Exosomes in Immune Regulation

Abstract
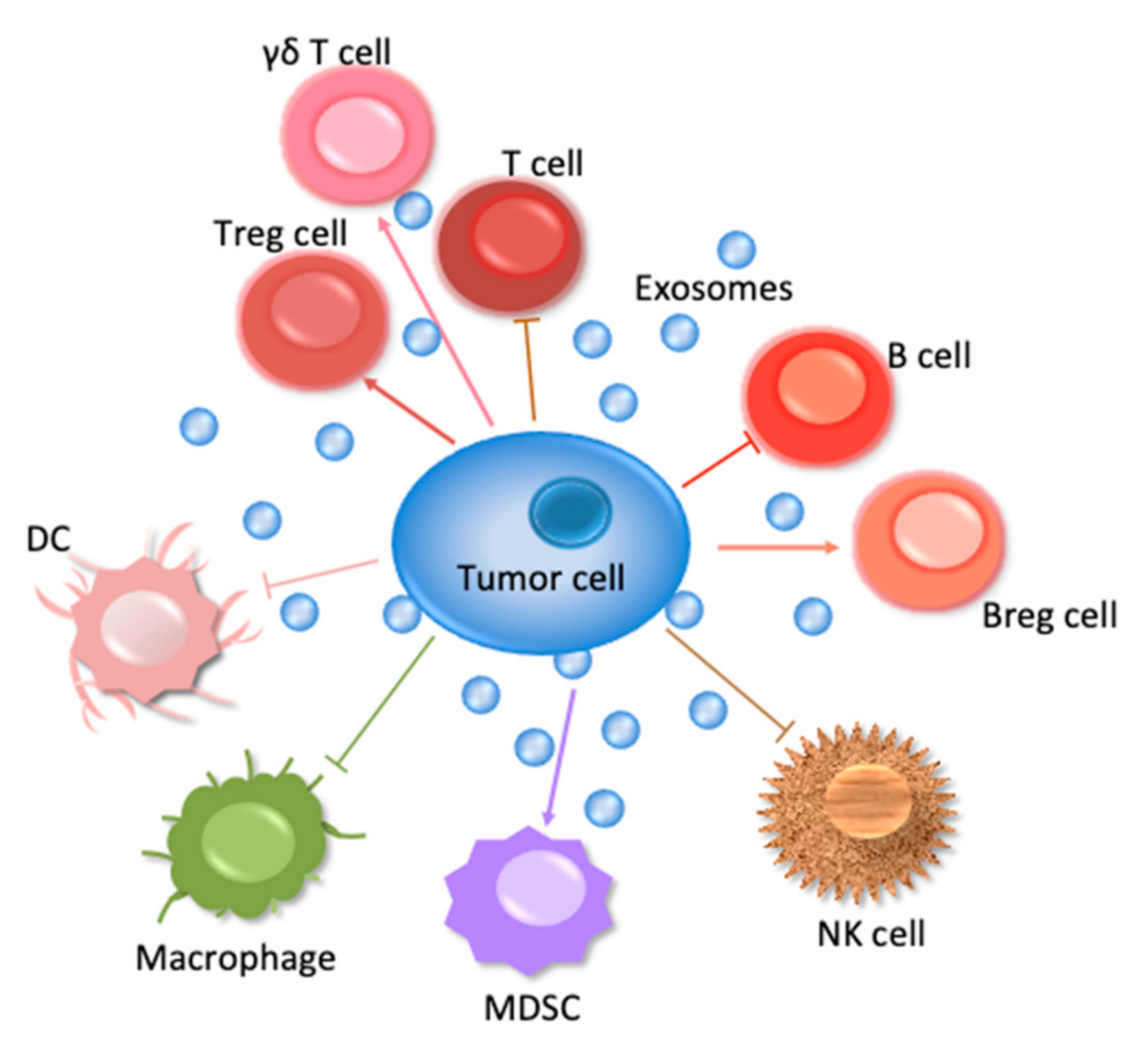
1. Introduction
2. Exosome Biogenesis and Function
3. Exosomal RNAs
3.1. Natural Occurring RNAs
3.1.1. miRNAs
3.1.2. Sponge circRNAs
3.1.3. lncRNAs
3.2. Artificial miRNAs
shRNAs
4. Exosomal Proteins
5. Exosomes and Lymphoid Cells
5.1. T Cells
5.2. B Cells
5.3. Natural Killer Cells
6. Exosomes and Myeloid Cells
6.1. Monocytes/Dendritic cells
6.2. Macrophages
7. Exosomes and Cancer Therapy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zijlstra, A.; Di Vizio, D. Size matters in nanoscale communication. Nat. Cell Biol. 2018, 20, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-Like Vesicles Are Present in Hu-man Blood Plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Grunewald, J.; Thyberg, J.; Gripenback, S.; Tornling, G.; Eklund, A.; Scheynius, A.; Gabrielsson, S. Exosomes with Major Histocompatibility Complex Class Ii and Co-Stimulatory Molecules Are Present in Human Bal Fluid. Eur. Respir. J. 2003, 22, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.-J.; Lahesmaa, R.; Norman, M.; Neve, E.P.A.; Scheynius, A.; Gabrielsson, S. Exosomes with Immune Modulatory Features Are Present in Human Breast Milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.E.C.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Pisitkun, T.; Shen, R.-F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef]
- Lässer, C.; O’Neil, S.E.; Shelke, G.V.; Sihlbom, C.; Hansson, S.F.; Gho, Y.S.; Lundbäck, B.; Lötvall, J. Exosomes in the nose induce immune cell trafficking and harbour an altered protein cargo in chronic airway inflammation. J. Transl. Med. 2016, 14, 181. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of Secretion and Uptake of Exosomes and Other Extra-cellular Vesicles for Cell-to-Cell Communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signaling in Breast Cancer Cell Migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Yakimchuk, K. Exosomes: Isolation and Characterization Methods and Specific Markers. Mater. Methods 2015, 5, 1450–1453. [Google Scholar] [CrossRef]
- Liu, F.; Vermesh, O.; Mani, V.; Ge, T.J.; Madsen, S.J.; Sabour, A.; Hsu, E.-C.; Gowrishankar, G.; Kanada, M.; Jokerst, J.V.; et al. The Exosome Total Isolation Chip. ACS Nano 2017, 11, 10712–10723. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Choi, M.; Lee, H.; Kim, Y.-H.; Han, J.-Y.; Lee, E.; Cho, Y. Direct isolation and characterization of circulating exosomes from biological samples using magnetic nanowires. J. Nanobiotechnology 2019, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- La Shu, S.; Yang, Y.; Allen, C.L.; Hurley, E.; Tung, K.H.; Minderman, H.; Wu, Y.; Ernstoff, M.S. Purity and yield of melanoma exosomes are dependent on isolation method. J. Extracell. Vesicles 2020, 9, 1692401. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, L.; Ouandaogo, Z.G.; Anakor, E.; Connolly, O.; Browne, G.B.; Laine, J.; Duddy, W.; Duguez, S. Optimized method for extraction of exosomes from human primary muscle cells. Skelet. Muscle 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Clayton, A.; Court, J.; Navabi, H.; Adams, M.; Mason, M.D.; Hobot, J.A.; Newman, G.R.; Jasani, B. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J. Immunol. Methods 2001, 247, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Magini, A.; Buratta, S.; Brozzi, A.; Sagini, K.; Polchi, A.; Tancini, B.; Emiliani, C. Signaling Pathways in Exosomes Biogenesis, Secretion and Fate. Genes 2013, 4, 152–170. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of Uptake. J. Circ. Biomark. 2015, 4, 7. [Google Scholar] [CrossRef]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.-A.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte–Neuron Communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Gahan, P.B. MicroRNA Shuttle from Cell-To-Cell by Exosomes and Its Impact in Cancer. Non-Coding RNA 2019, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, G.; Hu, W.; Yao, Y.; Yu, X.-F. Emerging roles and therapeutic value of exosomes in cancer metastasis. Mol. Cancer 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. Discovery of Double-Stranded Genomic DNA in Circulating Exosomes. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.J.; Du, M.; Liang, M.; Dittmar, R.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef]
- Wang, X.; Weidling, I.; Koppel, S.; Menta, B.; Ortiz, J.P.; Kalani, A.; Wilkins, H.M.; Swerdlow, R.H. Detection of mitochondria-pertinent components in exosomes. Mitochondrion 2020, 55, 100–110. [Google Scholar] [CrossRef]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Bortolin-Cavaillé, M.-L.; Dance, M.; Weber, M.; Cavaillé, J. C19MC microRNAs are processed from introns of large Pol-II, non-protein-coding transcripts. Nucleic Acids Res. 2009, 37, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Chen, G.; Zhu, Z.; Shen, Z.; Du, C.; Zang, R.; Su, Y.; Xie, H.; Li, H.; Xu, X.; et al. Correction: Circular RNA ZNF609 functions as a competitive endogenous RNA to regulate AKT3 expression by sponging miR-150-5p in Hirschsprung’s disease. Oncotarget 2019, 10, 3313–3314. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. Mirna-Dependent Gene Silencing Involving Ago2-Mediated Cleavage of a Circular Antisense RNA. EMBO J. 2011, 21, 4414–4422. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs Are a Large Class of Animal RNAs with Regulatory Potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Zeng, K.; Chen, X.; Xu, M.; Liu, X.; Hu, X.; Xu, T.; Sun, H.; Pan, Y.; He, B.; Wang, S. CircHIPK3 promotes colorectal cancer growth and metastasis by sponging miR-7. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Hentze, M.W.; Preiss, T. Circular RNAs: Splicing’s enigma variations. EMBO J. 2013, 32, 923–925. [Google Scholar] [CrossRef]
- Zhang, P.-F.; Wei, C.-Y.; Huang, X.-Y.; Peng, R.; Yang, X.; Lu, J.-C.; Zhang, C.; Gao, C.; Cai, J.-B.; Gao, P.-T.; et al. Circular RNA circTRIM33–12 acts as the sponge of MicroRNA-191 to suppress hepatocellular carcinoma progression. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef]
- Liu, K.-S.; Pan, F.; Mao, X.-D.; Liu, C.; Chen, Y.-J. Biological functions of circular RNAs and their roles in occurrence of reproduction and gynecological diseases. Am. J. Transl. Res. 2019, 11, 1–15. [Google Scholar]
- Zhang, H.; Shen, Y.; Li, Z.; Ruan, Y.; Li, T.; Xiao, B.; Sun, W. The biogenesis and biological functions of circular RNAs and their molecular diagnostic values in cancers. J. Clin. Lab. Anal. 2020, 34, e23049. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wu, Z.; Zhang, J.; Su, B. Salivary lncRNA as a potential marker for oral squamous cell carcinoma diagnosis. Mol. Med. Rep. 2012, 7, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wang, G.; Chen, L.; Jiang, J.; Liu, L.; Li, N.; Zhao, J.; Sun, X.; Zhou, P. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus). BMC Genom. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Ma, X.-Y.; Wang, J.; Wang, J.-L.; Ma, C.X.; Wang, X.-C.; Liu, F. Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells. BMC Genom. 2015, 16, 1–11. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Liu, A.; Tang, X. Long non-coding RNA UCA1 enhances tamoxifen resistance in breast cancer cells through a miR-18a-HIF1α feedback regulatory loop. Tumor Biol. 2016, 37, 14733–14743. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, M.; Xing, P.; Yan, X.; Xie, B. Increased Expression of Exosomal AGAP2-AS1 (AGAP2 Antisense RNA 1) In Breast Cancer Cells Inhibits Trastuzumab-Induced Cell Cytotoxicity. Med. Sci. Monit. 2019, 25, 2211–2220. [Google Scholar] [CrossRef]
- Dong, H.; Wang, W.; Chen, R.; Zhang, Y.; Zou, K.; Ye, M.; He, X.; Zhang, F.; Han, J. Exosome-mediated transfer of lncRNA-SNHG14 promotes trastuzumab chemoresistance in breast cancer. Int. J. Oncol. 2018, 53, 1013–1026. [Google Scholar] [CrossRef]
- Xu, C.-G.; Yang, M.-F.; Ren, Y.-Q.; Wu, C.-H.; Wang, L.-Q. Exosomes mediated transfer of lncRNA UCA1 results in increased tamoxifen resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4362–4368. [Google Scholar]
- Taxman, D.J.; Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. Comput. Biol. 2010, 629, 139–156. [Google Scholar] [CrossRef]
- Kutter, C.; Svoboda, P. miRNA, siRNA, piRNA: Knowns of the Unknown. RNA Biol. 2008, 5, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Duban, L.; Segura, E.; Véron, P.; Lantz, O.; Amigorena, S. Indirect activation of naïve CD4+ T cells by dendritic cell–derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019, 177, 414.e13–427.e13. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Nakagawa, Y.; Yokomuro, K.; Berzofsky, J.A. Induction of CD8+ cytotoxic T lymphocytes by immunization with syngeneic irradiated HIV-1 envelope derived peptide-pulsed dendritic cells. Int. Immunol. 1993, 5, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional Transfer of Microrna-Loaded Exosomes from T Cells to Antigen-Presenting Cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef]
- Hong, C.-S.; Danet-Desnoyers, G.; Shan, X.; Sharma, P.; Whiteside, T.L.; Boyiadzis, M. Human acute myeloid leukemia blast-derived exosomes in patient-derived xenograft mice mediate immune suppression. Exp. Hematol. 2019, 76, 60–66.e2. [Google Scholar] [CrossRef]
- Lindenbergh, M.F.S.; Koerhuis, D.G.J.; Borg, E.G.F.; van ’t Veld, E.M.; Driedonks, T.A.P.; Wubbolts, R.; Stoorvogel, W.; Boes, M. Bystander T-Cells Support Clonal T-Cell Activation by Controlling the Release of Dendritic Cell-Derived Immune-Stimulatory Extracellular Vesicles. Front. Immunol. 2019, 10, 448. [Google Scholar] [CrossRef]
- Shojaei, S.; Hashemi, S.M.; Ghanbarian, H.; Salehi, M.; Mohammadi-Yeganeh, S. Effect of mesenchymal stem cells-derived exosomes on tumor microenvironment: Tumor progression versus tumor suppression. J. Cell. Physiol. 2019, 234, 3394–3409. [Google Scholar] [CrossRef]
- Shen, Y.; Xue, C.; Li, X.; Ba, L.; Gu, J.; Sun, Z.; Han, Q.; Zhao, R.C. Effects of Gastric Cancer Cell-Derived Exosomes on the Immune Regulation of Mesenchymal Stem Cells by the Nf-Kb Signaling Pathway. Stem Cells Dev. 2019, 28, 464–476. [Google Scholar] [CrossRef]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-κB, IκB, and IKK: Integral Components of Immune System Signaling. Cannabinoids Neuropsychiatr. Disord. 2019, 1172, 207–226. [Google Scholar] [CrossRef]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.-E.; Sung, K.J.; Sung, Y.H.; Pack, C.-G.; Jung, M.-K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Suo, B.; Long, G.; Gao, Y.; Song, J.; Zhang, M.; Feng, B.; Shang, C.; Wang, D. Exosomal miRNA-16-5p Derived From M1 Macrophages Enhances T Cell-Dependent Immune Response by Regulating PD-L1 in Gastric Cancer. Front. Cell Dev. Biol. 2020, 8, 572689. [Google Scholar] [CrossRef]
- Dou, D.; Ren, X.; Han, M.; Xu, X.; Ge, X.; Gu, Y.; Wang, X. Cancer-Associated Fibroblasts-Derived Exosomes Suppress Immune Cell Function in Breast Cancer via the miR-92/PD-L1 Pathway. Front. Immunol. 2020, 11, 2026. [Google Scholar] [CrossRef]
- Peng, X.X.; Yu, R.; Wu, X.; Wu, S.Y.; Pi, C.; Chen, Z.H.; Zhang, X.C.; Gao, C.Y.; Shao, Y.W.; Liu, L.; et al. Correlation of Plasma Exosomal Micrornas with the Efficacy of Immunotherapy in Egfr / Alk Wild-Type Advanced Non-Small Cell Lung Cancer. J. Immunother Cancer 2020, 8. [Google Scholar] [CrossRef]
- Wu, F.; Yin, Z.; Yang, L.; Fan, J.; Xu, J.; Jin, Y.; Yu, J.; Zhang, D.; Yang, G. Smoking Induced Extracellular Vesicles Release and Their Distinct Properties in Non-Small Cell Lung Cancer. J. Cancer 2019, 10, 3435–3443. [Google Scholar] [CrossRef]
- Xu, J.; Yin, Z.; Yang, L.; Wu, F.; Fan, J.; Huang, Q.; Jin, Y.; Yang, G. Evidence that dysplasia related microRNAs in Barrett’s esophagus target PD-L1 expression and contribute to the development of esophageal adenocarcinoma. Aging 2020, 12, 17062–17078. [Google Scholar] [CrossRef]
- Mishra, V.; Pathak, C. Human Toll-Like Receptor 4 (hTLR4): Structural and functional dynamics in cancer. Int. J. Biol. Macromol. 2019, 122, 425–451. [Google Scholar] [CrossRef]
- Domenis, R.; Cifù, A.; Marinò, D.; Fabris, M.; Niazi, K.R.; Soon-Shiong, P.; Curcio, F. Toll-like Receptor-4 Activation Boosts the Immunosuppressive Properties of Tumor Cells-derived Exosomes. Sci. Rep. 2019, 9, 8457. [Google Scholar] [CrossRef] [PubMed]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [PubMed]
- Hellwinkel, J.E.; Redzic, J.S.; Harland, T.A.; Gunaydin, D.; Anchordoquy, T.J.; Graner, M.W. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro-Oncology 2016, 18, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Iorgulescu, J.B.; Ivan, M.E.; Safaee, M.; Parsa, A.T. The limited capacity of malignant glioma-derived exosomes to suppress peripheral immune effectors. J. Neuroimmunol. 2016, 290, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. OncoImmunology 2018, 7, e1478647. [Google Scholar] [CrossRef]
- Li, P.; Feng, J.; Liu, Y.; Liu, Q.; Fan, L.; Liu, Q.; She, X.; Liu, C.; Liu, T.; Zhao, C.; et al. Novel Therapy for Glioblastoma Multiforme by Restoring LRRC4 in Tumor Cells: LRRC4 Inhibits Tumor-Infitrating Regulatory T Cells by Cytokine and Programmed Cell Death 1-Containing Exosomes. Front. Immunol. 2017, 8, 1748. [Google Scholar] [CrossRef]
- Li, P.; Xu, G.; Li, G.Y.; Wu, M. Function and mechanism of tumor suppressor gene LRRC4/NGL-2. Mol. Cancer 2014, 13, 266. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4+ T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 Inhibitors: Targeting Mitochondrial Apoptotic Pathways in Cancer Therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Ii, D.J.K. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018, 285, 1033–1050. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, W.; McGinley, E.C.; Ii, D.J.K. Melanoma exosomes deliver a complex biological payload that upregulates PTPN11 to suppress T lymphocyte function. Pigment. Cell Melanoma Res. 2017, 30, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.M.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, X.; Zhao, T.; Li, W.; Xiang, J. Natural CD8+25+ regulatory T cell-secreted exosomes capable of suppressing cytotoxic T lymphocyte-mediated immunity against B16 melanoma. Biochem. Biophys. Res. Commun. 2013, 438, 152–155. [Google Scholar] [CrossRef]
- Guo, D.; Chen, Y.; Wang, S.; Yu, L.; Shen, Y.; Zhong, H.; Yang, Y. Exosomes from heat-stressed tumour cells inhibit tumour growth by converting regulatory T cells to Th17 cells via IL-6. Immunology 2018, 154, 132–143. [Google Scholar] [CrossRef]
- Raverdeau, M.; Cunningham, S.P.; Harmon, C.; Lynch, L. Gammadelta T Cells in Cancer: A Small Population of Lymphocytes with Big Implications. Clin. Transl. Immunol. 2019, 10, e01080. [Google Scholar] [CrossRef]
- Li, L.; Cao, B.; Liang, X.; Lu, S.; Luo, H.; Wang, Z.; Wang, S.; Jiang, J.; Lang, J.; Zhu, G. Microenvironmental Oxygen Pressure Orchestrates an Anti- and Pro-Tumoral Gammadelta T Cell Equilibrium Via Tumor-Derived Exosomes. Oncogene 2019, 38, 2830–2843. [Google Scholar] [CrossRef]
- Theodoraki, M.; Hoffmann, T.K.; Whiteside, T.L. Separation of plasma-derived exosomes into CD3(+) and CD3(-) fractions allows for association of immune cell and tumour cell markers with disease activity in HNSCC patients. Clin. Exp. Immunol. 2018, 192, 271–283. [Google Scholar] [CrossRef]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef]
- Theodoraki, M.; Hoffmann, T.K.; Jackson, E.K.; Whiteside, T.L. Exosomes in HNSCC plasma as surrogate markers of tumour progression and immune competence. Clin. Exp. Immunol. 2018, 194, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, C.M.; Gastman, B.R. Tumor-derived exosomes induce CD8+ T cell suppressors. J. Immunother. Cancer 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.-C.; Chen, H.-Y.; Kuo, C.-C.; Sytwu, H.-K. Role of Galectins in Tumors and in Clinical Immunotherapy. Int. J. Mol. Sci. 2018, 19, 430. [Google Scholar] [CrossRef]
- Ludwig, S.; Floros, T.; Theodoraki, M.-N.; Hong, C.-S.; Jackson, E.K.; Lang, S.; Whiteside, T.L. Suppression of Lymphocyte Functions by Plasma Exosomes Correlates with Disease Activity in Patients with Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 4843–4854. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar]
- Ludwig, S.; Marczak, L.; Sharma, P.; Abramowicz, A.; Gawin, M.; Widlak, P.; Whiteside, T.L.; Pietrowska, M. Proteomes of exosomes from HPV(+) or HPV(-) head and neck cancer cells: Differential enrichment in immunoregulatory proteins. OncoImmunology 2019, 8, e1593808. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Rodríguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in Cancer Cell Signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar] [CrossRef]
- Shenoy, G.N.; Loyall, J.; Berenson, C.S.; Kelleher, R.J.; Iyer, V.; Balu-Iyer, S.V.; Odunsi, K.; Bankert, R.B. Sialic Acid–Dependent Inhibition of T Cells by Exosomal Ganglioside GD3 in Ovarian Tumor Microenvironments. J. Immunol. 2018, 201, 3750–3758. [Google Scholar] [CrossRef]
- Shenoy, G.N.; Loyall, J.L.; Maguire, O.; Iyer, V.; Kelleher, R.J.; Minderman, H.; Wallace, P.K.; Odunsi, K.; Balu-Iyer, S.V.; Bankert, R.B. Exosomes Associated with Human Ovarian Tumors Harbor a Reversible Checkpoint of T-cell Responses. Cancer Immunol. Res. 2018, 6, 236–247. [Google Scholar] [CrossRef]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Ni Wong, K.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer–Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef]
- Rani, S.; Corcoran, C.; Shiels, L.; Germano, S.; Breslin, S.; Madden, S.; McDermott, M.S.; Browne, B.C.; O’Donovan, N.; Crown, J.; et al. Neuromedin U: A Candidate Biomarker and Therapeutic Target to Predict and Overcome Resistance to HER-Tyrosine Kinase Inhibitors. Cancer Res. 2014, 74, 3821–3833. [Google Scholar] [CrossRef]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. OncoImmunology 2017, 6, e1362530. [Google Scholar] [CrossRef]
- Li, R.; Chibbar, R.; Xiang, J. Novel EXO-T vaccine using polyclonal CD4+ T cells armed with HER2-specific exosomes for HER2-positive breast cancer. OncoTargets Ther. 2018, 11, 7089–7093. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, J.; Xu, A.; Ahmeqd, S.; Sami, A.; Chibbar, R.; Freywald, A.; Zheng, C.; Xiang, J. Heterologous human/rat HER2-specific exosome-targeted T cell vaccine stimulates potent humoral and CTL responses leading to enhanced circumvention of HER2 tolerance in double transgenic HLA-A2/HER2 mice. Vaccine 2018, 36, 1414–1422. [Google Scholar] [CrossRef]
- Ye, S.; Zhang, H.; Cai, T.-T.; Liu, Y.-N.; Ni, J.-J.; He, J.; Peng, J.-Y.; Chen, Q.-Y.; Mo, H.-Y.; Cui, J.-; et al. Exosomal miR-24-3p impedes T-cell function by targetingFGF11and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef]
- Ye, S.-B.; Li, Z.-L.; Luo, D.-H.; Huang, B.-J.; Chen, Y.-S.; Zhang, X.-S.; Cui, J.; Zeng, Y.-X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef]
- Mrizak, D.; Martin, N.; Barjon, C.; Jimenez-Pailhes, A.-S.; Mustapha, R.; Niki, T.; Guigay, J.; Pancré, V.; De Launoit, Y.; Busson, P.; et al. Effect of Nasopharyngeal Carcinoma-Derived Exosomes on Human Regulatory T Cells. J. Natl. Cancer Inst. 2014, 107, 363. [Google Scholar] [CrossRef]
- Salimu, J.; Webber, J.; Gurney, M.; Al-Taei, S.; Clayton, A.; Tabi, Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J. Extracell. Vesicles 2017, 6, 1368823. [Google Scholar] [CrossRef]
- Hendrix, A.; De Wever, O. Rab27 GTPases Distribute Extracellular Nanomaps for Invasive Growth and Metastasis: Implications for Prognosis and Treatment. Int. J. Mol. Sci. 2013, 14, 9883–9892. [Google Scholar] [CrossRef]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016, 6, 20254. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Luo, C.; Xia, Y.; Chen, H.; He, Y. Glycosyl-phosphatidylinositol-anchored interleukin-2 expressed on tumor-derived exosomes induces antitumor immune response in vitro. Tumori J. 2010, 96, 452–459. [Google Scholar] [CrossRef]
- Xu, H.; Li, N.; Yao, N.; Xu, X.; Wang, H.; Liu, X.; Zhang, Y. CD8+ T cells stimulated by exosomes derived from RenCa cells mediate specific immune responses through the FasL/Fas signaling pathway and, combined with GM-CSF and IL-12, enhance the anti-renal cortical adenocarcinoma effect. Oncol. Rep. 2019, 42, 866–879. [Google Scholar] [CrossRef]
- Linkermann, A.; Qian, J.; Lettau, M.; Kabelitz, D.; Janssen, O. Considering Fas ligand as a target for therapy. Expert Opin. Ther. Targets 2005, 9, 119–134. [Google Scholar] [CrossRef]
- Yang, M.-Q.; Du, Q.; Varley, P.R.; Goswami, J.; Liang, Z.; Wang, R.; Li, H.; Stolz, D.B.; Geller, D.A. Interferon regulatory factor 1 priming of tumour-derived exosomes enhances the antitumour immune response. Br. J. Cancer 2018, 118, 62–71. [Google Scholar] [CrossRef]
- Menay, F.; Herschlik, L.; De Toro, J.; Cocozza, F.; Tsacalian, R.; Gravisaco, M.J.; Di Sciullo, M.P.; Vendrell, A.; Waldner, C.I.; Mongini, C. Exosomes Isolated from Ascites of T-Cell Lymphoma-Bearing Mice Expressing Surface Cd24 and Hsp-90 In-duce a Tumor-Specific Immune Response. Front. Immunol. 2017, 8, 286. [Google Scholar] [CrossRef]
- Muntasell, A.; Berger, A.C.; Roche, P.A. T Cell-Induced Secretion of Mhc Class Ii-Peptide Complexes on B Cell Exo-somes. EMBO J. 2007, 26, 4263–4272. [Google Scholar] [CrossRef]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S.; et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, L.; Wei, W. Regulatory B cells in inflammatory diseases and tumor. Int. Immunopharmacol. 2019, 67, 281–286. [Google Scholar] [CrossRef]
- Du, P.; Xiong, R.; Li, X.; Jiang, J. Immune Regulation and Antitumor Effect of TIM-1. J. Immunol. Res. 2016, 2016, 1–6. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, Q.; Cheng, Y.; Chen, X.; Wang, G.; Shi, M.; Zhang, T.; Cao, Y.; Pan, H.; Zhang, L.; et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1+ regulatory B cell expansion. J. Immunother. Cancer 2018, 6, 145. [Google Scholar] [CrossRef]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from burkitt’s lymphoma cell lines induce proliferation, differentiation, and class-switch recombination in B cells. J. Immunol. 2014, 192, 5852–5862. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Huyan, T.; Du, Y.; Huang, Q.; Huang, Q.; Li, Q. Uptake Characterization of Tumor Cell-derived Exosomes by Natural Killer Cells. Iran. J. Public Heal. 2018, 47, 803–813. [Google Scholar]
- Mincheva-Nilsson, L.; Baranov, V. Cancer exosomes and NKG2D receptor–ligand interactions: Impairing NKG2D-mediated cytotoxicity and anti-tumour immune surveillance. Semin. Cancer Biol. 2014, 28, 24–30. [Google Scholar] [CrossRef]
- Ashiru, O.; Boutet, P.; Fernandez-Messina, L.; Aguera-Gonzalez, S.; Skepper, J.N.; Vales-Gomez, M.; Reyburn, H.T. Natu-ral Killer Cell Cytotoxicity Is Suppressed by Exposure to the Human Nkg2d Ligand Mica*008 That Is Shed by Tumor Cells in Exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef]
- Lundholm, M.; Schroder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikstrom, P. Prostate Tu-mor-Derived Exosomes Down-Regulate Nkg2d Expression on Natural Killer Cells and Cd8+ T Cells: Mechanism of Im-mune Evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef]
- Xiao, W.; Dong, W.; Zhang, C.; Saren, G.; Geng, P.; Zhao, H.; Li, Q.; Zhu, J.; Li, G.; Zhang, S.; et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur. J. Med. Res. 2013, 18, 61. [Google Scholar] [CrossRef]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.-H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural killer–derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Schmittgen, T.D. Exosomal miRNA Cargo as Mediator of Immune Escape Mechanisms in Neuroblastoma. Cancer Res. 2019, 79, 1293–1294. [Google Scholar] [CrossRef]
- Shoae-Hassani, A.; Hamidieh, A.A.; Behfar, M.; Mohseni, R.; Mortazavi-Tabatabaei, S.A.; Asgharzadeh, S. NK Cell–derived Exosomes From NK Cells Previously Exposed to Neuroblastoma Cells Augment the Antitumor Activity of Cytokine-activated NK Cells. J. Immunother. 2017, 40, 265–276. [Google Scholar] [CrossRef]
- Zhu, L.; Gangadaran, P.; Kalimuthu, S.; Oh, J.M.; Baek, S.H.; Jeong, S.Y.; Lee, S.B.; Lee, J.; Ahn, B.-C. Novel alternatives to extracellular vesicle-based immunotherapy—exosome mimetics derived from natural killer cells. Cells Nanomed. Biotechnol. 2018, 46, S166–S179. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, C.; Ricci, B.; Vulpis, E.; Fionda, C.; Ricciardi, M.R.; Petrucci, M.T.; Masuelli, L.; Peri, A.; Cippitelli, M.; Zingoni, A.; et al. Drug-Induced Senescent Multiple Myeloma Cells Elicit NK Cell Proliferation by Direct or Exosome-Mediated IL15 Trans-Presentation. Cancer Immunol. Res. 2018, 6, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, Q.; Zhen, Q.; Zhao, Y.; Liu, N.; Li, T.; Hao, Y.; Zhang, Y.; Luo, C.; Wu, X. Negative Regulation of Tu-mor-Infiltrating Nk Cell in Clear Cell Renal Cell Carcinoma Patients through the Exosomal Pathway. Oncotarget 2017, 8, 37783–37795. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Xiang, J.Y.; Ding, G.P.; Cao, L.P. Cholangiocarcinoma-Derived Exosomes Inhibit the Antitumor Activity of Cytokine-Induced Killer Cells by Down-Regulating the Secretion of Tumor Necrosis Factor-Alpha and Perforin. J. Zhejiang Univ. Sci. B 2016, 17, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic Tumor-Derived Microvesicles Negatively Regulate Nk Cell Function by a Mechanism Involving Tgf-Beta and Mir23a Transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef] [PubMed]
- Colaco, C.A.; Bailey, C.R.; Walker, K.B.; Keeble, J. Heat Shock Proteins: Stimulators of Innate and Acquired Immuni-ty. Biomed. Res. Int. 2013, 2013, 461230. [Google Scholar] [CrossRef]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer Drugs Cause Release of Exosomes with Heat Shock Proteins from Human Hepatocellular Carcinoma Cells That Elicit Effective Natural Killer Cell Antitumor Responses in Vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef]
- Katsiougiannis, S.; Chia, D.; Kim, Y.; Singh, R.P.; Wong, D.T. Saliva exosomes from pancreatic tumor–bearing mice modulate NK cell phenotype and antitumor cytotoxicity. FASEB J. 2017, 31, 998–1010. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.G.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, L.; Wei, W.; Deng, X.; Ma, L.; Hao, S. Tumor cell-derived exosome-targeted dendritic cells stimulate stronger CD8+ CTL responses and antitumor immunities. Biochem. Biophys. Res. Commun. 2013, 436, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, G.G.; Zelante, B.B.; Toniolo, P.A.; Migliori, I.K.; Barbuto, J.A.M. Dendritic Cell-Derived Exosomes may be a Tool for Cancer Immunotherapy by Converting Tumor Cells into Immunogenic Targets. Front. Immunol. 2015, 5, 692. [Google Scholar] [CrossRef] [PubMed]
- Leone, D.A.; Peschel, A.; Brown, M.; Schachner, H.; Ball, M.J.; Gyuraszova, M.; Salzer-Muhar, U.; Fukuda, M.; Vizzardelli, C.; Bohle, B.; et al. Surface Lamp-2 Is an Endocytic Receptor That Diverts Antigen Internalized by Human Den-dritic Cells into Highly Immunogenic Exosomes. J. Immunol. 2017, 199, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Valenti, R.; Huber, V.; Filipazzi, P.; Pilla, L.; Sovena, G.; Villa, A.; Corbelli, A.; Fais, S.; Parmiani, G.; Rivoltini, L. Human Tumor-Released Microvesicles Promote the Differentiation of Myeloid Cells with Transforming Growth Fac-tor-Beta-Mediated Suppressive Activity on T Lymphocytes. Cancer Res. 2006, 66, 9290–9298. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; De Veirman, K.; De Beule, N.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Vanderkerken, K.; Menu, E. The Bone Marrow Microenvironment Enhances Multiple Myeloma Progression by Exosome-Mediated Activation of Mye-loid-Derived Suppressor Cells. Oncotarget 2015, 6, 43992–44004. [Google Scholar] [CrossRef] [PubMed]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D.; et al. Pre-Metastatic Cancer Exosomes Induce Immune Sur-veillance by Patrolling Monocytes at the Metastatic Niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef]
- Schuldner, M.; Dörsam, B.; Shatnyeva, O.; Reiners, K.S.; Kubarenko, A.; Hansen, H.P.; Finkernagel, F.; Roth, K.; Theurich, S.; Nist, A.; et al. Exosome-dependent immune surveillance at the metastatic niche requires BAG6 and CBP/p300-dependent acetylation of p53. Theranostics 2019, 9, 6047–6062. [Google Scholar] [CrossRef]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple Myeloma Exosomes Estab-lish a Favourable Bone Marrow Microenvironment with Enhanced Angiogenesis and Immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Harshyne, L.A.; Nasca, B.J.; Kenyon, L.C.; Andrews, D.W.; Hooper, D.C. Serum exosomes and cytokines promote a T-helper cell type 2 environment in the peripheral blood of glioblastoma patients. Neuro-Oncology 2015, 18, 206–215. [Google Scholar] [CrossRef]
- Liu, H.; Chen, L.; Liu, J.; Meng, H.; Zhang, R.; Ma, L.; Wu, L.; Yu, S.; Shi, F.; Li, Y.; et al. Co-Delivery of Tumor-Derived Exosomes with Al-pha-Galactosylceramide on Dendritic Cell-Based Immunotherapy for Glioblastoma. Cancer Lett. 2017, 411, 182–190. [Google Scholar] [CrossRef]
- Bu, N.; Wu, H.; Zhang, G.; Ma, X.; Zhao, P.; Zhai, N.; Xiang, L.; Cao, H.; Yang, X.; Liu, J. Exosome from chaperone-rich cell lysates-loaded dendritic cells produced by CELLine 1000 culture system exhibits potent immune activity. Biochem. Biophys. Res. Commun. 2015, 456, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Bu, N.; Wu, H.; Zhang, G.; Zhan, S.; Zhang, R.; Sun, H.; Du, Y.; Yao, L.; Wang, H. Exosomes from Dendritic Cells Loaded with Chaperone-Rich Cell Lysates Elicit a Potent T Cell Immune Response Against Intracranial Glioma in Mice. J. Mol. Neurosci. 2015, 56, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive Effects of Hy-poxia-Induced Glioma Exosomes through Myeloid-Derived Suppressor Cells Via the Mir-10a/Rora and Mir-21/Pten Path-ways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, W.; Wang, J.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Guo, Q.; et al. Glioma Ex-osomes Mediate the Expansion and Function of Myeloid-Derived Suppressor Cells through Microrna-29a/Hbp1 and Mi-crorna-92a/Prkar1a Pathways. Int. J. Cancer 2019, 144, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Javeed, N.; Gustafson, M.P.; Dutta, S.K.; Lin, Y.; Bamlet, W.R.; Oberg, A.L.; Petersen, G.M.; Chari, S.T.; Dietz, A.B.; Mukhopadhyay, D. Immunosuppressive Cd14(+)Hla-Dr(Lo/Neg) Monocytes Are Elevated in Pancreatic Cancer and ‘Primed’ by Tumor-Derived Exosomes. Oncoimmunology 2017, 6, e1252013. [Google Scholar] [CrossRef] [PubMed]
- Que, R.S.; Lin, C.; Ding, G.P.; Wu, Z.R.; Cao, L.P. Increasing the Immune Activity of Exosomes: The Effect of Mir-na-Depleted Exosome Proteins on Activating Dendritic Cell/Cytokine-Induced Killer Cells against Pancreatic Cancer. J. Zhejiang Univ. Sci. B 2016, 17, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, S.; Jia, S.; Ding, G.; Jiang, G.; Cao, L. Integrated Analysis of Long Non-Coding RNA and mRNA Expression Profile in Pancreatic Cancer Derived Exosomes Treated Dendritic Cells by Microarray Analysis. J. Cancer 2018, 9, 21–31. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell. Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Mo, Y.; Zeng, Z.; Wei, P.; Li, T. Effect of hyperthermic CO2-treated dendritic cell-derived exosomes on the human gastric cancer AGS cell line. Oncol. Lett. 2015, 10, 71–76. [Google Scholar] [CrossRef]
- Behzadi, E.; Hosseini, H.M.; Halabian, R.; Fooladi, A.A.I. Macrophage Cell-Derived Exosomes/Staphylococcal Entero-toxin B against Fibrosarcoma Tumor. Microb. Pathog. 2017, 111, 132–138. [Google Scholar] [CrossRef]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring Anticancer Immune Response by Targeting Tumor-Derived Exosomes With a HSP70 Peptide Aptamer. J. Natl. Cancer Inst. 2016, 108, djv330. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, Y.; Ahmed, K.A.; Ahmed, S.; Sami, A.; Chibbar, R.; Xu, Q.; Kane, S.E.; Hao, S.; Mulligan, S.J.; et al. Exo-somal Pmhc-I Complex Targets T Cell-Based Vaccine to Directly Stimulate Ctl Responses Leading to Antitumor Immunity in Transgenic Fvbneun and Hla-A2/Her2 Mice and Eradicating Trastuzumab-Resistant Tumor in Athymic Nude Mice. Breast Cancer Res. Treat. 2013, 140, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a Sting-Dependent Pathway and Reinforce Anti-tumor Immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; DeMaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Taghikhani, A.; Hassan, Z.M.; Ebrahimi, M.; Moazzeni, S. microRNA modified tumor-derived exosomes as novel tools for maturation of dendritic cells. J. Cell. Physiol. 2019, 234, 9417–9427. [Google Scholar] [CrossRef]
- Li, J.; Huang, S.; Zhou, Z.; Lin, W.; Chen, S.; Chen, M.; Ye, Y. Exosomes Derived from Raav/Afp-Transfected Dendritic Cells Elicit Specific T Cell-Mediated Immune Responses against Hepatocellular Carcinoma. Cancer Manag. Res. 2018, 10, 4945–4957. [Google Scholar] [CrossRef]
- Rao, Q.; Zuo, B.; Lu, Z.; Gao, X.; You, A.; Wu, C.; Du, Z.; Yin, H. Tumor-Derived Exosomes Elicit Tumor Suppression in Murine Hepatocellular Carcinoma Models and Humans in Vitro. Hepatology 2016, 64, 456–472. [Google Scholar] [CrossRef]
- Lu, Z.; Zuo, B.; Jing, R.; Gao, X.; Rao, Q.; Liu, Z.; Qi, H.; Guo, H.; Yin, H. Dendritic Cell-Derived Exosomes Elicit Tumor Re-gression in Autochthonous Hepatocellular Carcinoma Mouse Models. J. Hepatol. 2017, 67, 739–748. [Google Scholar] [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. OncoImmunology 2016, 5, e1071008. [Google Scholar] [CrossRef]
- Li, W.; Mu, D.; Tian, F.; Hu, Y.; Jiang, T.; Han, Y.; Chen, J.; Han, G.; Li, X. Exosomes derived from Rab27a-overexpressing tumor cells elicit efficient induction of antitumor immunity. Mol. Med. Rep. 2013, 8, 1876–1882. [Google Scholar] [CrossRef]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor Exosomes Block Dendritic Cells Maturation to De-crease the T Cell Immune Response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Bala, S. Extracellular Vesicles in Oral Squamous Carcinoma Carry Oncogenic Mirna Profile and Reprogram Monocytes Via Nf-Kappab Pathway. Oncotarget 2018, 9, 34838–34854. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Guo, D.; Weng, L.; Wang, S.; Ma, Z.; Yang, Y.; Wang, P.; Wang, J.; Cai, Z. Tumor-Derived Exosomes Educate Den-dritic Cells to Promote Tumor Metastasis Via Hsp72/Hsp105-Tlr2/Tlr4 Pathway. Oncoimmunology 2017, 6, e1362527. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor Exosomes Inhibit Differentiation of Bone Marrow Dendritic Cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, C.; Wei, W.; Shen, C.; Deng, X.; Chen, L.; Ma, L.; Hao, S. Dendritic Cells Pulsed with Leukemia Cell-Derived Exosomes More Efficiently Induce Antileukemic Immunities. PLoS ONE 2014, 9, e91463. [Google Scholar] [CrossRef]
- Huang, F.; Wan, J.; Hao, S.; Deng, X.; Chen, L.; Ma, L. TGF-Beta1-Silenced Leukemia Cell-Derived Exosomes Target Den-dritic Cells to Induce Potent Anti-Leukemic Immunity in a Mouse Model. Cancer Immunol. Immunother. 2017, 66, 1321–1331. [Google Scholar] [CrossRef]
- Ren, G.; Wang, Y.; Yuan, S.; Wang, B. Dendritic Cells Loaded with Hela-Derived Exosomes Simulate an Antitumor Im-mune Response. Oncol. Lett. 2018, 15, 6636–6640. [Google Scholar] [CrossRef]
- Chen, S.; Lv, M.; Fang, S.; Ye, W.; Gao, Y.; Xu, Y. Poly(I:C) Enhanced Anti-Cervical Cancer Immunities Induced by Den-dritic Cells-Derived Exosomes. Int. J. Biol. Macromol. 2018, 113, 1182–1187. [Google Scholar] [CrossRef]
- Damo, M.; Wilson, D.S.; Simeoni, E.; Hubbell, J.A. Tlr-3 Stimulation Improves Anti-Tumor Immunity Elicited by Den-dritic Cell Exosome-Based Vaccines in a Murine Model of Melanoma. Sci. Rep. 2015, 5, 17622. [Google Scholar] [CrossRef]
- Cianciaruso, C.; Beltraminelli, T.; Duval, F.; Nassiri, S.; Hamelin, R.; Mozes, A.; Gallart-Ayala, H.; Ceada Torres, G.; Torchia, B.; Ries, C.H.; et al. Molecular Profiling and Functional Analysis of Macrophage-Derived Tumor Ex-tracellular Vesicles. Cell Rep. 2019, 27, 3062–3080. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypox-ia-Induced Tumor Exosomes Promote M2-Like Macrophage Polarization of Infiltrating Myeloid Cells and Mi-crorna-Mediated Metabolic Shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lei, Y.; Wu, M.; Li, N. Regulation of Macrophage Activation and Polarization by HCC-Derived Exosomal lncRNA TUC339. Int. J. Mol. Sci. 2018, 19, 2958. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Liu, J.; Liu, Q.; Liu, Y.; Fan, L.; Wang, F.; Yu, H.; Li, Y.; Bu, L.; Li, X.; et al. Exosomes from Melatonin Treated Hepatocellularcarcinoma Cells Alter the Immunosupression Status through Stat3 Pathway in Macro-phages. Int. J. Biol. Sci. 2017, 13, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Zhou, W.; Liu, L.; Fong, M.Y.; Champer, J.; Van Haute, D.; Chin, A.R.; Ren, X.; Gugiu, B.G.; Meng, Z.; et al. Macrophage Immunomodulation by Breast Cancer-Derived Exosomes Requires Toll-Like Receptor 2-Mediated Activation of Nf-Kappab. Sci. Rep. 2014, 4, 5750. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-Y.; Lee, J.-K.; Jeon, Y.K.; Kim, C.-W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Wang, F.; Li, B.; Wei, Y.; Zhao, Y.; Wang, L.; Zhang, P.; Yang, J.; He, W.; Chen, H.; Jiao, Z.; et al. Tumor-derived exosomes induce PD1+ macrophage population in human gastric cancer that promotes disease progression. Oncogenesis 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. OncoImmunology 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Tai, S.K.; Yang, M.H. Snail-Overexpressing Cancer Cells Promote M2-Like Polarization of Tu-mor-Associated Macrophages by Delivering Mir-21-Abundant Exosomes. Neoplasia 2018, 20, 775–788. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal Mirna Confers Chemo Resistance Via Targeting Cav1/P-Gp/M2-Type Macrophage Axis in Ovarian Cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Chen, K.; Lu, X.; Li, A.; Liu, C.; Wu, B. Dual Targeting of Pd-L1 and Pd-L2 by Pced1b-As1 Via Sponging Hsa-Mir-194-5p Induces Immunosuppression in Hepatocellular Carcinoma. Hepatol. Int. 2020, 21. [Google Scholar] [CrossRef]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic Reticulum Stress-Induced Exosomal Mir-27a-3p Promotes Immune Escape in Breast Cancer Via Regulating Pd-L1 Expression in Macrophages. J. Cell Mol. Med. 2020, 24, 9560–9573. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, L.; Dai, T.; Jin, K.; Zhang, Z.; Wang, S.; Xie, F.; Fang, P.; Yang, B.; Huang, H.; et al. Tumor-derived exosomes antagonize innate antiviral immunity. Nat. Immunol. 2018, 19, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-inflammatory Drugs From the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Antimisiaris, S.G.; Mourtas, S.; Marazioti, A. Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics 2018, 10, 218. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef]
- Familtseva, A.; Jeremic, N.S.; Tyagi, S.C. Exosomes: Cell-created drug delivery systems. Mol. Cell. Biochem. 2019, 459, 1–6. [Google Scholar] [CrossRef]
- Shigekawa, K.; Dower, W.J. Electroporation of eukaryotes and prokaryotes: A general approach to the introduction of macromolecules into cells. Biotechniques 1988, 6, 742–751. [Google Scholar]
- Thabet, E.; Yusuf, A.; Abdelmonsif, D.A.; Nabil, I.; Mourad, G.; Mehanna, R.A. Extracellular Vesicles Mirna-21: A Poten-tial Therapeutic Tool in Premature Ovarian Dysfunction. Mol. Hum. Reprod. 2020, 206, 906–919. [Google Scholar] [CrossRef]
- Munir, J.; Yoon, J.K.; Ryu, S. Therapeutic Mirna-Enriched Extracellular Vesicles: Current Approaches and Future Pro-spects. Cells 2020, 9, 2271. [Google Scholar] [CrossRef] [PubMed]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Pérez-López, A.M.; Rubio-Ruiz, B.; Sebastián, V.; Hamilton, L.; Adam, C.; Bray, T.L.; Irusta, S.; Brennan, P.M.; Lloyd-Jones, G.C.; Sieger, D.; et al. Gold-Triggered Uncaging Chemistry in Living Systems. Angew. Chem. Int. Ed. 2017, 56, 12548–12552. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Albero, M.; Rubio-Ruiz, B.; Perez-Lopez, A.M.; Sebastian, V.; Martin-Duque, P.; Arruebo, M.; Santamaria, J.; Un-citi-Broceta, A. Cancer-Derived Exosomes Loaded with Ultrathin Palladium Nanosheets for Targeted Bioorthogonal Cataly-sis. Nat. Catal. 2019, 2, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Pérez-López, A.M.; Hamilton, L.; Rubio-Ruiz, B.; Bray, T.L.; Sieger, D.; Brennan, P.M.; Unciti-Broceta, A. Frontispiece: Bioorthogonal Uncaging of the Active Metabolite of Irinotecan by Palladium-Functionalized Microdevices. Chemistry 2018, 24, 16783–16790. [Google Scholar] [CrossRef] [PubMed]
- Pérez-López, A.M.; Rubio-Ruiz, B.; Valero, T.; Contreras-Montoya, R.; Álvarez De Cienfuegos, L.; Sebastián, V.; Santamaría, J.; Unciti-Broceta, A. Bioorthogonal Uncaging of Cytotoxic Paclitaxel through Pd Nanosheet–Hydrogel Frameworks. J. Med. Chem. 2020, 63, 9650–9659. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Kalluri, R. Emerging role of bacterial extracellular vesicles in cancer. Oncogene 2020, 39, 6951–6960. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- De La Peña, H.; Madrigal, J.A.; Rusakiewicz, S.; Bencsik, M.; Cave, G.W.V.; Selman, A.; Rees, R.C.; Travers, P.J.; Dodi, I.A. Artificial exosomes as tools for basic and clinical immunology. J. Immunol. Methods 2009, 344, 121–132. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic Prospects of Extracellular Vesicles in Cancer Treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Zhang, K.L.; Wang, Y.J.; Sun, J.; Zhou, J.; Xing, C.; Huang, G.; Li, J.; Yang, H. Artificial Chimeric Exosomes for An-ti-Phagocytosis and Targeted Cancer Therapy. Chem. Sci. 2019, 10, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.P.; Moon, M.J.; Park, R.; Jeong, Y.Y. Bioactive Nanoparticles for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3877. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| miRNA | ExV Release | Induction | Tumor | Reference |
|---|---|---|---|---|
| miR-155 | bystander T-cell activated DCs | Antigen-specific T cells | AML | [52] |
| let-7i | cells | Expression of MHCII, CD80 and CD40, DC maturation, | breast | [53] |
| miR-16 | EGCG-treated cells | Inhibition of TAM infiltration and M2 polarization. | breast | [54] |
| miR-92 | CAFs | higher cellular levels of PD-L1 and miR-92; apoptosis, impaired proliferation of T cells | breast | [55] |
| miR-27a-3p | cells | immune evasion by up-regulating PD-L1 via MAGI2/PTEN/PI3K axis in macrophages | breast | [56] |
| miR-15a, miR-15b, miR-16, miR-193a-3p | PD-L1 targets | BE, EAC | [57] | |
| miR-1246 | mutant TP53 cells | Increase in TGF-β, reprogramming of macrophages | colon | [58] |
| miR-29a-3p, miR-21-5p | TAMs | Suppression of STAT3 in CD4+T cells | EOC | [59] |
| miR-101 | cells | Targeting of EZH2, EED, DNMT3, reversion of promoter methylation, re-expression of LRRC4 in GBM cells | GBM | [60] |
| miR-16-5p | M1 macrophage | T cell immune response that in turn inhibits tumor formation by decreasing the expression of PD-L1 | GC | [61] |
| miR-10a, miR-21 | hypoxic cells | Targeting of RORA and PTEN Expansion and activation of MDSCs | glioma | [62] |
| miR-29a, miR-92a | hypoxic cells | Targeting of Hbp1 and Prkar1a, differentiation of MDSCs | glioma | [63] |
| miR-21 | cells | Suppression of M1, increase in M2 markers in CD14+monocytes | HNC | [64] |
| miR-690 | melanoma | Activation of caspase-3, -7 -9, down-regulation of BCL-2, MCL-1, BCL-xL in CD4+T cells | melanoma | [65] |
| miR-150-5p, miR-142-3p | Tregs | Increase in IL-10, decrease in IL-6 following LPS stimulation in DCs | mice | [66] |
| miR-186 | NK cells | Cytotoxicity against neuroblastoma cell, inhibition of NK cells | neuro-blastoma | [67,68] |
| miR-24-3p | serum | repression of FGF11, Suppression of T-cells | NPC | [69] |
| miR-24-3p, miR-891a, miR-106a-5p, miR-20a-5p, miR-1908, | cells | Down-regulation of MARK1 signaling pathway in T cells | NPC | [70] |
| miR-210, miR-23a | hypoxic cells | Release of TGF-β1 in NK cells, decrease in expression of NKG2D, inhibition of NK cell function | NSCLC | [71] |
| miR-320d, miR-320c, miR-320b miR-125b-5p | cells | efficacy of immunotherapy | NSCLC | [72] |
| miR-1246 | cells | transfer to M2-type macrophages, but not to M0-type macrophages | OC | [73] |
| miR-21, miR-27 | plasma | Activation of NF-κB pathway increase in IL-6, CCL2, prostaglandin E2 and MMP 9 in monocytes | OSCC | [74] |
| miR-203 | cells | Downregulation of TLR4, TNF-α and IL-12 in DCs | pancreas | [75] |
| lncRNA | ||||
| PCED1B-AS1 | cells | Interaction of PCED1B-AS1 with miR-194-5p, increased PD-L1 and PD-L2 expression, inhibition of T cells and macrophages, cell proliferation, colony formation, inhibited apoptosis, tumor formation | HCC | [76] |
| TUC339 | cells | Toll-like receptor signaling, regulation of the actin cytoskeleton, regulation of macrophage M1/M2 polarization | HCC | [77] |
| MALAT1, HOTAIR, HOTTIP, AGAP2-AS1, ATB, TCF7, FOXD2-AS1, HOXA11-AS, PCAF1, BCAR4 | cells | Cancer development | NSCLC | [78] |
| ENST00000560647 | cells | Differential expression in DCs, immune escape | pancreas | [79] |
| PD-L1 | ||||
| draining lymph node | Suppression of T cell activation | [51] | ||
| HER2-positive cells | immunosuppressive | breast | [80] | |
| cells | Inhibition of maturation and migration of DCs, decrease in CD4+IFN-γ+Th1 cell differentiation, increase in Tregs. | breast, LLC, | [81] | |
| cells | Inflammation, immune escape | CLL | [82] | |
| cells | Shift of monocytes towards M2 phenotype. | glioblastoma, | [83] | |
| cells | Secretion of IL-6, IL-10, IL-1β and TNF-α, reduction of activation of STAT3 in macrophages | HCC | [84] | |
| cells | immune cell reprogramming by the adenosine pathway | HNSCC | [85] | |
| cells | Suppressing of CD8+T cells, positive correlation with IFN-γ | metastatic melanoma | [86] | |
| plasma | Reduction of the secretion of IFN-γ by T cells, apoptosis in CD8+T cells | NSCL | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarzenbach, H.; Gahan, P.B. Exosomes in Immune Regulation. Non-Coding RNA 2021, 7, 4. https://doi.org/10.3390/ncrna7010004
Schwarzenbach H, Gahan PB. Exosomes in Immune Regulation. Non-Coding RNA. 2021; 7(1):4. https://doi.org/10.3390/ncrna7010004
Chicago/Turabian StyleSchwarzenbach, Heidi, and Peter B. Gahan. 2021. "Exosomes in Immune Regulation" Non-Coding RNA 7, no. 1: 4. https://doi.org/10.3390/ncrna7010004
APA StyleSchwarzenbach, H., & Gahan, P. B. (2021). Exosomes in Immune Regulation. Non-Coding RNA, 7(1), 4. https://doi.org/10.3390/ncrna7010004