.; Tome, J.M.; Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 2020, 21, 292–310. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930.e922–944.e922. [Google Scholar] [CrossRef]
- Wutz, G.; Varnai, C.; Nagasaka, K.; Cisneros, D.A.; Stocsits, R.R.; Tang, W.; Schoenfelder, S.; Jessberger, G.; Muhar, M.; Hossain, M.J.; et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017, 36, 3573–3599. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Kim, S.; Yu, N.K.; Kaang, B.K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Kim, T.K.; Hemberg, M.; Gray, J.M. Enhancer RNAs: A class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb. Perspect. Biol. 2015, 7, a018622. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Chen, S.; Tan, Y.; Li, Y.; Tang, F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp. Mol. Med. 2020, 52, 713–723. [Google Scholar] [CrossRef]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Lauberth, S.M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 2020, 27, 521–528. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.H.; Ruan, H.; Ye, Y.; Krakowiak, J.; Hu, Q.; Xiang, Y.; Gong, J.; Zhou, B.; Wang, L.; et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019, 10, 4562. [Google Scholar] [CrossRef]
- Sakabe, N.J.; Savic, D.; Nobrega, M.A. Transcriptional enhancers in development and disease. Genome Biol. 2012, 13, 238. [Google Scholar] [CrossRef]
- Xia, J.H.; Wei, G.H. Enhancer Dysfunction in 3D Genome and Disease. Cells 2019, 8, 1281. [Google Scholar] [CrossRef]
- Chakravarty, D.; Solit, D.B. Clinical cancer genomic profiling. Nat. Rev. Genet. 2021. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Beckmann, J.S.; Estivill, X.; Antonarakis, S.E. Copy number variants and genetic traits: Closer to the resolution of phenotypic to genotypic variability. Nat. Rev. Genet. 2007, 8, 639–646. [Google Scholar] [CrossRef]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerbo, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3.0: Integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012, 40, D210–D215. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Carrot-Zhang, J.; Chambwe, N.; Damrauer, J.S.; Knijnenburg, T.A.; Robertson, A.G.; Yau, C.; Zhou, W.; Berger, A.C.; Huang, K.L.; Newberg, J.Y.; et al. Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 2020, 37, 639.e636–654.e636. [Google Scholar] [CrossRef]
- Hong, E.L.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; et al. Principles of metadata organization at the ENCODE data coordination center. Database 2016, 2016. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, D.; Dhiman, V.; Jiang, P.; Xu, J.; McGillivray, P.; Yang, H.; Liu, J.; Meyerson, W.; Clarke, D.; et al. An integrative ENCODE resource for cancer genomics. Nat. Commun. 2020, 11, 3696. [Google Scholar] [CrossRef]
- Liu, C.; Bai, B.; Skogerbo, G.; Cai, L.; Deng, W.; Zhang, Y.; Bu, D.; Zhao, Y.; Chen, R. NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005, 33, D112–D115. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu Rev. Genom. Hum. Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef]
- Kalender Atak, Z.; Imrichova, H.; Svetlichnyy, D.; Hulselmans, G.; Christiaens, V.; Reumers, J.; Ceulemans, H.; Aerts, S. Identification of cis-regulatory mutations generating de novo edges in personalized cancer gene regulatory networks. Genome Med. 2017, 9, 80. [Google Scholar] [CrossRef]
- Erson-Bensan, A.E. RNA-biology ruling cancer progression? Focus on 3’UTRs and splicing. Cancer Metastasis Rev. 2020, 39, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef]
- Bashyam, M.D.; Animireddy, S.; Bala, P.; Naz, A.; George, S.A. The Yin and Yang of cancer genes. Gene 2019, 704, 121–133. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Yong, S.Y.; Raben, T.G.; Lello, L.; Hsu, S.D.H. Genetic architecture of complex traits and disease risk predictors. Sci. Rep. 2020, 10, 12055. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Soong, D.; Stratford, J.; Avet-Loiseau, H.; Bahlis, N.; Davies, F.; Dispenzieri, A.; Sasser, A.K.; Schecter, J.M.; Qi, M.; Brown, C.; et al. CNV Radar: An improved method for somatic copy number alteration characterization in oncology. BMC Bioinform. 2020, 21, 98. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef]
- Kumaran, M.; Cass, C.E.; Graham, K.; Mackey, J.R.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Germline copy number variations are associated with breast cancer risk and prognosis. Sci. Rep. 2017, 7, 14621. [Google Scholar] [CrossRef]
- Granados-Soler, J.L.; Bornemann-Kolatzki, K.; Beck, J.; Brenig, B.; Schutz, E.; Betz, D.; Junginger, J.; Hewicker-Trautwein, M.; Murua Escobar, H.; Nolte, I. Analysis of Copy-Number Variations and Feline Mammary Carcinoma Survival. Sci. Rep. 2020, 10, 1003. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Grade, M.; Nguyen, Q.T.; Hormann, P.; Becker, S.; Hummon, A.B.; Rodriguez, V.; Chandrasekharappa, S.; Chen, Y.; Difilippantonio, M.J.; et al. Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer Res. 2008, 68, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Lahortiga, I.; De Keersmaecker, K.; Van Vlierberghe, P.; Graux, C.; Cauwelier, B.; Lambert, F.; Mentens, N.; Beverloo, H.B.; Pieters, R.; Speleman, F.; et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 2007, 39, 593–595. [Google Scholar] [CrossRef]
- Basecke, J.; Whelan, J.T.; Griesinger, F.; Bertrand, F.E. The MLL partial tandem duplication in acute myeloid leukaemia. Br. J. Haematol. 2006, 135, 438–449. [Google Scholar] [CrossRef]
- Kumaran, M.; Krishnan, P.; Cass, C.E.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Breast cancer associated germline structural variants harboring small noncoding RNAs impact post-transcriptional gene regulation. Sci Rep. 2018, 8, 7529. [Google Scholar] [CrossRef]
- Li, Y.R.; Glessner, J.T.; Coe, B.P.; Li, J.; Mohebnasab, M.; Chang, X.; Connolly, J.; Kao, C.; Wei, Z.; Bradfield, J.; et al. Rare copy number variants in over 100,000 European ancestry subjects reveal multiple disease associations. Nat. Commun. 2020, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, R.C.; Brown, K.M. Cancer regulatory variation. Curr Opin Genet. Dev. 2021, 66, 41–49. [Google Scholar] [CrossRef]
- Spielmann, M.; Klopocki, E. CNVs of noncoding cis-regulatory elements in human disease. Curr. Opin. Genet. Dev. 2013, 23, 249–256. [Google Scholar] [CrossRef]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.F.; et al. RADICL-seq identifies general and cell type-specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 2020, 11, 1018. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Deng, L.; Xia, Y.; Wei, X.; Cao, Y.; Guo, R.; Zhang, R.; Guo, J.; Liang, D.; Wu, L. A clear bias in parental origin of de novo pathogenic CNVs related to intellectual disability, developmental delay and multiple congenital anomalies. Sci. Rep. 2017, 7, 44446. [Google Scholar] [CrossRef]
- Wang, B.; Ji, T.; Zhou, X.; Wang, J.; Wang, X.; Wang, J.; Zhu, D.; Zhang, X.; Sham, P.C.; Zhang, X.; et al. CNV analysis in Chinese children of mental retardation highlights a sex differentiation in parental contribution to de novo and inherited mutational burdens. Sci. Rep. 2016, 6, 25954. [Google Scholar] [CrossRef]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshoj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 2018, 174, 564.e518–575.e518. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, R.; Liu, B.; Kong, J.; Lin, H.; Yu, X.; Wang, R.; Li, L.; Gao, M.; Zhou, B.; et al. SNP rs17079281 decreases lung cancer risk through creating an YY1-binding site to suppress DCBLD1 expression. Oncogene 2020, 39, 4092–4102. [Google Scholar] [CrossRef]
- Motawi, T.M.K.; Sadik, N.A.H.; Sabry, D.; Shahin, N.N.; Fahim, S.A. rs2267531, a promoter SNP within glypican-3 gene in the X chromosome, is associated with hepatocellular carcinoma in Egyptians. Sci. Rep. 2019, 9, 6868. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, N.; Zhang, L.; Song, Y.; Liu, J.; Yu, J.; Yang, M. Oncogene HSPH1 modulated by the rs2280059 genetic variant diminishes EGFR-TKIs efficiency in advanced lung adenocarcinoma. Carcinogenesis 2020, 41, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ahmed, M.; Zhang, F.; Yao, C.Q.; Li, S.; Liang, Y.; Hua, J.; Soares, F.; Sun, Y.; Langstein, J.; et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat. Genet. 2016, 48, 1142–1150. [Google Scholar] [CrossRef]
- Grampp, S.; Platt, J.L.; Lauer, V.; Salama, R.; Kranz, F.; Neumann, V.K.; Wach, S.; Stohr, C.; Hartmann, A.; Eckardt, K.U.; et al. Genetic variation at the 8q24.21 renal cancer susceptibility locus affects HIF binding to a MYC enhancer. Nat. Commun. 2016, 7, 13183. [Google Scholar] [CrossRef]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef]
- Walker, L.C.; BCFR; Marquart, L.; Pearson, J.F.; Wiggins, G.A.R.; O’Mara, T.A.; Parsons, M.T.; Barrowdale, D.; McGuffog, L.; Dennis, J.; et al. Evaluation of copy-number variants as modifiers of breast and ovarian cancer risk for BRCA1 pathogenic variant carriers. Eur. J. Hum. Genet. 2017, 25, 432–438. [Google Scholar] [CrossRef]
- Wang, J.; Zou, Y.; Du, B.; Li, W.; Yu, G.; Li, L.; Zhou, L.; Gu, X.; Song, S.; Liu, Y.; et al. SNP-mediated lncRNA-ENTPD3-AS1 upregulation suppresses renal cell carcinoma via miR-155/HIF-1alpha signaling. Cell Death Dis. 2021, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Ostrovsky, O.; Grushchenko-Polaq, A.H.; Beider, K.; Mayorov, M.; Canaani, J.; Shimoni, A.; Vlodavsky, I.; Nagler, A. Identification of strong intron enhancer in the heparanase gene: Effect of functional rs4693608 variant on HPSE enhancer activity in hematological and solid malignancies. Oncogenesis 2018, 7, 51. [Google Scholar] [CrossRef]
- Painter, J.N.; Kaufmann, S.; O’Mara, T.A.; Hillman, K.M.; Sivakumaran, H.; Darabi, H.; Cheng, T.H.T.; Pearson, J.; Kazakoff, S.; Waddell, N.; et al. A Common Variant at the 14q32 Endometrial Cancer Risk Locus Activates AKT1 through YY1 Binding. Am. J. Hum. Genet. 2016, 98, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Zheng, R.; Ma, G.; Chu, H.; Lu, J.; Li, S.; Xin, J.; Tong, N.; Zhang, G.; Wang, W.; et al. Remote modulation of lncRNA GCLET by risk variant at 16p13 underlying genetic susceptibility to gastric cancer. Sci. Adv. 2020, 6, eaay5525. [Google Scholar] [CrossRef]
- Helmsauer, K.; Valieva, M.E.; Ali, S.; Chamorro Gonzalez, R.; Schopflin, R.; Roefzaad, C.; Bei, Y.; Dorado Garcia, H.; Rodriguez-Fos, E.; Puiggros, M.; et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nat. Commun. 2020, 11, 5823. [Google Scholar] [CrossRef]
- El Hajj, P.; Gilot, D.; Migault, M.; Theunis, A.; van Kempen, L.C.; Sales, F.; Fayyad-Kazan, H.; Badran, B.; Larsimont, D.; Awada, A.; et al. SNPs at miR-155 binding sites of TYRP1 explain discrepancy between mRNA and protein and refine TYRP1 prognostic value in melanoma. Br. J. Cancer 2015, 113, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zandi, R.; Shao, R.; Gu, J.; Ye, Y.; Wang, J.; Zhao, Y.; Pertsemlidis, A.; Wistuba, I.I.; Wu, X.; et al. A miR-SNP biomarker linked to an increased lung cancer survival by miRNA-mediated down-regulation of FZD4 expression and Wnt signaling. Sci. Rep. 2017, 7, 9029. [Google Scholar] [CrossRef] [PubMed]
- Gilam, A.; Conde, J.; Weissglas-Volkov, D.; Oliva, N.; Friedman, E.; Artzi, N.; Shomron, N. Local microRNA delivery targets Palladin and prevents metastatic breast cancer. Nat. Commun. 2016, 7, 12868. [Google Scholar] [CrossRef]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.; Cordiner, R.A.; Young, R.S.; Hug, N.; Macias, S.; Caceres, J.F. Genetic variation and RNA structure regulate microRNA biogenesis. Nat. Commun. 2017, 8, 15114. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.P.; Su, X.L. Polymorphism in pre-miR-30c contributes to gastric cancer risk in a Chinese population. Med. Oncol. 2012, 29, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, X.; Meng, F.; Zhang, Y.; Wang, M.; Chen, Y.; Guo, X.; Chen, W.; Wang, W. The rs7911488-T allele promotes the growth and metastasis of colorectal cancer through modulating miR-1307/PRRX1. Cell Death Dis. 2020, 11, 651. [Google Scholar] [CrossRef]
- Yang, Q.; Jie, Z.; Ye, S.; Li, Z.; Han, Z.; Wu, J.; Yang, C.; Jiang, Y. Genetic variations in miR-27a gene decrease mature miR-27a level and reduce gastric cancer susceptibility. Oncogene 2014, 33, 193–202. [Google Scholar] [CrossRef]
- Redis, R.S.; Vela, L.E.; Lu, W.; Ferreira de Oliveira, J.; Ivan, C.; Rodriguez-Aguayo, C.; Adamoski, D.; Pasculli, B.; Taguchi, A.; Chen, Y.; et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol. Cell 2016, 61, 520–534. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A Novel Genetic Variant in Long Non-coding RNA Gene NEXN-AS1 is Associated with Risk of Lung Cancer. Sci. Rep. 2016, 6, 34234. [Google Scholar] [CrossRef]
- Wu, S.; Sun, H.; Wang, Y.; Yang, X.; Meng, Q.; Yang, H.; Zhu, H.; Tang, W.; Li, X.; Aschner, M.; et al. MALAT1 rs664589 Polymorphism Inhibits Binding to miR-194-5p, Contributing to Colorectal Cancer Risk, Growth, and Metastasis. Cancer Res. 2019, 79, 5432–5441. [Google Scholar] [CrossRef]
- Shen, C.; Yan, T.; Wang, Z.; Su, H.C.; Zhu, X.; Tian, X.; Fang, J.Y.; Chen, H.; Hong, J. Variant of SNP rs1317082 at CCSlnc362 (RP11-362K14.5) creates a binding site for miR-4658 and diminishes the susceptibility to CRC. Cell Death Dis. 2018, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zheng, J.; Deng, J.; Hu, M.; You, Y.; Li, N.; Li, W.; Lu, J.; Zhou, Y. A genetic polymorphism in lincRNA-uc003opf.1 is associated with susceptibility to esophageal squamous cell carcinoma in Chinese populations. Carcinogenesis 2013, 34, 2908–2917. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, X.; Tan, W.; Yu, D.; Du, Z.; Chang, J.; Wei, L.; Han, Y.; Wang, C.; Che, X.; et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016, 48, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Dhamodharan, S.; Rose, M.M.; Chakkarappan, S.R.; Umadharshini, K.V.; Arulmurugan, R.; Subbiah, S.; Inoue, I.; Munirajan, A.K. Genetic variant rs10251977 (G > A) in EGFR-AS1 modulates the expression of EGFR isoforms A and D. Sci. Rep. 2021, 11, 8808. [Google Scholar] [CrossRef]
- Lee, D.S.M.; Park, J.; Kromer, A.; Baras, A.; Rader, D.J.; Ritchie, M.D.; Ghanem, L.R.; Barash, Y. Disrupting upstream translation in mRNAs is associated with human disease. Nat. Commun. 2021, 12, 1515. [Google Scholar] [CrossRef] [PubMed]
- Steri, M.; Idda, M.L.; Whalen, M.B.; Orru, V. Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA 2018, 9, e1474. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.P. The ever-changing world of gene fusions in cancer: A secondary gene fusion and progression. Oncogene 2019, 38, 7197–7199. [Google Scholar] [CrossRef]
- Rashkin, S.R.; Graff, R.E.; Kachuri, L.; Thai, K.K.; Alexeeff, S.E.; Blatchins, M.A.; Cavazos, T.B.; Corley, D.A.; Emami, N.C.; Hoffman, J.D.; et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun. 2020, 11, 4423. [Google Scholar] [CrossRef] [PubMed]
- Giral, H.; Landmesser, U.; Kratzer, A. Into the Wild: GWAS Exploration of Non-coding RNAs. Front. Cardiovasc. Med. 2018, 5, 181. [Google Scholar] [CrossRef]
- Hrdlickova, B.; de Almeida, R.C.; Borek, Z.; Withoff, S. Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta 2014, 1842, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.P.; Zhang, B.X.; Chu, L. Small Nucleolar RNAs: Insight into Their Function in Cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Zhu, H.; Uuskula-Reimand, L.; Isaev, K.; Wadi, L.; Alizada, A.; Shuai, S.; Huang, V.; Aduluso-Nwaobasi, D.; Paczkowska, M.; Abd-Rabbo, D.; et al. Candidate Cancer Driver Mutations in Distal Regulatory Elements and Long-Range Chromatin Interaction Networks. Mol. Cell 2020, 77, 1307.e1310–1321.e1310. [Google Scholar] [CrossRef]
- Cheng, Y.; Quinn, J.F.; Weiss, L.A. An eQTL mapping approach reveals that rare variants in the SEMA5A regulatory network impact autism risk. Hum. Mol. Genet. 2013, 22, 2960–2972. [Google Scholar] [CrossRef]
- Li, X.; Kim, Y.; Tsang, E.K.; Davis, J.R.; Damani, F.N.; Chiang, C.; Hess, G.T.; Zappala, Z.; Strober, B.J.; Scott, A.J.; et al. The impact of rare variation on gene expression across tissues. Nature 2017, 550, 239–243. [Google Scholar] [CrossRef]
- Huyghe, J.R.; Bien, S.A.; Harrison, T.A.; Kang, H.M.; Chen, S.; Schmit, S.L.; Conti, D.V.; Qu, C.; Jeon, J.; Edlund, C.K.; et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019, 51, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Luo, Y.; Sun, Y.; Guo, W.; Zhao, X.; Xi, Y.; Ma, Y.; Shao, M.; Tan, W.; Gao, G.; et al. Genomic and transcriptomic alterations associated with drug vulnerabilities and prognosis in adenocarcinoma at the gastroesophageal junction. Nat. Commun. 2020, 11, 6091. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Tian, T.; Zhu, L.; Zhou, H.; Hu, H.; Liu, Y.; Li, X.; Hu, F.; Zhao, Y.; Wang, G. Copy number variation of E3 ubiquitin ligase genes in peripheral blood leukocyte and colorectal cancer. Sci. Rep. 2016, 6, 29869. [Google Scholar] [CrossRef][Green Version]
- Mamlouk, S.; Childs, L.H.; Aust, D.; Heim, D.; Melching, F.; Oliveira, C.; Wolf, T.; Durek, P.; Schumacher, D.; Blaker, H.; et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017, 8, 14093. [Google Scholar] [CrossRef] [PubMed]
- Fagny, M.; Platig, J.; Kuijjer, M.L.; Lin, X.; Quackenbush, J. Nongenic cancer-risk SNPs affect oncogenes, tumour-suppressor genes, and immune function. Br. J. Cancer 2020, 122, 569–577. [Google Scholar] [CrossRef]
- Liu, X.Q.; Liu, X.S.; Rong, J.Y.; Gao, F.; Wu, Y.D.; Deng, C.H.; Jiang, H.Y.; Li, X.F.; Chen, Y.Q.; Zhao, Z.G.; et al. Precise diagnosis of three top cancers using dbGaP data. Sci. Rep. 2021, 11, 823. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Song, J.; Lu, Y.; Fall, K.; Chen, X.; Fang, F.; Landen, M.; Hultman, C.M.; Czene, K.; Sullivan, P.; et al. A shared genetic contribution to breast cancer and schizophrenia. Nat. Commun. 2020, 11, 4637. [Google Scholar] [CrossRef]
- Zhong, Q.; Lu, M.; Yuan, W.; Cui, Y.; Ouyang, H.; Fan, Y.; Wang, Z.; Wu, C.; Qiao, J.; Hang, J. Eight-lncRNA signature of cervical cancer were identified by integrating DNA methylation, copy number variation and transcriptome data. J. Transl. Med. 2021, 19, 58. [Google Scholar] [CrossRef]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Frose, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Poulos, R.C.; Sloane, M.A.; Hesson, L.B.; Wong, J.W. The search for cis-regulatory driver mutations in cancer genomes. Oncotarget 2015, 6, 32509–32525. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Muratani, M.; Deng, N.; Ooi, W.F.; Lin, S.J.; Xing, M.; Xu, C.; Qamra, A.; Tay, S.T.; Malik, S.; Wu, J.; et al. Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nat. Commun. 2014, 5, 4361. [Google Scholar] [CrossRef]
- Liu, J.; Adhav, R.; Miao, K.; Su, S.M.; Mo, L.; Chan, U.I.; Zhang, X.; Xu, J.; Li, J.; Shu, X.; et al. Characterization of BRCA1-deficient premalignant tissues and cancers identifies Plekha5 as a tumor metastasis suppressor. Nat. Commun. 2020, 11, 4875. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Estecio, M.R.; Chen, R.; Reuben, A.; Wang, L.; Fujimoto, J.; Carrot-Zhang, J.; McGranahan, N.; Ying, L.; Fukuoka, J.; et al. Evolution of DNA methylome from precancerous lesions to invasive lung adenocarcinomas. Nat. Commun. 2021, 12, 687. [Google Scholar] [CrossRef]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Cancer Genome Atlas Research, N.; Liang, H. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386.e312–399.e312. [Google Scholar] [CrossRef]
- Lidschreiber, K.; Jung, L.A.; von der Emde, H.; Dave, K.; Taipale, J.; Cramer, P.; Lidschreiber, M. Transcriptionally active enhancers in human cancer cells. Mol. Syst. Biol. 2021, 17, e9873. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liao, Z.; Gritsch, D.; Hadzhiev, Y.; Bai, Y.; Locascio, J.J.; Guennewig, B.; Liu, G.; Blauwendraat, C.; Wang, T.; et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat. Neurosci. 2018, 21, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Garieri, M.; Delaneau, O.; Santoni, F.; Fish, R.J.; Mull, D.; Carninci, P.; Dermitzakis, E.T.; Antonarakis, S.E.; Fort, A. The effect of genetic variation on promoter usage and enhancer activity. Nat. Commun. 2017, 8, 1358. [Google Scholar] [CrossRef]
- Elkon, R.; Agami, R. Characterization of noncoding regulatory DNA in the human genome. Nat. Biotechnol. 2017, 35, 732–746. [Google Scholar] [CrossRef]
- Thandapani, P. Super-enhancers in cancer. Pharmacol. Ther. 2019, 199, 129–138. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Peng, J.; Su, Z.; Li, Z.; Zhang, B.; Ma, J.; Zhuo, M.; Zou, D.; Liu, X.; et al. Mutant Kras co-opts a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells to initiate pancreatic cancer. Nature Cancer 2021, 2, 49–65. [Google Scholar] [CrossRef]
- Chen, R.; Ren, S.; Sun, Y. Genome-wide association studies on prostate cancer: The end or the beginning? Protein Cell 2013, 4, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet. 2013, 22, 408–415. [Google Scholar] [CrossRef]
- Shui, I.M.; Lindstrom, S.; Kibel, A.S.; Berndt, S.I.; Campa, D.; Gerke, T.; Penney, K.L.; Albanes, D.; Berg, C.; Bueno-de-Mesquita, H.B.; et al. Prostate cancer (PCa) risk variants and risk of fatal PCa in the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. Eur. Urol. 2014, 65, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and Clinical Implications of the 19q13 Aggressive Prostate Cancer Susceptibility Locus. Cell 2018, 174, 576.e518–589.e518. [Google Scholar] [CrossRef]
- Shang, Z.; Yu, J.; Sun, L.; Tian, J.; Zhu, S.; Zhang, B.; Dong, Q.; Jiang, N.; Flores-Morales, A.; Chang, C.; et al. LncRNA PCAT1 activates AKT and NF-kappaB signaling in castration-resistant prostate cancer by regulating the PHLPP/FKBP51/IKKalpha complex. Nucleic Acids Res. 2019, 47, 4211–4225. [Google Scholar] [CrossRef]
- Song, Y.H.; Shiota, M.; Kuroiwa, K.; Naito, S.; Oda, Y. The important role of glycine N-methyltransferase in the carcinogenesis and progression of prostate cancer. Mod. Pathol 2011, 24, 1272–1280. [Google Scholar] [CrossRef]
- Bonaccorsi, L.; Luciani, P.; Nesi, G.; Mannucci, E.; Deledda, C.; Dichiara, F.; Paglierani, M.; Rosati, F.; Masieri, L.; Serni, S.; et al. Androgen receptor regulation of the seladin-1/DHCR24 gene: Altered expression in prostate cancer. Lab. Investig. 2008, 88, 1049–1056. [Google Scholar] [CrossRef]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010, 20, 1191–1197. [Google Scholar] [CrossRef]
- Wright, J.B.; Brown, S.J.; Cole, M.D. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Mol. Cell Biol. 2010, 30, 1411–1420. [Google Scholar] [CrossRef]
- Doni Jayavelu, N.; Jajodia, A.; Mishra, A.; Hawkins, R.D. Candidate silencer elements for the human and mouse genomes. Nat. Commun. 2020, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Snyder, M.P. Systematic identification of silencers in human cells. Nat. Genet. 2020, 52, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Dunning, A.M.; Michailidou, K.; Kuchenbaecker, K.B.; Thompson, D.; French, J.D.; Beesley, J.; Healey, C.S.; Kar, S.; Pooley, K.A.; Lopez-Knowles, E.; et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 2016, 48, 374–386. [Google Scholar] [CrossRef]
- Jing, H.; Vakoc, C.R.; Ying, L.; Mandat, S.; Wang, H.; Zheng, X.; Blobel, G.A. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol. Cell 2008, 29, 232–242. [Google Scholar] [CrossRef]
- Grotsch, B.; Brachs, S.; Lang, C.; Luther, J.; Derer, A.; Schlotzer-Schrehardt, U.; Bozec, A.; Fillatreau, S.; Berberich, I.; Hobeika, E.; et al. The AP-1 transcription factor Fra1 inhibits follicular B cell differentiation into plasma cells. J. Exp. Med. 2014, 211, 2199–2212. [Google Scholar] [CrossRef]
- Hadjiagapiou, C.; Borthakur, A.; Dahdal, R.Y.; Gill, R.K.; Malakooti, J.; Ramaswamy, K.; Dudeja, P.K. Role of USF1 and USF2 as potential repressor proteins for human intestinal monocarboxylate transporter 1 promoter. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1118–G1126. [Google Scholar] [CrossRef]
- Nakayama, A.; Murakami, H.; Maeyama, N.; Yamashiro, N.; Sakakibara, A.; Mori, N.; Takahashi, M. Role for RFX transcription factors in non-neuronal cell-specific inactivation of the microtubule-associated protein MAP1A promoter. J. Biol. Chem 2003, 278, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Timblin, G.A.; Schlissel, M.S. Ebf1 and c-Myb repress rag transcription downstream of Stat5 during early B cell development. J. Immunol. 2013, 191, 4676–4687. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, S.; Unno, M.; Muto, A.; Takeuchi, A.; Kometani, K.; Kurosaki, T.; Igarashi, K.; Saito, T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. USA 2013, 110, 10735–10740. [Google Scholar] [CrossRef]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-binding specificities of human transcription factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef]
- Gaszner, M.; Felsenfeld, G. Insulators: Exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 2006, 7, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Maurano, M.T.; Wang, H.; Qi, H.; Song, C.Z.; Navas, P.A.; Emery, D.W.; Stamatoyannopoulos, J.A.; Stamatoyannopoulos, G. Genomic discovery of potent chromatin insulators for human gene therapy. Nat. Biotechnol. 2015, 33, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lou, D.; Wang, Z. Crosstalk of Genetic Variants, Allele-Specific DNA Methylation, and Environmental Factors for Complex Disease Risk. Front. Genet. 2018, 9, 695. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635–110649. [Google Scholar] [CrossRef]
- Dai, J.; Zhu, M.; Wang, C.; Shen, W.; Zhou, W.; Sun, J.; Liu, J.; Jin, G.; Ma, H.; Hu, Z.; et al. Systematical analyses of variants in CTCF-binding sites identified a novel lung cancer susceptibility locus among Chinese population. Sci. Rep. 2015, 5, 7833. [Google Scholar] [CrossRef]
- Wang, J.; Guan, X.; Zhang, Y.; Ge, S.; Zhang, L.; Li, H.; Wang, X.; Liu, R.; Ning, T.; Deng, T.; et al. Exosomal miR-27a Derived from Gastric Cancer Cells Regulates the Transformation of Fibroblasts into Cancer-Associated Fibroblasts. Cell Physiol. Biochem. 2018, 49, 869–883. [Google Scholar] [CrossRef]
- Zhou, L.; Liang, X.; Zhang, L.; Yang, L.; Nagao, N.; Wu, H.; Liu, C.; Lin, S.; Cai, G.; Liu, J. MiR-27a-3p functions as an oncogene in gastric cancer by targeting BTG2. Oncotarget 2016, 7, 51943–51954. [Google Scholar] [CrossRef]
- Kim, J.H.; Hwang, J.; Jung, J.H.; Lee, H.J.; Lee, D.Y.; Kim, S.H. Molecular networks of FOXP family: Dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol. Cancer 2019, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Qi, Y.; Lai, Y.L.; Shen, P.C.; Chen, F.H.; Lin, L.J.; Wu, H.H.; Peng, P.H.; Hsu, K.W.; Cheng, W.C. Identification and validation of a miRNA-based prognostic signature for cervical cancer through an integrated bioinformatics approach. Sci. Rep. 2020, 10, 22270. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Robles, A.I.; Harris, C.C. Genetic variation in microRNA networks: The implications for cancer research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Scott, J.G.; Harris, A.L.; Buffa, F.M. Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat. Commun. 2018, 9, 5228. [Google Scholar] [CrossRef]
- Lin, W.C.; Shih, P.H.; Wang, W.; Wu, C.H.; Hsia, S.M.; Wang, H.J.; Hwang, P.A.; Wang, C.Y.; Chen, S.H.; Kuo, Y.T. Inhibitory effects of high stability fucoxanthin on palmitic acid-induced lipid accumulation in human adipose-derived stem cells through modulation of long non-coding RNA. Food Funct. 2015, 6, 2215–2223. [Google Scholar] [CrossRef]
- Lee, A.R.; Park, J.; Jung, K.J.; Jee, S.H.; Kim-Yoon, S. Genetic variation rs7930 in the miR-4273-5p target site is associated with a risk of colorectal cancer. Onco. Targets Ther. 2016, 9, 6885–6895. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, M.; Du, M.; Ma, L.; Chu, H.; Lv, Q.; Ye, D.; Guo, J.; Gu, C.; Xia, G.; Zhu, Y.; et al. A functional variant in TP63 at 3q28 associated with bladder cancer risk by creating an miR-140-5p binding site. Int. J. Cancer 2016, 139, 65–74. [Google Scholar] [CrossRef]
- Wynendaele, J.; Bohnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3’ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef]
- Jacinta-Fernandes, A.; Xavier, J.M.; Magno, R.; Lage, J.G.; Maia, A.T. Allele-specific miRNA-binding analysis identifies candidate target genes for breast cancer risk. NPJ Genom. Med. 2020, 5, 4. [Google Scholar] [CrossRef]
- Hua, K.T.; Liu, Y.F.; Hsu, C.L.; Cheng, T.Y.; Yang, C.Y.; Chang, J.S.; Lee, W.J.; Hsiao, M.; Juan, H.F.; Chien, M.H.; et al. 3’UTR polymorphisms of carbonic anhydrase IX determine the miR-34a targeting efficiency and prognosis of hepatocellular carcinoma. Sci. Rep. 2017, 7, 4466. [Google Scholar] [CrossRef]
- Hogg, D.R.; Harries, L.W. Human genetic variation and its effect on miRNA biogenesis, activity and function. Biochem. Soc. Trans. 2014, 42, 1184–1189. [Google Scholar] [CrossRef]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef]
- Gould, P.S.; Bird, H.; Easton, A.J. Translation toeprinting assays using fluorescently labeled primers and capillary electrophoresis. Biotechniques 2005, 38, 397–400. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ambrosone, C.B.; Zhao, H. Novel genetic variants in microRNA genes and familial breast cancer. Int. J. Cancer 2009, 124, 1178–1182. [Google Scholar] [CrossRef] [PubMed]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1393. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, H.; Maheronnaghsh, M.; Molaei, F.; Mashouri, L.; Reza Aref, A.; Momeny, M.; Alahari, S.K. Long noncoding RNAs and exosomal lncRNAs: Classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene 2020, 39, 953–974. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.S.; Somvanshi, S.; Patel, E.; Chen, T.W.; Singh, V.P.; Zorman, B.; Patil, S.L.; Pan, Y.; Chatterjee, S.S.; Cancer Genome Atlas Research, N.; et al. Pan-Cancer Analysis of lncRNA Regulation Supports Their Targeting of Cancer Genes in Each Tumor Context. Cell Rep. 2018, 23, 297.e212–312.e212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Z.; Liu, H.; Chen, S.R. Mechanisms of Long Non-Coding RNAs in Cancers and Their Dynamic Regulations. Cancers 2020, 12, 1245. [Google Scholar] [CrossRef]
- Begolli, R.; Sideris, N.; Giakountis, A. LncRNAs as Chromatin Regulators in Cancer: From Molecular Function to Clinical Potential. Cancers 2019, 11, 1524. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Kanhere, A. The Missing Link Between Cancer-Associated Variants and LncRNAs. Trends Genet. 2021, 37, 410–413. [Google Scholar] [CrossRef]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef]
- Johnsson, P.; Lipovich, L.; Grander, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef]
- Pisignano, G.; Ladomery, M. Post-Transcriptional Regulation through Long Non-Coding RNAs (lncRNAs). Noncoding RNA 2021, 7, 29. [Google Scholar] [CrossRef]
- Liz, J.; Portela, A.; Soler, M.; Gomez, A.; Ling, H.; Michlewski, G.; Calin, G.A.; Guil, S.; Esteller, M. Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol. Cell 2014, 55, 138–147. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Proudfoot, N.J.; Jopling, C.L. Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat. Struct. Mol. Biol. 2015, 22, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long Non-coding RNA Structure and Function: Is There a Link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Aznaourova, M.; Schmerer, N.; Schmeck, B.; Schulte, L.N. Disease-Causing Mutations and Rearrangements in Long Non-coding RNA Gene Loci. Front. Genet. 2020, 11, 527484. [Google Scholar] [CrossRef]
- Gao, P.; Wei, G.H. Genomic Insight into the Role of lncRNA in Cancer Susceptibility. Int. J. Mol. Sci. 2017, 18, 1239. [Google Scholar] [CrossRef]
- Tang, X.; Feng, D.; Li, M.; Zhou, J.; Li, X.; Zhao, D.; Hao, B.; Li, D.; Ding, K. Transcriptomic Analysis of mRNA-lncRNA-miRNA Interactions in Hepatocellular Carcinoma. Sci. Rep. 2019, 9, 16096. [Google Scholar] [CrossRef]
- Xia, T.; Liao, Q.; Jiang, X.; Shao, Y.; Xiao, B.; Xi, Y.; Guo, J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Cai, G.; Kong, L.; Zhang, T.; Ren, Y.; Wu, Y.; Mei, M.; Zhang, L.; Wang, X. Long Non Coding RNA MALAT1 Promotes Tumor Growth and Metastasis by inducing Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma. Sci. Rep. 2015, 5, 15972. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, X.; Qiao, L.; Liu, W.; Xu, C.; Wang, X. MALAT1 regulates miR-34a expression in melanoma cells. Cell Death Dis. 2019, 10, 389. [Google Scholar] [CrossRef]
- Han, Y.; Wu, Z.; Wu, T.; Huang, Y.; Cheng, Z.; Li, X.; Sun, T.; Xie, X.; Zhou, Y.; Du, Z. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016, 7, e2123. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Z.; Zhao, Y.; Jin, Y.; An, L.; Wu, B.; Liu, Z.; Chen, X.; Chen, X.; Zhou, H.; et al. Genetic polymorphisms of lncRNA-p53 regulatory network genes are associated with concurrent chemoradiotherapy toxicities and efficacy in nasopharyngeal carcinoma patients. Sci. Rep. 2017, 7, 8320. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Liu, C.; Li, H.; Zhang, L.; Luo, G.; Liang, S.; Lu, M. Research progress on the interactions between long non-coding RNAs and microRNAs in human cancer. Oncol. Lett. 2020, 19, 595–605. [Google Scholar] [CrossRef]
- Jin, N.; Bi, A.; Lan, X.; Xu, J.; Wang, X.; Liu, Y.; Wang, T.; Tang, S.; Zeng, H.; Chen, Z.; et al. Identification of metabolic vulnerabilities of receptor tyrosine kinases-driven cancer. Nat. Commun. 2019, 10, 2701. [Google Scholar] [CrossRef]
- Lo, R.S. Receptor tyrosine kinases in cancer escape from BRAF inhibitors. Cell Res. 2012, 22, 945–947. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Brown, B.P.; Kim, S.; Ferguson, D.; Pavlick, D.C.; Jayakumaran, G.; Benayed, R.; Gallant, J.N.; Zhang, Y.K.; Yan, Y.; et al. Structure-function analysis of oncogenic EGFR Kinase Domain Duplication reveals insights into activation and a potential approach for therapeutic targeting. Nat. Commun. 2021, 12, 1382. [Google Scholar] [CrossRef] [PubMed]
- Banys-Paluchowski, M.; Witzel, I.; Riethdorf, S.; Rack, B.; Janni, W.; Fasching, P.A.; Solomayer, E.F.; Aktas, B.; Kasimir-Bauer, S.; Pantel, K.; et al. Evaluation of serum epidermal growth factor receptor (EGFR) in correlation to circulating tumor cells in patients with metastatic breast cancer. Sci. Rep. 2017, 7, 17307. [Google Scholar] [CrossRef]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Nanjo, S.; Ishikawa, D.; Sano, T.; Kita, K.; Nakamura, T.; Matsumoto, K.; Suda, K.; et al. Combined therapy with mutant-selective EGFR inhibitor and Met kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol. Cancer Ther. 2012, 11, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Neto, J.M.; Nadal, E.; Bosdriesz, E.; Ooft, S.N.; Farre, L.; McLean, C.; Klarenbeek, S.; Jurgens, A.; Hagen, H.; Wang, L.; et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat. Commun. 2020, 11, 3157. [Google Scholar] [CrossRef]
- Gyorffy, B.; Pongor, L.; Bottai, G.; Li, X.; Budczies, J.; Szabo, A.; Hatzis, C.; Pusztai, L.; Santarpia, L. An integrative bioinformatics approach reveals coding and non-coding gene variants associated with gene expression profiles and outcome in breast cancer molecular subtypes. Br. J. Cancer 2018, 118, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Soltis, A.R.; Dalgard, C.L.; Pollard, H.B.; Wilkerson, M.D. MutEnricher: A flexible toolset for somatic mutation enrichment analysis of tumor whole genomes. BMC Bioinform. 2020, 21, 338. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Fulco, C.P.; Nasser, J.; Jones, T.R.; Munson, G.; Bergman, D.T.; Subramanian, V.; Grossman, S.R.; Anyoha, R.; Doughty, B.R.; Patwardhan, T.A.; et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019, 51, 1664–1669. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Furey, T.S. Identifying and characterizing regulatory sequences in the human genome with chromatin accessibility assays. Genes 2012, 3, 651–670. [Google Scholar] [CrossRef] [PubMed]
- Fadason, T.; Schierding, W.; Lumley, T.; O’Sullivan, J.M. Chromatin interactions and expression quantitative trait loci reveal genetic drivers of multimorbidities. Nat. Commun. 2018, 9, 5198. [Google Scholar] [CrossRef] [PubMed]
- Keele, G.R.; Quach, B.C.; Israel, J.W.; Chappell, G.A.; Lewis, L.; Safi, A.; Simon, J.M.; Cotney, P.; Crawford, G.E.; Valdar, W.; et al. Integrative QTL analysis of gene expression and chromatin accessibility identifies multi-tissue patterns of genetic regulation. PLoS Genet. 2020, 16, e1008537. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Rendon, A.; Wernisch, L. Transcription factor and chromatin features predict genes associated with eQTLs. Nucleic Acids Res. 2013, 41, 1450–1463. [Google Scholar] [CrossRef]
- Kim, K.; Jang, I.; Kim, M.; Choi, J.; Kim, M.S.; Lee, B.; Jung, I. 3DIV update for 2021: A comprehensive resource of 3D genome and 3D cancer genome. Nucleic Acids Res. 2021, 49, D38–D46. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 1980, 286, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Chen, D.; Chen, Y. Advances of DNase-seq for mapping active gene regulatory elements across the genome in animals. Gene 2018, 667, 83–94. [Google Scholar] [CrossRef]
- Kumar, V.; Muratani, M.; Rayan, N.A.; Kraus, P.; Lufkin, T.; Ng, H.H.; Prabhakar, S. Uniform, optimal signal processing of mapped deep-sequencing data. Nat. Biotechnol. 2013, 31, 615–622. [Google Scholar] [CrossRef]
- Lu, F.; Liu, Y.; Inoue, A.; Suzuki, T.; Zhao, K.; Zhang, Y. Establishing Chromatin Regulatory Landscape during Mouse Preimplantation Development. Cell 2016, 165, 1375–1388. [Google Scholar] [CrossRef]
- Yan, H.; Tian, S.; Slager, S.L.; Sun, Z.; Ordog, T. Genome-Wide Epigenetic Studies in Human Disease: A Primer on -Omic Technologies. Am. J. Epidemiol. 2016, 183, 96–109. [Google Scholar] [CrossRef]
- Zentner, G.E.; Henikoff, S. Surveying the epigenomic landscape, one base at a time. Genome Biol. 2012, 13, 250. [Google Scholar] [CrossRef]
- Guo, F.; Li, L.; Li, J.; Wu, X.; Hu, B.; Zhu, P.; Wen, L.; Tang, F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017, 27, 967–988. [Google Scholar] [CrossRef] [PubMed]
- D’antonio, M.; Weghorn, D.; D’antonio-Chronowska, A.; Coulet, F.; Olson, K.M.; DeBoever, C.; Drees, F.; Arias, A.; Alakus, H.; Richardson, A.L.; et al. Identifying DNase I hypersensitive sites as driver distal regulatory elements in breast cancer. Nat. Commun. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Tang, Q.; Wan, M.; Cui, K.; Zhang, Y.; Ren, G.; Ni, B.; Sklar, J.; Przytycka, T.M.; Childs, R.; et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528, 142–146. [Google Scholar] [CrossRef]
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267. [Google Scholar] [CrossRef]
- Schones, D.E.; Cui, K.; Cuddapah, S.; Roh, T.Y.; Barski, A.; Wang, Z.; Wei, G.; Zhao, K. Dynamic regulation of nucleosome positioning in the human genome. Cell 2008, 132, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.C.; Hainer, S.J. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res. 2020, 28, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Cui, K.; Zhao, K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol. Biol. 2012, 833, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Shashikant, T.; Ettensohn, C.A. Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 2019, 151, 219–235. [Google Scholar] [CrossRef]
- Bryois, J.; Garrett, M.E.; Song, L.; Safi, A.; Giusti-Rodriguez, P.; Johnson, G.D.; Shieh, A.W.; Buil, A.; Fullard, J.F.; Roussos, P.; et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun. 2018, 9, 3121. [Google Scholar] [CrossRef]
- Kheradpour, P.; Ernst, J.; Melnikov, A.; Rogov, P.; Wang, L.; Zhang, X.; Alston, J.; Mikkelsen, T.S.; Kellis, M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013, 23, 800–811. [Google Scholar] [CrossRef]
- Kwasnieski, J.C.; Mogno, I.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. USA 2012, 109, 19498–19503. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.Y.; Patwardhan, R.P.; Kim, M.J.; Findlay, G.M.; Martin, B.; Zhao, J.; Bell, R.J.; Smith, R.P.; Ku, A.A.; Shendure, J.; et al. Systematic dissection of coding exons at single nucleotide resolution supports an additional role in cell-specific transcriptional regulation. PLoS Genet. 2014, 10, e1004592. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.; Murugan, A.; Zhang, X.; Tesileanu, T.; Wang, L.; Rogov, P.; Feizi, S.; Gnirke, A.; Callan, C.G., Jr.; Kinney, J.B.; et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012, 30, 271–277. [Google Scholar] [CrossRef]
- Patwardhan, R.P.; Hiatt, J.B.; Witten, D.M.; Kim, M.J.; Smith, R.P.; May, D.; Lee, C.; Andrie, J.M.; Lee, S.I.; Cooper, G.M.; et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012, 30, 265–270. [Google Scholar] [CrossRef]
- White, M.A.; Myers, C.A.; Corbo, J.C.; Cohen, B.A. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc. Natl. Acad. Sci. USA 2013, 110, 11952–11957. [Google Scholar] [CrossRef]
- Arnold, C.D.; Gerlach, D.; Stelzer, C.; Boryn, L.M.; Rath, M.; Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013, 339, 1074–1077. [Google Scholar] [CrossRef]
- Muerdter, F.; Boryn, L.M.; Arnold, C.D. STARR-seq - principles and applications. Genomics 2015, 106, 145–150. [Google Scholar] [CrossRef]
- Klein, J.C.; Keith, A.; Rice, S.J.; Shepherd, C.; Agarwal, V.; Loughlin, J.; Shendure, J. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019, 10, 2434. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, J.H.; Zhu, J.; Gao, P.; Tian, Y.J.; Du, M.; Guo, Y.C.; Suleman, S.; Zhang, Q.; Kohli, M.; et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 2022. [Google Scholar] [CrossRef] [PubMed]
- Vanhille, L.; Griffon, A.; Maqbool, M.A.; Zacarias-Cabeza, J.; Dao, L.T.; Fernandez, N.; Ballester, B.; Andrau, J.C.; Spicuglia, S. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat. Commun. 2015, 6, 6905. [Google Scholar] [CrossRef]
- Arnold, C.D.; Gerlach, D.; Spies, D.; Matts, J.A.; Sytnikova, Y.A.; Pagani, M.; Lau, N.C.; Stark, A. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat. Genet. 2014, 46, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.D.; Barrera, A.; McDowell, I.C.; D’Ippolito, A.M.; Majoros, W.H.; Vockley, C.M.; Wang, X.; Allen, A.S.; Reddy, T.E. Human genome-wide measurement of drug-responsive regulatory activity. Nat. Commun. 2018, 9, 5317. [Google Scholar] [CrossRef]
- Gong, H.; Yang, Y.; Zhang, S.; Li, M.; Zhang, X. Application of Hi-C and other omics data analysis in human cancer and cell differentiation research. Comput. Struct. Biotechnol. J. 2021, 19, 2070–2083. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Ruan, Y. ChIP-based methods for the identification of long-range chromatin interactions. J. Cell Biochem. 2009, 107, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, K. The Toolbox for Untangling Chromosome Architecture in Immune Cells. Front. Immunol. 2021, 12, 670884. [Google Scholar] [CrossRef]
- Li, G.; Cai, L.; Chang, H.; Hong, P.; Zhou, Q.; Kulakova, E.V.; Kolchanov, N.A.; Ruan, Y. Chromatin Interaction Analysis with Paired-End Tag (ChIA-PET) sequencing technology and application. BMC Genom. 2014, 15 (Suppl. S12), S11. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Luo, O.J.; Wang, P.; Zheng, M.; Wang, D.; Piecuch, E.; Zhu, J.J.; Tian, S.Z.; Tang, Z.; Li, G.; et al. Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nat. Protoc. 2017, 12, 899–915. [Google Scholar] [CrossRef]
- Al Bkhetan, Z.; Plewczynski, D. Three-dimensional Epigenome Statistical Model: Genome-wide Chromatin Looping Prediction. Sci. Rep. 2018, 8, 5217. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef]
- Xi, W.; Beer, M.A. Loop competition and extrusion model predicts CTCF interaction specificity. Nat. Commun. 2021, 12, 1046. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Kuchenbaecker, K.B.; Michailidou, K.; Mendoza-Fandino, G.A.; Nord, S.; Lilyquist, J.; Olswold, C.; Hallberg, E.; Agata, S.; Ahsan, H.; et al. Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat. Commun. 2016, 7, 11375. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Kuchenbaecker, K.B.; Michailidou, K.; Beesley, J.; Kar, S.; Lindstrom, S.; Hui, S.; Lemacon, A.; Soucy, P.; Dennis, J.; et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 2017, 49, 1767–1778. [Google Scholar] [CrossRef]
- Cai, L.; Chang, H.; Fang, Y.; Li, G. A Comprehensive Characterization of the Function of LincRNAs in Transcriptional Regulation Through Long-Range Chromatin Interactions. Sci. Rep. 2016, 6, 36572. [Google Scholar] [CrossRef] [PubMed]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Chandra, V.; Vijayanand, P.; Ay, F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 2019, 10, 4221. [Google Scholar] [CrossRef]
- Ray, J.P.; de Boer, C.G.; Fulco, C.P.; Lareau, C.A.; Kanai, M.; Ulirsch, J.C.; Tewhey, R.; Ludwig, L.S.; Reilly, S.K.; Bergman, D.T.; et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat. Commun. 2020, 11, 1237. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Spurdle, A.B.; Glubb, D.M.; Endometrial Cancer Association, C. Analysis of Promoter-Associated Chromatin Interactions Reveals Biologically Relevant Candidate Target Genes at Endometrial Cancer Risk Loci. Cancers 2019, 11, 1440. [Google Scholar] [CrossRef]
- Fang, R.; Yu, M.; Li, G.; Chee, S.; Liu, T.; Schmitt, A.D.; Ren, B. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 2016, 26, 1345–1348. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Fresard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Deng, X.; Kong, F.; Li, S.; Jiang, H.; Dong, L.; Xu, X.; Zhang, X.; Yuan, H.; Xu, Y.; Chu, Y.; et al. A KLF4/PiHL/EZH2/HMGA2 regulatory axis and its function in promoting oxaliplatin-resistance of colorectal cancer. Cell Death Dis. 2021, 12, 485. [Google Scholar] [CrossRef]
- Feng, Y.C.; Liu, X.Y.; Teng, L.; Ji, Q.; Wu, Y.; Li, J.M.; Gao, W.; Zhang, Y.Y.; La, T.; Tabatabaee, H.; et al. c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis. Nat. Commun. 2020, 11, 4980. [Google Scholar] [CrossRef]
- Xu, L.; Huan, L.; Guo, T.; Wu, Y.; Liu, Y.; Wang, Q.; Huang, S.; Xu, Y.; Liang, L.; He, X. LncRNA SNHG11 facilitates tumor metastasis by interacting with and stabilizing HIF-1alpha. Oncogene 2020, 39, 7005–7018. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed]
- Feretzaki, M.; Pospisilova, M.; Valador Fernandes, R.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Laffleur, B.; Lim, J.; Zhang, W.; Chen, Y.; Pefanis, E.; Bizarro, J.; Batista, C.R.; Wu, L.; Economides, A.N.; Wang, J.; et al. Noncoding RNA processing by DIS3 regulates chromosomal architecture and somatic hypermutation in B cells. Nat. Genet. 2021, 53, 230–242. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Nguyen, T.C.; Zaleta-Rivera, K.; Huang, X.; Dai, X.; Zhong, S. RNA, Action through Interactions. Trends Genet. 2018, 34, 867–882. [Google Scholar] [CrossRef]
- Kato, M.; Carninci, P. Genome-Wide Technologies to Study RNA-Chromatin Interactions. Noncoding RNA 2020, 6, 20. [Google Scholar] [CrossRef]
- Machyna, M.; Simon, M.D. Catching RNAs on chromatin using hybridization capture methods. Brief. Funct. Genom. 2018, 17, 96–103. [Google Scholar] [CrossRef]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Yan, Z.; Nguyen, T.C.; Bouman Chen, Z.; Chien, S.; Zhong, S. Mapping RNA-chromatin interactions by sequencing with iMARGI. Nat. Protoc. 2019, 14, 3243–3272. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.T.; Li, H.; Fu, X.D. GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef]
- Zhou, B.; Li, X.; Luo, D.; Lim, D.H.; Zhou, Y.; Fu, X.D. GRID-seq for comprehensive analysis of global RNA-chromatin interactions. Nat. Protoc. 2019, 14, 2036–2068. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Jukam, D.; Teran, N.A.; Risca, V.I.; Smith, O.K.; Johnson, W.L.; Skotheim, J.M.; Greenleaf, W.J.; Straight, A.F. Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.; Lander, E.S.; Guttman, M. RNA antisense purification (RAP) for mapping RNA interactions with chromatin. Methods Mol. Biol. 2015, 1262, 183–197. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; D’Onorio De Meo, P.; Pallocca, M.; Picardi, E.; D’Erchia, A.M.; Calogero, R.A.; Castrignano, T.; Pesole, G. RAP: RNA-Seq Analysis Pipeline, a new cloud-based NGS web application. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef]
- Grillone, K.; Riillo, C.; Scionti, F.; Rocca, R.; Tradigo, G.; Guzzi, P.H.; Alcaro, S.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Non-coding RNAs in cancer: Platforms and strategies for investigating the genomic “dark matter”. J. Exp. Clin. Cancer Res. 2020, 39, 117. [Google Scholar] [CrossRef]
- Weidmann, C.A.; Mustoe, A.M.; Jariwala, P.B.; Calabrese, J.M.; Weeks, K.M. Analysis of RNA-protein networks with RNP-MaP defines functional hubs on RNA. Nat. Biotechnol. 2021, 39, 347–356. [Google Scholar] [CrossRef]
- Yi, W.; Li, J.; Zhu, X.; Wang, X.; Fan, L.; Sun, W.; Liao, L.; Zhang, J.; Li, X.; Ye, J.; et al. CRISPR-assisted detection of RNA-protein interactions in living cells. Nat. Methods 2020, 17, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, K.A.; Chaudhari, H.G.; Cohen, B.A.; Djuranovic, S. PTRE-seq reveals mechanism and interactions of RNA binding proteins and miRNAs. Nat. Commun. 2018, 9, 301. [Google Scholar] [CrossRef]
- Wan, Y.; Qu, K.; Ouyang, Z.; Chang, H.Y. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat. Protoc. 2013, 8, 849–869. [Google Scholar] [CrossRef]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Solomon, O.; Di Segni, A.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1440. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Hou, G.; Harley, I.T.W.; Lu, X.; Zhou, T.; Xu, N.; Yao, C.; Qin, Y.; Ouyang, Y.; Ma, J.; Zhu, X.; et al. SLE non-coding genetic risk variant determines the epigenetic dysfunction of an immune cell specific enhancer that controls disease-critical microRNA expression. Nat. Commun. 2021, 12, 135. [Google Scholar] [CrossRef]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef]
- Palin, K.; Pitkanen, E.; Turunen, M.; Sahu, B.; Pihlajamaa, P.; Kivioja, T.; Kaasinen, E.; Valimaki, N.; Hanninen, U.A.; Cajuso, T.; et al. Contribution of allelic imbalance to colorectal cancer. Nat. Commun. 2018, 9, 3664. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Soares, F.; Xia, J.H.; Yang, Y.; Li, J.; Guo, H.; Su, P.; Tian, Y.; Lee, H.J.; Wang, M.; et al. CRISPRi screens reveal a DNA methylation-mediated 3D genome dependent causal mechanism in prostate cancer. Nat. Commun. 2021, 12, 1781. [Google Scholar] [CrossRef]
- Nichols, C.A.; Gibson, W.J.; Brown, M.S.; Kosmicki, J.A.; Busanovich, J.P.; Wei, H.; Urbanski, L.M.; Curimjee, N.; Berger, A.C.; Gao, G.F.; et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020, 11, 2517. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Smargon, A.A.; Shi, Y.J.; Yeo, G.W. RNA-targeting CRISPR systems from metagenomic discovery to transcriptomic engineering. Nat. Cell Biol. 2020, 22, 143–150. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Wagner, A.H.; Walsh, B.; Mayfield, G.; Tamborero, D.; Sonkin, D.; Krysiak, K.; Deu-Pons, J.; Duren, R.P.; Gao, J.; McMurry, J.; et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat. Genet. 2020, 52, 448–457. [Google Scholar] [CrossRef]
- Piraino, S.W.; Furney, S.J. Beyond the exome: The role of non-coding somatic mutations in cancer. Ann. Oncol. 2016, 27, 240–248. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Shen, S.; Chen, X.; Szlachta, K.; Edmonson, M.N.; Shao, Y.; Ma, X.; Hyle, J.; Wright, S.; et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat. Genet. 2020, 52, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Drubay, D.; Michiels, S.; Gautheret, D. Mining the coding and non-coding genome for cancer drivers. Cancer Lett. 2015, 369, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wu, L.X.; Tan, L.; Shang, F.F.; Zhou, H.H. Significance of Single-Nucleotide Variants in Long Intergenic Non-protein Coding RNAs. Front. Cell Dev. Biol. 2020, 8, 347. [Google Scholar] [CrossRef]
- Kikutake, C.; Yoshihara, M.; Suyama, M. Pan-cancer analysis of non-coding recurrent mutations and their possible involvement in cancer pathogenesis. NAR Cancer 2021, 3. [Google Scholar] [CrossRef]
- Pamula-Pilat, J.; Tecza, K.; Kalinowska-Herok, M.; Grzybowska, E. Genetic 3’UTR variations and clinical factors significantly contribute to survival prediction and clinical response in breast cancer patients. Sci. Rep. 2020, 10, 5736. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Hu, Y.; Jing, F.; Li, Y.; Gu, S.; Jiang, X.; Mao, Y.; Li, Q.; Jin, M.; Chen, K. A novel SNP in promoter region of RP11-3N2.1 is associated with reduced risk of colorectal cancer. J. Hum. Genet. 2018, 63, 47–54. [Google Scholar] [CrossRef]
- Wu, E.R.; Chou, Y.E.; Liu, Y.F.; Hsueh, K.C.; Lee, H.L.; Yang, S.F.; Su, S.C. Association of lncRNA H19 Gene Polymorphisms with the Occurrence of Hepatocellular Carcinoma. Genes 2019, 10, 506. [Google Scholar] [CrossRef]
- Wang, B.G.; Jiang, L.Y.; Xu, Q. Comprehensive assessment for miRNA polymorphisms in hepatocellular cancer risk: A systematic review and meta-analysis. BioSci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Endo, S.; Oguri, H.; Segawa, J.; Kawai, M.; Hu, D.; Xia, S.; Okada, T.; Irie, K.; Fujii, S.; Gouda, H.; et al. Development of Novel AKR1C3 Inhibitors as New Potential Treatment for Castration-Resistant Prostate Cancer. J. Med. Chem. 2020, 63, 10396–10411. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Mao, S.; Li, Q.; Sameni, M.; Krawetz, S.A.; Sloane, B.F.; Mattingly, R.R. RNA-Seq of human breast ductal carcinoma in situ models reveals aldehyde dehydrogenase isoform 5A1 as a novel potential target. PLoS ONE 2012, 7, e50249. [Google Scholar] [CrossRef]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Rawlings-Goss, R.A.; Campbell, M.C.; Tishkoff, S.A. Global population-specific variation in miRNA associated with cancer risk and clinical biomarkers. BMC Med. Genom. 2014, 7, 53. [Google Scholar] [CrossRef]
- Hoffman, A.E.; Liu, R.; Fu, A.; Zheng, T.; Slack, F.; Zhu, Y. Targetome profiling, pathway analysis and genetic association study implicate miR-202 in lymphomagenesis. Cancer Epidemiol. Biomark. Prev. 2013, 22, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Pipan, V.; Zorc, M.; Kunej, T. MicroRNA Polymorphisms in Cancer: A Literature Analysis. Cancers 2015, 7, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Feng, Y.; Zheng, Y.; Ogundiran, T.O.; Ojengbede, O.; Zheng, W.; Blot, W.; Ambrosone, C.B.; John, E.M.; Bernstein, L.; et al. Genetic variants in microRNA and microRNA biogenesis pathway genes and breast cancer risk among women of African ancestry. Hum. Genet. 2016, 135, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef] [PubMed]
- Belin, L.; Kamal, M.; Mauborgne, C.; Plancher, C.; Mulot, F.; Delord, J.P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; et al. Randomized phase II trial comparing molecularly targeted therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer: Cross-over analysis from the SHIVA trial. Ann. Oncol 2017, 28, 590–596. [Google Scholar] [CrossRef]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Rosello, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef] [PubMed]
- Vasconcellos, V.F.; Colli, L.M.; Awada, A.; de Castro Junior, G. Precision oncology: As much expectations as limitations. Ecancermedicalscience 2018, 12, ed86. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cowie, P.; Hay, E.A.; MacKenzie, A. The noncoding human genome and the future of personalised medicine. Expert Rev. Mol. Med. 2015, 17, e4. [Google Scholar] [CrossRef]
- Zhang, Z.; Gu, M.; Gu, Z.; Lou, Y.R. Role of Long Non-Coding RNA Polymorphisms in Cancer Chemotherapeutic Response. J. Pers. Med. 2021, 11, 513. [Google Scholar] [CrossRef]
- Lin, A.; Hu, Q.; Li, C.; Xing, Z.; Ma, G.; Wang, C.; Li, J.; Ye, Y.; Yao, J.; Liang, K.; et al. The LINK-A lncRNA interacts with PtdIns(3,4,5)P3 to hyperactivate AKT and confer resistance to AKT inhibitors. Nat. Cell Biol. 2017, 19, 238–251. [Google Scholar] [CrossRef]
- Meddens, C.A.; van der List, A.C.J.; Nieuwenhuis, E.E.S.; Mokry, M. Non-coding DNA in IBD: From sequence variation in DNA regulatory elements to novel therapeutic potential. Gut 2019, 68, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Yuan, S.; Xie, W.; Liu, Y.; Xiang, Y.; Wu, N.; Wu, L.; Ma, X.; Cai, T.; et al. Genetic predisposition to lung cancer: Comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci. Rep. 2017, 7, 8371. [Google Scholar] [CrossRef]
- Yan, T.; Shen, C.; Jiang, P.; Yu, C.; Guo, F.; Tian, X.; Zhu, X.; Lu, S.; Han, B.; Zhong, M.; et al. Risk SNP-induced lncRNA-SLCC1 drives colorectal cancer through activating glycolysis signaling. Signal Transduct. Target. Ther. 2021, 6, 70. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021, 184, 2239.e2239–2254.e2239. [Google Scholar] [CrossRef]
- Lu, Y.; Kweon, S.S.; Tanikawa, C.; Jia, W.H.; Xiang, Y.B.; Cai, Q.; Zeng, C.; Schmit, S.L.; Shin, A.; Matsuo, K.; et al. Large-Scale Genome-Wide Association Study of East Asians Identifies Loci Associated with Risk for Colorectal Cancer. Gastroenterology 2019, 156, 1455–1466. [Google Scholar] [CrossRef]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef]
- Lin, Y.; Nakatochi, M.; Hosono, Y.; Ito, H.; Kamatani, Y.; Inoko, A.; Sakamoto, H.; Kinoshita, F.; Kobayashi, Y.; Ishii, H.; et al. Genome-wide association meta-analysis identifies GP2 gene risk variants for pancreatic cancer. Nat. Commun. 2020, 11, 3175. [Google Scholar] [CrossRef]
- Pashayan, N.; Antoniou, A.C.; Ivanus, U.; Esserman, L.J.; Easton, D.F.; French, D.; Sroczynski, G.; Hall, P.; Cuzick, J.; Evans, D.G.; et al. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat. Rev. Clin. Oncol. 2020, 17, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, H.C.; Chanock, S.J. SNPs in cancer research and treatment. Br. J. Cancer 2004, 90, 747–751. [Google Scholar] [CrossRef]
- Fu, X.; Shi, Y.; Qi, T.; Qiu, S.; Huang, Y.; Zhao, X.; Sun, Q.; Lin, G. Precise design strategies of nanomedicine for improving cancer therapeutic efficacy using subcellular targeting. Signal Transduct. Target. Ther. 2020, 5, 262. [Google Scholar] [CrossRef] [PubMed]
- Muthuirulan, P.; Zhao, D.; Young, M.; Richard, D.; Liu, Z.; Emami, A.; Portilla, G.; Hosseinzadeh, S.; Cao, J.; Maridas, D.; et al. Joint disease-specificity at the regulatory base-pair level. Nat. Commun. 2021, 12, 4161. [Google Scholar] [CrossRef]
- Russ, A.P.; Lampel, S. The druggable genome: An update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Rogge, M. Targeting RNA: A Transformative Therapeutic Strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Kopechek, J.A.; McTiernan, C.F.; Chen, X.; Zhu, J.; Mburu, M.; Feroze, R.; Whitehurst, D.A.; Lavery, L.; Cyriac, J.; Villanueva, F.S. Ultrasound and Microbubble-targeted Delivery of a microRNA Inhibitor to the Heart Suppresses Cardiac Hypertrophy and Preserves Cardiac Function. Theranostics 2019, 9, 7088–7098. [Google Scholar] [CrossRef] [PubMed]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; He, Z.; Chen, P.; Jiang, X.; Chen, Y.; Liu, X.; Jiang, J. LncRNA CERS6-AS1 promotes proliferation and metastasis through the upregulation of YWHAG and activation of ERK signaling in pancreatic cancer. Cell Death Dis. 2021, 12, 648. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, R.; Shen, D.; Cheng, S.; Wang, H.; Lu, Z.; Zheng, Q.; Wang, L.; Xia, L.; Li, G. Role of noncoding RNA in drug resistance of prostate cancer. Cell Death Dis. 2021, 12, 590. [Google Scholar] [CrossRef]




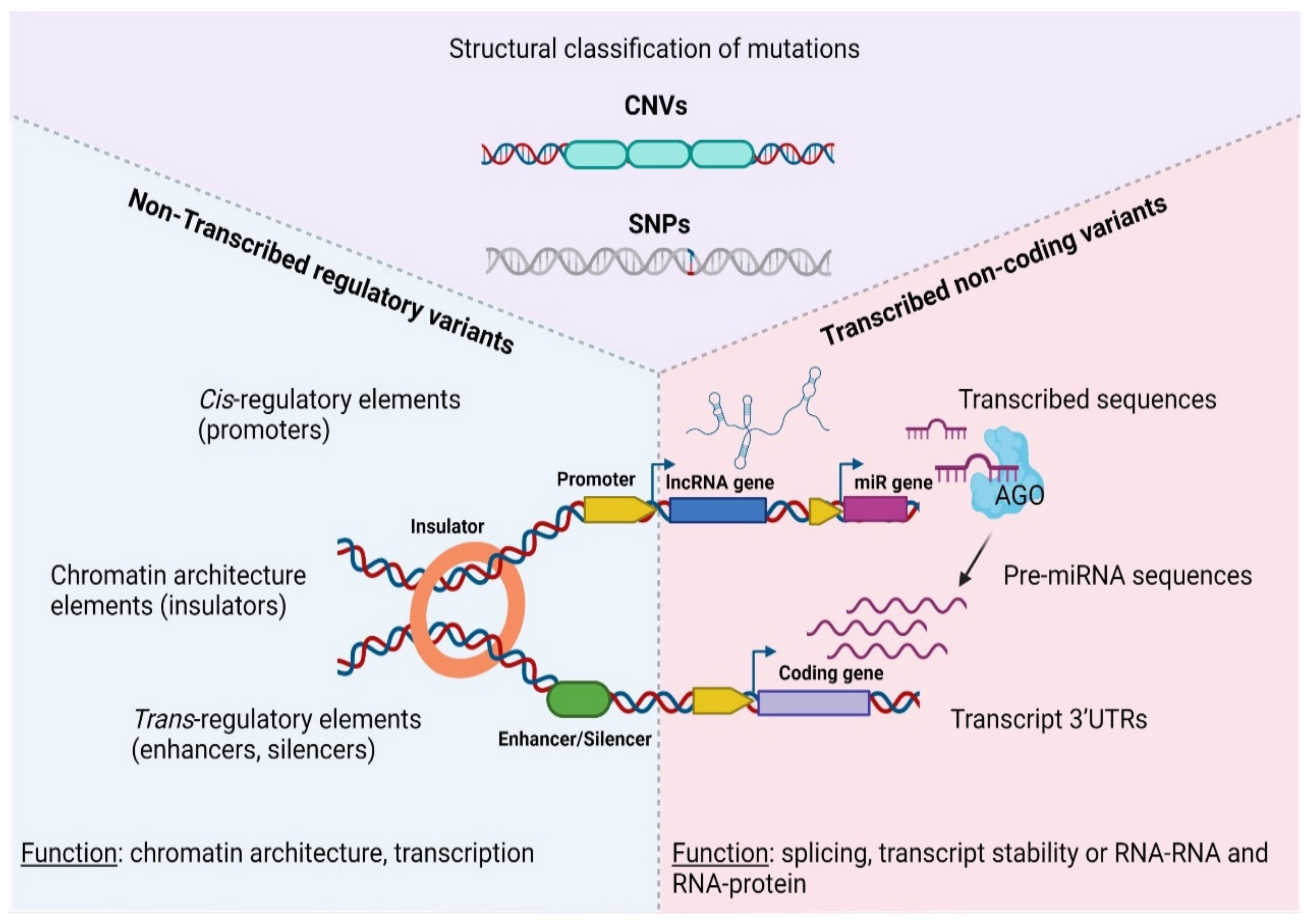{kind=link}
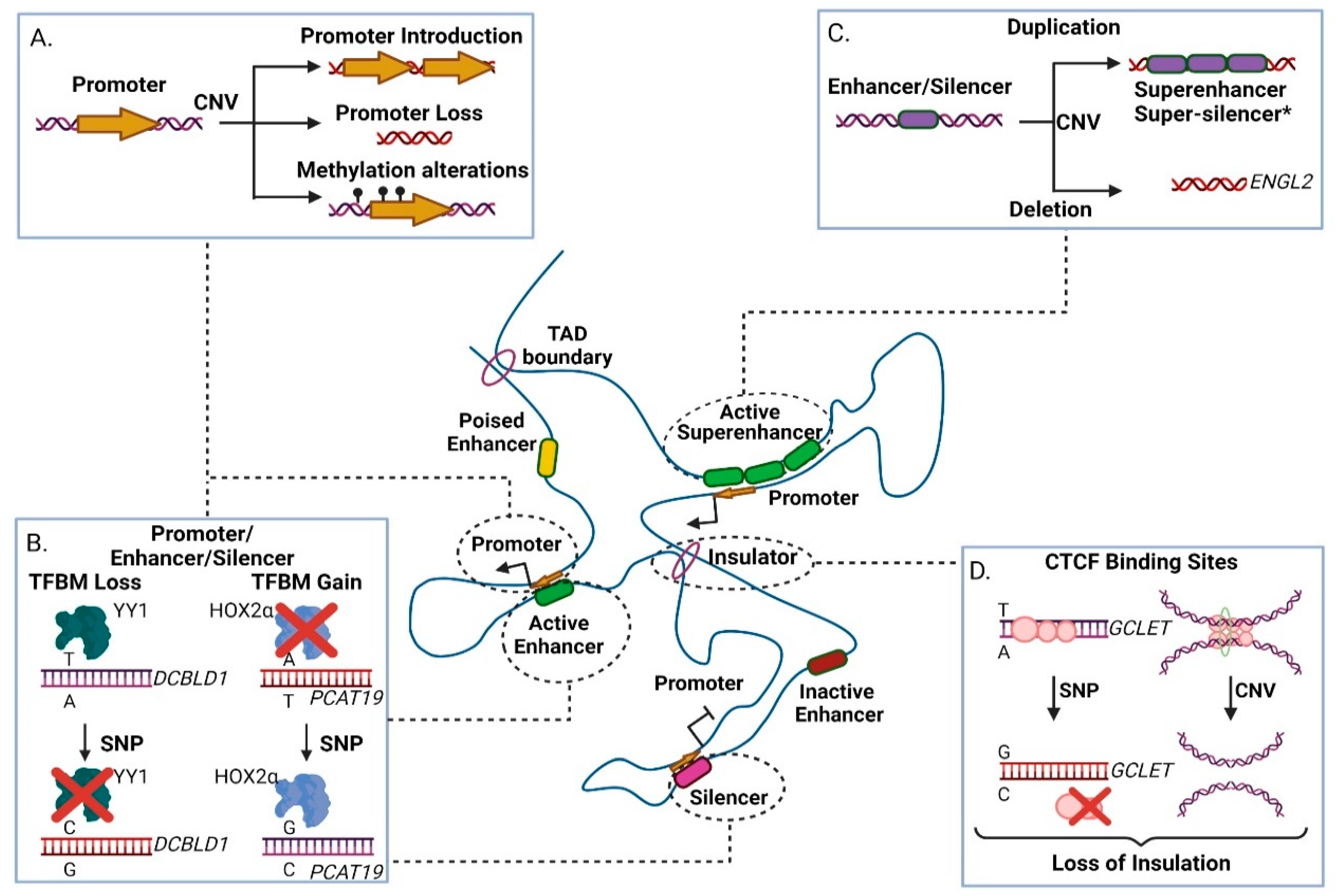{kind=link}
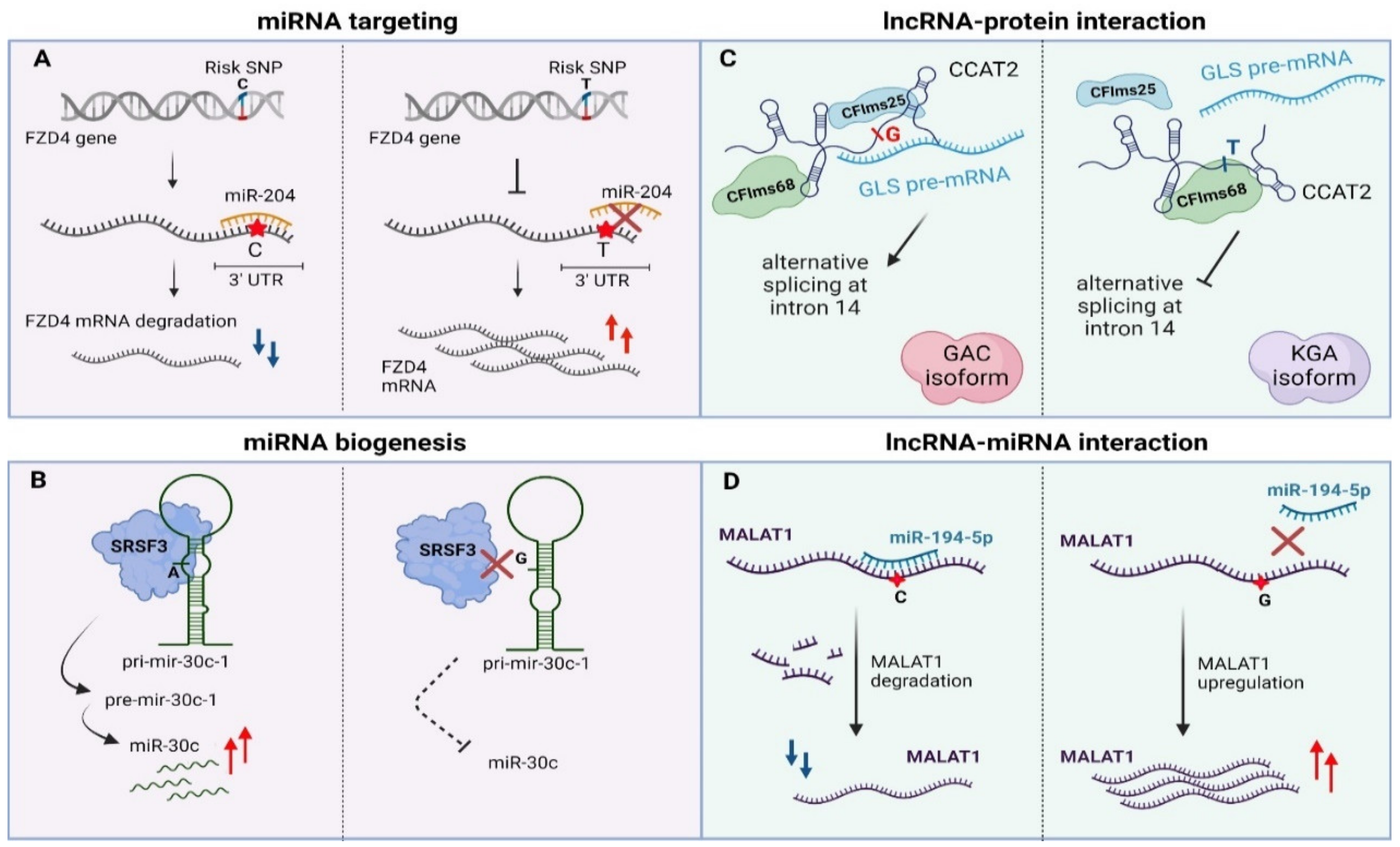{kind=link}
| Non-Coding Type | Variant ID | Target Locus | Mechanism | Cancer Type | Citation |
|---|---|---|---|---|---|
| Non-transcribed regulatory variants | |||||
| Promoters | rs11672691 | PCAT19 promoter | NKX3.1, YY1 binding | Prostate | [68] |
| rs887391 | PCAT19 promoter | NKX3.1, YY1 binding | Prostate | ||
| rs17079281 | DCBLD1 promoter | YY1 binding | Lung | [69] | |
| rs2267531 | Glypican-3 promoter | - | HCC | [70] | |
| rs2280059 | HSPH1 promoter | HSPH1 increased expression | NSCLC | [71] | |
| Enhancer | rs11672691 | PCAT19 Enhancer | NKX3.1, YY1, HOXA2 interaction with PCAT19 | Prostate | [68] |
| rs7463708 | PCAT1 Enhancer | ONECUT, AR interaction with PCAT1 | Prostate | [72] | |
| rs35252396 | Enhancer between MYC and PVT1 genes | Binding of HIFs | RCC | [73] | |
| rs6983267 | Enhancer between MYC and PVT1 | Binding of HIFs | Prostate, Colorectal | [74] | |
| EGLN2 CNV | Enhancer | Genomic deletion | Ovaries | [75] | |
| rs67311347 | Enhancer of ENTPD3-AS1 | Binding of ZNF8 | RCC | [76] | |
| rs4693608 | Enhancer of HPSE | Regulation of HPSE | ALL | [77] | |
| Silencer | rs249473 | Silencer in AKT locus | Binding of AKT | Endometrial | [78] |
| Insulator | rs3850997 | Insulator at GCLET intron | CTCF binding | Gastric | [79] |
| MYCN CNV | Insulator of MYCN | Deletion, Loss of CTCF binding | Neuroblastoma | [80] | |
| Transcribed regulatory variants | |||||
| miRNA | rs683/rs910 SNPs | 3′UTR region of TYRP1 | miRNA targeting | Μelanoma | [81] |
| rs713065 | miR-204 | miRNA targeting of FZD4 | NSCLC | [82] | |
| rs1071738 | 3′UTR of Palladin | miR-96/miR-182 targeting of Palladin | Breast | [83] | |
| rs1048638 | 3′UTR of CA9 | miR-34a targeting of CA9 | HCC | [84] | |
| rs928508 | miR-30c | pri-mir-30c-1 biogenesis miR-30c interaction with SRSF3 | Breast, Gastric | [85,86] | |
| rs6983267 | Pre-miR-1307 | pre-miR-1307 maturation | Colorectal | [87] | |
| rs11671784 | Maturation process of miR-27a | miR-27a HOXA | Gastric | [88] | |
| lncRNA | rs6983267 | CCAT2 | lncRNA interaction with CFIms25 | Colorectal | [89] |
| rs114020893 | lncRNA NEXN-AS1 | LncRNA secondary structure | Lung | [90] | |
| rs664589 | miR-194-5p | miR-194-5p interaction with MALAT1 | Colorectal | [91] | |
| rs1317082 | CCSlnc362 | miR-4658 interaction with CCSlnc362 | Colorectal | [92] | |
| rs11752942 | LINC00951 | miRNA-149 interaction with LINC00951 | ESCC | [93] | |
| rs11655237 | LINC00673 | miR-1231 interaction with LINC00673 | PDCA | [94] | |
| rs10251977 | EGFR-AS1 | Isoform selection via miR-891b and EGFR-AS interaction | Oral | [95] | |
| Experimental Approach | Advantages | Disadvantages | Publicly Available Databases/Software |
|---|---|---|---|
| Methodologies to study genomic areas in open-chromatin state | |||
| DNase-seq | • Enrich in cis-acting Res • No need for specific TF targeting • scDNase-seq improves sensitivity | • Biased in favor of promoters | • HOMER (Hypergeometric Optimization of Motif EnRichment) http://homer.ucsd.edu/homer/download.html |
| FAIRE-seq | • Simple application • Low bias • Sensitivity for intronic | • Low signal-to-noise ratio. • Requires high fixation efficiency. | • ENCODE: Wiggler https://sites.google.com/site/anshulkundaje/projects/wiggler |
| MNase-seq | • Less noise from mtDNA | • Laborious protocol • Digestion-based | • http://compbio-zhanglab.org/NUCOME/ |
| ATAC-seq | • Efficiency • Simple, cost-efficient application • Nucleosome and TF occupancy | • Demands coupling with other techniques | • ENCODE-DCC version 10 https://github.com/ENCODE-DCC/encoded/releases/tag/v101.0 |
| Methodologies for non-transcribed functional variant identification | |||
| MPRAs/CRE-seq | • High-throughput examination of enhancer activity • Allows multiple independent examinations | • Episomal assay • Cell-type specific enhancer activation profile • False-positive ratio | • Shendurelab/MPRAflow https://github.com/shendurelab/MPRAflow |
| STARR-seq | • High-throughput examination of enhancer activity • Reduced false-positive ratio • No barcoding | • Episomal assay • Cell-type specific enhancer activation profile • Reporter transcript stability | • Gersteinlab/starrpeaker https://github.com/gersteinlab/starrpeaker • hyulab/eSTARR https://github.com/hyulab/eSTARR |
| ChIA-PET | • Precise global interaction map • Long-read ChIA-PETS has improved mapping efficiency | • Complex data analysis • Inefficient • Demands coupling with RNA-targeted methodology | • ChIA-PET Utilities-CPU https://github.com/cheehongsg/CPU • Mango https://github.com/dphansti/mango • TheJacksonLaboratory/ChIA-PIPE https://github.com/TheJacksonLaboratory/ChIA-PIPE |
| HiChIP | • Efficiency • Low false-positive ratio • Simple workflow | • Not available | • FitHiChIP https://github.com/ay-lab/FitHiChIP |
| PLAC-seq | • Efficiency • Specificity • Simple workflow | • Not available | • HPRep https://github.com/yunliUNC/HPRep • MAPS https://github.com/HuMingLab/MAPS |
| ChIRP-seq | • Commonly used | • Increased false-positive ratio • Targets known RNA | • Not available |
| Experimental Approach | Advantages | Disadvantages | Publicly Available Databases/Software |
|---|---|---|---|
| Methodologies for transcribed functional variant identification | |||
| RAP-seq | • Genome-wide RNA:DNA interaction maps • Low background noise | • Known RNA sequence | • SPRITE https://github.com/GuttmanLab/sprite-pipeline |
| RNP-MaP | • Efficient • Resolution • Unbiased • Study protein networks | • Coupling with protein-targeted methodology | • Not available |
| CARPID | • Specificity • Low background noise • Determine allelic expression | • Coupling with protein-targeted methodology | • Not available |
| PTRE-seq | • High-throughput examination of 3′UTR regulatory activity | • Not available | • Not available |
| PARS-seq | • RNA structural information • Distinguish paired/unpaired bases • Alternative to MS, NMR, crystallography | • Non-specific enzyme digestion • RNA over-digestion • Need for optimization • Only in vitro applications | • RNAFramework https://github.com/dincarnato/RNAFramework |
| Experimental Approach | Advantages | Disadvantages | Publicly Available Databases/Software |
|---|---|---|---|
| CRISPR-Cas Systems | • Allows variant correction/creation • Low off-target effects (especially when using nickase Cas9) • Activation and inhibition of regulatory element function • Alteration in methylation status • RNA targeting | • Needs fine-tuning to avoid off-target effects • Efficiency differs between systems | • Design http://www.rgenome.net/be-designer/ http://zifit.partners.org/ZiFiT/ http://www.e-crisp.org/E-CRISP/ https://chopchop.cbu.uib.no/ http://crispr-era.stanford.edu/ https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design • Analysis http://www.rgenome.net/be-analyzer/# |
| SNP ID | Target Locus | Clinical Trait | Cancer Type | Citation |
|---|---|---|---|---|
| rs291593 | DPYD 3′UTR | Drug toxicity | Breast cancer | [316] |
| rs3209896, rs1824125 | AKR1C3 3′UTR, PGR 3′UTR | Progression-free Survival | ||
| rs1054899 | ALDH5A1 3′UTR | Chemotherapeutic response to FAC | ||
| rs7756222, rs9487402 | SLC22A16 3′UTR | Overall survival | ||
| rs13230517 | RP11-3N2.1 promoter | Cancer Risk | Colorectal cancer | [317] |
| rs531564 | pri-miR-124 | Lymph node metastasis | ||
| rs3741219, rs2910164, rs4938723 | H19 lncRNA, hsa-mir-146a, hsa-mir-34b/c | Cancer risk | Hepatocellular carcinoma | [318] |
| rs11614913, rs2292832 | hsa-mir-196a-2, hsa-mir-149 | Cancer risk | HBV-related HCC | [319] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lange, M.; Begolli, R.; Giakountis, A. Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA 2021, 7, 47. https://doi.org/10.3390/ncrna7030047
Lange M, Begolli R, Giakountis A. Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA. 2021; 7(3):47. https://doi.org/10.3390/ncrna7030047
Chicago/Turabian StyleLange, Marios, Rodiola Begolli, and Antonis Giakountis. 2021. "Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine" Non-Coding RNA 7, no. 3: 47. https://doi.org/10.3390/ncrna7030047
APA StyleLange, M., Begolli, R., & Giakountis, A. (2021). Non-Coding Variants in Cancer: Mechanistic Insights and Clinical Potential for Personalized Medicine. Non-Coding RNA, 7(3), 47. https://doi.org/10.3390/ncrna7030047