Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer



Abstract
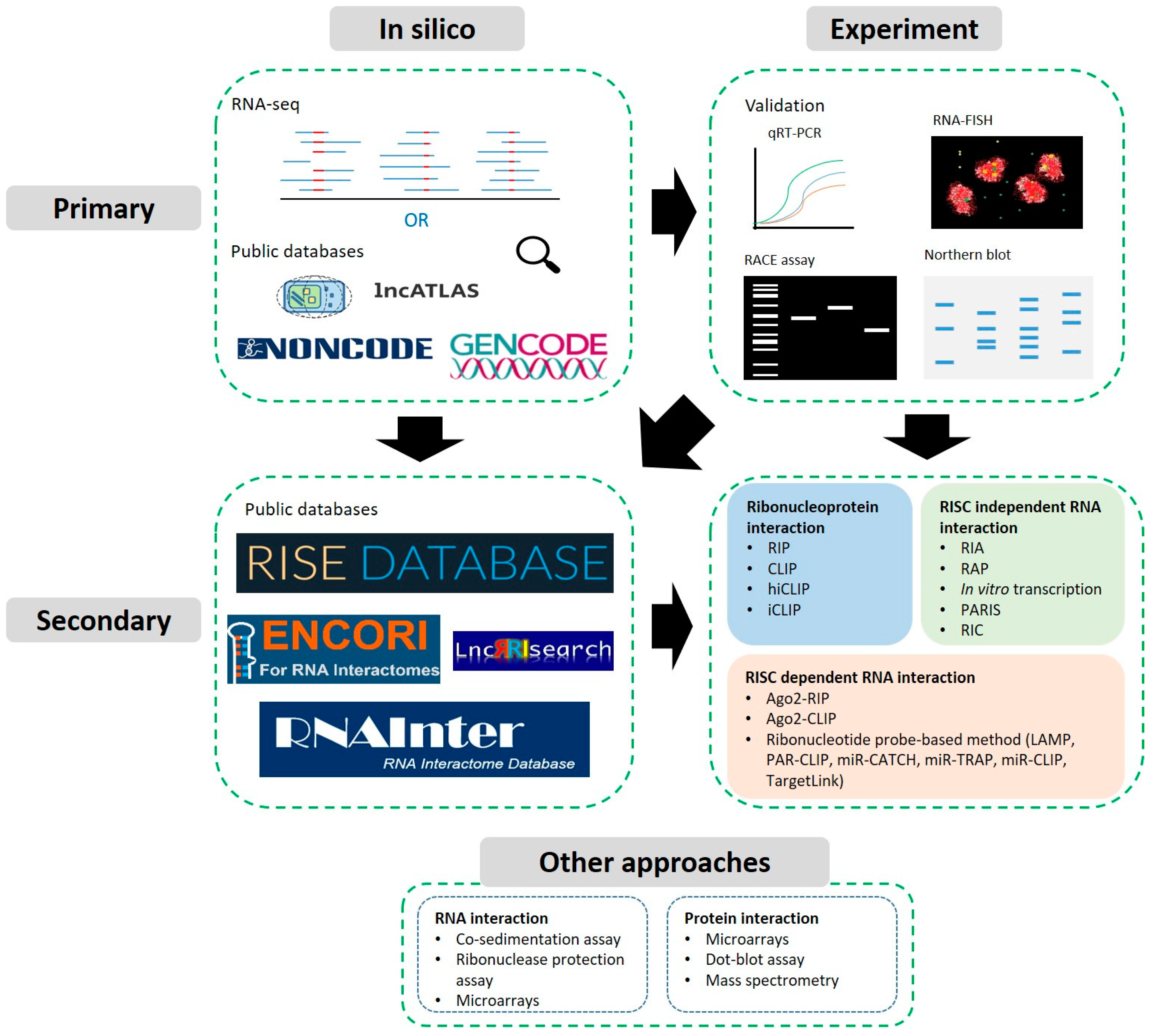
1. Introduction
2. Identification and Primary Characterisation
2.1. Predictions, Identification from High-Throughput Data and Databases
2.2. Experimental Approaches: Validation of Expression, Localisation & Structure
3. Secondary Characterisation: Predicting and Detecting Interactions
3.1. Predictions and Databases

{kind=link}
| Database | Link | Interaction Type | Primary Source | Additional Sources |
|---|---|---|---|---|
| NPInter v4 (2019) [160] | http://bigdata.ibp.ac.cn/npinter4 (accessed on 8 March 2021) | miRNA-RNA; ncRNA-DNA; ncRNA-Protein; circRNA | EXP: Re-processing and integration of experimental data (GEO; ENCODE; RISE) | CPU: miRNA binding (miRanda, TargetScan); Disease association (LncRNADisease, MNDR, eDGAR and circRNADisease)EXP: Literature mining |
| lncRRIsearch (2019) [163] | http://rtools.cbrc.jp/LncRRIsearch/ (accessed on 8 March 2021) | lncRNA-mRNA | CPU: RIBlast | EXP: Tissue expression |
| DIANA-LncBase v3 (2020) [164] | https://diana.e-ce.uth.gr/lncbasev3 (accessed on 8 March 2021) | miRNA-lncRNA | EXP: Re-processing and integration of experimental data (miRNA, AGO2-CLIP-Seq and CLIP-Seq) | CPU: Correlation with lncRNA expression |
| SPONGEdb v1 (2021) [165] | https://exbio.wzw.tum.de/sponge/home (accessed on 8 March 2021) | miRNA-lncRNA | CPU: DIANA-LncBase, TargetScan, miRcode, miRTarBase | EXP: TCGA expression |
| LnCeVar v1 (2020) [166] | http://www.bio-bigdata.net/LnCeVar/ (accessed on 8 March 2021) | miRNA-lncRNA | EXP: SNP and mutation data from TCGA, COSMIC, 1000 Genomes Project | CPU: Integration from miRanda, mirBase, miRTarBase, TargetScan |
| miRSponge v1 (2015) [167] | http://bio-bigdata.hrbmu.edu.cn/miRSponge/ (accessed on 8 March 2021) | miRNA-lncRNA miRNA-circRNA | EXP: Manual curation from literature | CPU: Integration from TarBase, miRTarBase, miRanda, miRecord |
| starBase/ENCORI v2 (2014/2021) [79] | http://starbase.sysu.edu.cn/ (accessed on 8 March 2021) | miRNA-ncRNA; RBP-RNA;RNA-RNA | EXP: Re-processing and integration of experimental data (CLIP-Seq & variations) | CPU: Correlation of RBP somatic mutation with diseases EXP: Pan-Cancer networks from expression profiles (TCGA) |
| RAID v3/RNAInter (2020) [168] | http://www.rna-society.org/raid/ (accessed on 8 March 2021) | RNA-Protein; RNA-RNA; RNA-Histone; RNA-Drug | EXP/CPU: Integration of literature sources and 35 databases. | EXP: Methylation, localisation and editing data from other databases. |
| RISE (2018) [161] | http://rise.life.tsinghua.edu.cn/index.html (accessed on 8 March 2021) | RNA-RNA | EXP: Integration from sequencing based studies | CPU: Integration with several other databases (RAIN, RAID, NPInter) |
| LncRNA2Target v2 (2019) [169] | http://123.59.132.21/lncrna2target/ (accessed on 8 March 2021) | lncRNA-RNA | EXP: Manual extraction of interaction associations from literature | EXP: Re-processing of lncRNA perturbation RNA-Seq datasets |
| LncExpDB (2020) [170] | https://bigd.big.ac.cn/lncexpdb/interactions (accessed on 8 March 2021) | lncRNA-mRNA | CPU: Co-expression network analysis and prediction | EXP: Expression extracted from public repositories (GEO, SRA and ArrayExpress) |
| LncACTdb v2 (2019) [171] | http://www.bio-bigdata.net/LncACTdb (accessed on 8 March 2021) | miRNA-lncRNA-mRNAmiRNA-circRNA | EXP: Manual curation from literature | CPU: Predictions from networks and integration with Pan-Cancer data (TCGA) |
3.2. Sequencing Compatible Approaches
3.2.1. Ribonucleoprotein Complex Interaction Detection
3.2.2. RISC Dependent RNA Interactions
3.2.3. RISC Independent RNA Interactions
3.3. Other Approaches and Biochemical Assays
3.3.1. Protein Interaction Assays
3.3.2. RNA Interaction Assays
4. Closing Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- 6 Non-coding RNA characterization. Nature 2019. [CrossRef]
- Fabbri, M.; Girnita, L.; Varani, G.; Calin, G.A. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019, 29, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Lin, S.; Li, Y.; Zhao, W.; Mills, G.B.; Sahni, N. Functional variomics and network perturbation: Connecting genotype to phenotype in cancer. Nat. Rev. Genet. 2017, 18, 395–410. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Jewer, M.; Findlay, S.D.; Postovit, L.-M. Post-transcriptional regulation in cancer progression: Microenvironmental control of alternative splicing and translation. J. Cell Commun. Signal. 2012, 6, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Blume, S.W.; Grizzle, W.E. Translational Dysregulation in Cancer: Molecular Insights and Potential Clinical Applications in Biomarker Development. Front. Oncol. 2017, 7, 158. [Google Scholar] [CrossRef]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.L.; Cenik, C.; Reuter, J.A.; Kiss, G.; Pande, V.S.; Snyder, M.P.; Greenleaf, W.J. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat. Genet. 2016, 48, 117–125. [Google Scholar] [CrossRef]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Zhou, S.; Treloar, A.E.; Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discov. 2016, 6, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Pratt, H.; Weng, Z. Decoding the non-coding genome: Opportunities and challenges of genomic and epigenomic consortium data. Curr. Opin. Syst. Biol. 2018, 11, 82–90. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Gloss, B.S.; Dinger, M.E. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta 2016, 1859, 16–22. [Google Scholar] [CrossRef]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef]
- Suzuki, H.; Zuo, Y.; Wang, J.; Zhang, M.Q.; Malhotra, A.; Mayeda, A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006, 34, e63. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- St; St. Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef]
- Wenric, S.; ElGuendi, S.; Caberg, J.-H.; Bezzaou, W.; Fasquelle, C.; Charloteaux, B.; Karim, L.; Hennuy, B.; Frères, P.; Collignon, J.; et al. Transcriptome-wide analysis of natural antisense transcripts shows their potential role in breast cancer. Sci. Rep. 2017, 7, 17452. [Google Scholar] [CrossRef]
- Sanchez de Groot, N.; Armaos, A.; Graña-Montes, R.; Alriquet, M.; Calloni, G.; Vabulas, R.M.; Tartaglia, G.G. RNA structure drives interaction with proteins. Nat. Commun. 2019, 10, 3246. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Matouk, I.J.; DeGroot, N.; Mezan, S.; Ayesh, S.; Abu-lail, R.; Hochberg, A.; Galun, E. The H19 non-coding RNA is essential for human tumor growth. PLoS ONE 2007, 2, e845. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef]
- Sirchia, S.M.; Tabano, S.; Monti, L.; Recalcati, M.P.; Gariboldi, M.; Grati, F.R.; Porta, G.; Finelli, P.; Radice, P.; Miozzo, M. Misbehaviour of XIST RNA in breast cancer cells. PLoS ONE 2009, 4, e5559. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Tang, S.; Li, M.; Feng, A.; Qin, L.; Liu, Z.; Wang, X. The role of long noncoding RNA HOTAIR in the acquired multidrug resistance to imatinib in chronic myeloid leukemia cells. Hematology 2017, 22, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Yang, Y.A.; Zhang, A.; Fong, K.-W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 2016, 35, 2746–2755. [Google Scholar] [CrossRef]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. Adv. Exp. Med. Biol. 2017, 1008, 1–46. [Google Scholar] [CrossRef]
- Wang, W.-T.; Han, C.; Sun, Y.-M.; Chen, T.-Q.; Chen, Y.-Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55. [Google Scholar] [CrossRef]
- Chi, Y.; Wang, D.; Wang, J.; Yu, W.; Yang, J. Long Non-Coding RNA in the Pathogenesis of Cancers. Cells 2019, 8, 1015. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Du, W.W.; Wu, N.; Yang, W.; Awan, F.M.; Fang, L.; Ma, J.; Li, X.; Zeng, Y.; Yang, Z.; et al. A circular RNA promotes tumorigenesis by inducing c-myc nuclear translocation. Cell Death Differ. 2017, 24, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Xiao, Y.; Ma, J.; Tang, Y.; Tian, B.; Zhang, Y.; Li, X.; Wu, Z.; Yang, D.; Zhou, Y.; et al. Circular RNAs in Cancer: Emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol. Cancer 2019, 18, 90. [Google Scholar] [CrossRef]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Zhang, M.; Matyunina, L.V.; Walker, L.D.; Chen, W.; Xiao, H.; Benigno, B.B.; Wu, R.; McDonald, J.F. Evidence for the importance of post-transcriptional regulatory changes in ovarian cancer progression and the contribution of miRNAs. Sci. Rep. 2017, 7, 8171. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef]
- Huang, B.; Song, J.H.; Cheng, Y.; Abraham, J.M.; Ibrahim, S.; Sun, Z.; Ke, X.; Meltzer, S.J. Long non-coding antisense RNA KRT7-AS is activated in gastric cancers and supports cancer cell progression by increasing KRT7 expression. Oncogene 2016, 35, 4927–4936. [Google Scholar] [CrossRef] [PubMed]
- He, R.-Z.; Luo, D.-X.; Mo, Y.-Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis. 2019, 6, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. J. Hum. Genet. 2020, 65, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Gardini, A. Global Run-On Sequencing (GRO-Seq). Methods Mol. Biol. 2017, 1468, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Mahat, D.B.; Kwak, H.; Booth, G.T.; Jonkers, I.H.; Danko, C.G.; Patel, R.K.; Waters, C.T.; Munson, K.; Core, L.J.; Lis, J.T. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 2016, 11, 1455–1476. [Google Scholar] [CrossRef]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Magnuson, B.; Wilson, T.E.; Ljungman, M. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014, 67, 45–54. [Google Scholar] [CrossRef]
- Plessy, C.; Bertin, N.; Takahashi, H.; Simone, R.; Salimullah, M.; Lassmann, T.; Vitezic, M.; Severin, J.; Olivarius, S.; Lazarevic, D.; et al. Linking promoters to functional transcripts in small samples with nanoCAGE and CAGEscan. Nat. Methods 2010, 7, 528–534. [Google Scholar] [CrossRef]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef]
- Raha, D.; Hong, M.; Snyder, M. ChIP-Seq: A method for global identification of regulatory elements in the genome. Curr. Protoc. Mol. Biol. 2010, 21. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef]
- Antonov, I.V.; Mazurov, E.; Borodovsky, M.; Medvedeva, Y.A. Prediction of lncRNAs and their interactions with nucleic acids: Benchmarking bioinformatics tools. Brief. Bioinform. 2019, 20, 551–564. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Liu, C. The computational approaches of lncRNA identification based on coding potential: Status quo and challenges. Comput. Struct. Biotechnol. J. 2020, 18, 3666–3677. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Salzman, J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Zou, D.; Ma, L.; Yu, J.; Zhang, Z. Biological databases for human research. Genom. Proteom. Bioinform. 2015, 13, 55–63. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tassé, A.-M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Geo NCBI-GEO. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 29 January 2021).
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 29 January 2021).
- GTEx Portal. Available online: https://gtexportal.org/home/ (accessed on 29 January 2021).
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; de Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute Cancer Cell Line Encyclopedia (CCLE). Available online: https://portals.broadinstitute.org/ccle (accessed on 29 January 2021).
- Schriml, L.M.; Mitraka, E.; Munro, J.; Tauber, B.; Schor, M.; Nickle, L.; Felix, V.; Jeng, L.; Bearer, C.; Lichenstein, R.; et al. Human Disease Ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Res. 2019, 47, D955–D962. [Google Scholar] [CrossRef]
- Gene Ontology Consortium the Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- NCBI-MeSH. Available online: https://www.ncbi.nlm.nih.gov/mesh/ (accessed on 29 January 2021).
- GenomeOC Therapeutically Applicable Research to Generate Effective Treatments. Available online: https://ocg.cancer.gov/programs/target (accessed on 29 January 2021).
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef]
- Blin, K.; Dieterich, C.; Wurmus, R.; Rajewsky, N.; Landthaler, M.; Akalin, A. DoRiNA 2.0--upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2015, 43, D160–D167. [Google Scholar] [CrossRef]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gu, J.; Wang, T.; Ding, Z. OncomiRDB: A database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics 2014, 30, 2237–2238. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. dbDEMC 2.0: Updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49, D165–D171. [Google Scholar] [CrossRef]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, A.; Zou, D.; Xu, X.; Xia, L.; Yu, J.; Bajic, V.B.; Zhang, Z. LncRNAWiki: Harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 2015, 43, D187–D192. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Shang, S.; Guo, S.; Li, X.; Zhou, H.; Liu, H.; Sun, Y.; Wang, J.; Wang, P.; Zhi, H.; et al. Lnc2Cancer 3.0: An updated resource for experimentally supported lncRNA/circRNA cancer associations and web tools based on RNA-seq and scRNA-seq data. Nucleic Acids Res. 2021, 49, D1251–D1258. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wang, Z.; Pan, T.; Sahni, N.; Jin, X.; Wang, G.; Li, J.; Zheng, X.; Zhang, Y.; et al. LncMAP: Pan-cancer atlas of long noncoding RNA-mediated transcriptional network perturbations. Nucleic Acids Res. 2018, 46, 1113–1123. [Google Scholar] [CrossRef]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal ncRNA-disease repository with increased coverage and annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Liu, W.; Zhang, Q.; Guo, A.-Y. lncRNASNP2: An updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018, 46, D276–D280. [Google Scholar] [CrossRef]
- Chan, W.-L.; Huang, H.-D.; Chang, J.-G. lncRNAMap: A map of putative regulatory functions in the long non-coding transcriptome. Comput. Biol. Chem. 2014, 50, 41–49. [Google Scholar] [CrossRef]
- Zhao, H.; Shi, J.; Zhang, Y.; Xie, A.; Yu, L.; Zhang, C.; Lei, J.; Xu, H.; Leng, Z.; Li, T.; et al. LncTarD: A manually-curated database of experimentally-supported functional lncRNA-target regulations in human diseases. Nucleic Acids Res. 2020, 48, D118–D126. [Google Scholar] [CrossRef]
- Zhou, B.; Zhao, H.; Yu, J.; Guo, C.; Dou, X.; Song, F.; Hu, G.; Cao, Z.; Qu, Y.; Yang, Y.; et al. EVLncRNAs: A manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 2018, 46, D100–D105. [Google Scholar] [CrossRef]
- Lv, D.; Xu, K.; Jin, X.; Li, J.; Shi, Y.; Zhang, M.; Jin, X.; Li, Y.; Xu, J.; Li, X. LncSpA: LncRNA Spatial Atlas of Expression across Normal and Cancer Tissues. Cancer Res. 2020, 80, 2067–2071. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef]
- Maass, P.G.; Glažar, P.; Memczak, S.; Dittmar, G.; Hollfinger, I.; Schreyer, L.; Sauer, A.V.; Toka, O.; Aiuti, A.; Luft, F.C.; et al. A map of human circular RNAs in clinically relevant tissues. J. Mol. Med. 2017, 95, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Ma, X.-K.; Li, G.-W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genom. Proteom. Bioinform. 2018, 16, 226–233. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, K.; Wu, F.; Wang, W.; Zhang, K.; Hu, H.; Liu, Y.; Jiang, T. circRNA disease: A manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis. 2018, 9, 475. [Google Scholar] [CrossRef]
- Fan, C.; Lei, X.; Fang, Z.; Jiang, Q.; Wu, F.-X. CircR2Disease: A manually curated database for experimentally supported circular RNAs associated with various diseases. Database 2018, 2018. [Google Scholar] [CrossRef]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef]
- Rophina, M.; Sharma, D.; Poojary, M.; Scaria, V. Circad: A comprehensive manually curated resource of circular RNA associated with diseases. Database 2020, 2020. [Google Scholar] [CrossRef]
- Zhao, M.; Qu, H. circVAR database: Genome-wide archive of genetic variants for human circular RNAs. BMC Genom. 2020, 21, 750. [Google Scholar] [CrossRef]
- Xia, S.; Feng, J.; Chen, K.; Ma, Y.; Gong, J.; Cai, F.; Jin, Y.; Gao, Y.; Xia, L.; Chang, H.; et al. CSCD: A database for cancer-specific circular RNAs. Nucleic Acids Res. 2018, 46, D925–D929. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Yao, D.; Zhang, L.; Zheng, M.; Sun, X.; Lu, Y.; Liu, P. Circ2Disease: A manually curated database of experimentally validated circRNAs in human disease. Sci. Rep. 2018, 8, 11018. [Google Scholar] [CrossRef]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kevil, C.G.; Walsh, L.; Laroux, F.S.; Kalogeris, T.; Grisham, M.B.; Alexander, J.S. An improved, rapid Northern protocol. Biochem. Biophys. Res. Commun. 1997, 238, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Feng, Y.; Hu, Z.; Zhang, Y.; Yuan, C.-X.; Xu, X.; Zhang, L. Detection of Long Noncoding RNA Expression by Nonradioactive Northern Blots. In Long Non-Coding RNAs: Methods and Protocols; Feng, Y., Zhang, L., Eds.; Springer: New York, NY, USA, 2016; pp. 177–188. ISBN 9781493933785. [Google Scholar]
- Streit, S.; Michalski, C.W.; Erkan, M.; Kleeff, J.; Friess, H. Northern blot analysis for detection and quantification of RNA in pancreatic cancer cells and tissues. Nat. Protoc. 2009, 4, 37–43. [Google Scholar] [CrossRef]
- Zang, S.; Lin, R.-J. Northwestern Blot Analysis: Detecting RNA–Protein Interaction After Gel Separation of Protein Mixture. In RNA-Protein Complexes and Interactions: Methods and Protocols; Lin, R.-J., Ed.; Springer: New York, NY, USA, 2016; pp. 111–125. ISBN 9781493935918. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Jo, J.; Choi, S.; Oh, J.; Lee, S.-G.; Choi, S.Y.; Kim, K.K.; Park, C. Conventionally used reference genes are not outstanding for normalization of gene expression in human cancer research. BMC Bioinform. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Olivarius, S.; Plessy, C.; Carninci, P. High-throughput verification of transcriptional starting sites by Deep-RACE. Biotechniques 2009, 46, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, J.; Uszczynska-Ratajczak, B.; Santoyo-Lopez, J.; Gonzalez, J.M.; Tapanari, E.; Mudge, J.M.; Steward, C.A.; Wilming, L.; Tanzer, A.; Howald, C.; et al. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat. Commun. 2016, 7, 12339. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.R.; Orjalo, A.V.; Semaan, S.J.; Johansson, H.E. Simultaneous Detection of Nuclear and Cytoplasmic RNA Variants Utilizing Stellaris® RNA Fluorescence In Situ Hybridization in Adherent Cells. In In Situ Hybridization Protocols; Nielsen, B.S., Ed.; Springer: New York, NY, USA, 2014; pp. 189–199. ISBN 9781493914593. [Google Scholar]
- Orjalo, A.V.; Johansson, H.E. Stellaris® RNA Fluorescence In Situ Hybridization for the Simultaneous Detection of Immature and Mature Long Noncod-ing RNAs in Adherent Cells. In Long Non-Coding RNAs: Methods and Protocols; Feng, Y., Zhang, L., Eds.; Springer: New York, NY, USA, 2016; pp. 119–134. ISBN 9781493933785. [Google Scholar]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Cabili, M.N.; Rinn, J.; Raj, A. linc-HOXA1 is a noncoding RNA that represses Hoxa1 transcription in cis. Genes Dev. 2013, 27, 1260–1271. [Google Scholar] [CrossRef]
- Dunagin, M.; Cabili, M.N.; Rinn, J.; Raj, A. Visualization of lncRNA by Single-Molecule Fluorescence In Situ Hybridization. In Nuclear Bodies and Noncoding RNAs: Methods and Protocols; Nakagawa, S., Hirose, T., Eds.; Springer: New York, NY, USA, 2015; pp. 3–19. ISBN 9781493922536. [Google Scholar]
- Mortimer, S.A.; Weeks, K.M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 2007, 129, 4144–4145. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Smola, M.J.; Rice, G.M.; Busan, S.; Siegfried, N.A.; Weeks, K.M. Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 2015, 10, 1643–1669. [Google Scholar] [CrossRef]
- Schmidt, K.; Weidmann, C.A.; Hilimire, T.A.; Yee, E.; Hatfield, B.M.; Schneekloth, J.S., Jr.; Weeks, K.M.; Novina, C.D. Targeting the Oncogenic Long Non-coding RNA SLNCR1 by Blocking Its Sequence-Specific Binding to the Androgen Receptor. Cell Rep. 2020, 30, 541–554.e5. [Google Scholar] [CrossRef]
- Shields, E.J.; Petracovici, A.F.; Bonasio, R. lncRedibly versatile: Biochemical and biological functions of long noncoding RNAs. Biochem. J. 2019, 476, 1083–1104. [Google Scholar] [CrossRef]
- Mishra, K.; Kanduri, C. Understanding Long Noncoding RNA and Chromatin Interactions: What We Know So Far. Noncoding RNA 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, Y.; Kwoh, C.K. Deep learning based DNA:RNA triplex forming potential prediction. BMC Bioinform. 2020, 21, 522. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C.; Hänzelmann, S.; Sentürk Cetin, N.; Frank, S.; Zajzon, B.; Derks, J.-P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I.; et al. Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.; Al-Absi, H.R.H.; Schmeier, S. Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives. Noncoding RNA 2020, 6. [Google Scholar] [CrossRef]
- Iwakiri, J.; Hamada, M.; Asai, K. Bioinformatics tools for lncRNA research. Biochim. Biophys. Acta 2016, 1859, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Terai, G.; Iwakiri, J.; Kameda, T.; Hamada, M.; Asai, K. Comprehensive prediction of lncRNA-RNA interactions in human transcriptome. BMC Genom. 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, J.; Terai, G.; Hamada, M. Computational prediction of lncRNA-mRNA interactionsby integrating tissue specificity in human transcriptome. Biol. Direct 2017, 12, 15. [Google Scholar] [CrossRef]
- Mar-Aguilar, F.; Rodríguez-Padilla, C.; Reséndez-Pérez, D. Web-based tools for microRNAs involved in human cancer (Review). Oncol. Lett. 2016, 11, 3563–3570. [Google Scholar] [CrossRef][Green Version]
- Antonov, I.; Marakhonov, A.; Zamkova, M.; Medvedeva, Y. ASSA: Fast identification of statistically significant interactions between long RNAs. J. Bioinform. Comput. Biol. 2018, 16, 1840001. [Google Scholar] [CrossRef]
- Fukunaga, T.; Hamada, M. RIblast: An ultrafast RNA-RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics 2017, 33, 2666–2674. [Google Scholar] [CrossRef]
- Zhang, J.; Le, T.D.; Liu, L.; Li, J. Inferring and analyzing module-specific lncRNA-mRNA causal regulatory networks in human cancer. Brief. Bioinform. 2019, 20, 1403–1419. [Google Scholar] [CrossRef]
- Pyfrom, S.C.; Luo, H.; Payton, J.E. PLAIDOH: A novel method for functional prediction of long non-coding RNAs identifies cancer-specific LncRNA activities. BMC Genom. 2019, 20, 137. [Google Scholar] [CrossRef]
- Gawronski, A.R.; Uhl, M.; Zhang, Y.; Lin, Y.-Y.; Niknafs, Y.S.; Ramnarine, V.R.; Malik, R.; Feng, F.; Chinnaiyan, A.M.; Collins, C.C.; et al. MechRNA: Prediction of lncRNA mechanisms from RNA-RNA and RNA-protein interactions. Bioinformatics 2018, 34, 3101–3110. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- NCBI-SRA. Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 29 January 2021).
- Babbi, G.; Martelli, P.L.; Profiti, G.; Bovo, S.; Savojardo, C.; Casadio, R. eDGAR: A database of Disease-Gene Associations with annotated Relationships among genes. BMC Genom. 2017, 18, 554. [Google Scholar] [CrossRef]
- Junge, A.; Refsgaard, J.C.; Garde, C.; Pan, X.; Santos, A.; Alkan, F.; Anthon, C.; von Mering, C.; Workman, C.T.; Jensen, L.J.; et al. RAIN: RNA-protein Association and Interaction Networks. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhao, Y.; Li, C.; Zhang, L.; Huang, H.; Li, Y.; Liu, L.; Hou, P.; Cui, T.; Tan, P.; et al. RAID v2.0: An updated resource of RNA-associated interactions across organisms. Nucleic Acids Res. 2017, 45, D115–D118. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Shao, D.; Xu, K.; Lu, Z.; Lu, Z.J.; Yang, Y.T.; Zhang, Q.C. RISE: A database of RNA interactome from sequencing experiments. Nucleic Acids Res. 2018, 46, D194–D201. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, T.; Iwakiri, J.; Ono, Y.; Hamada, M. LncRRIsearch: A Web Server for lncRNA-RNA Interaction Prediction Integrated With Tissue-Specific Expression and Subcellular Localization Data. Front. Genet. 2019, 10, 462. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Tastsoglou, S.; Skoufos, G.; Karavangeli, A.; Pierros, V.; Zacharopoulou, E.; Hatzigeorgiou, A.G. DIANA-LncBase v3: Indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 2020, 48, D101–D110. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Pachl, E.; Hartung, M.; Stiegler, V.; Baumbach, J.; Schulz, M.H.; List, M. SPONGEdb: A pan-cancer resource for competing endogenous RNA interactions. NAR Cancer 2021, 3. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Gao, Y.; Guo, Q.; Ning, S.; Zhang, Y.; Shang, S.; Wang, J.; Wang, Y.; Zhi, H.; et al. LnCeVar: A comprehensive database of genomic variations that disturb ceRNA network regulation. Nucleic Acids Res. 2020, 48, D111–D117. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhi, H.; Zhang, Y.; Liu, Y.; Zhang, J.; Gao, Y.; Guo, M.; Ning, S.; Li, X. miRSponge: A manually curated database for experimentally supported miRNA sponges and ceRNAs. Database 2015, 2015. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, T.; Cui, T.; Wang, Z.; Zhang, Y.; Tan, P.; Huang, Y.; Yu, J.; Wang, D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020, 48, D189–D197. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G.; Jiang, H.; Jiang, Q. LncRNA2Target v2.0: A comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019, 47, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, L.; Jiang, S.; Li, Q.; Feng, C.; Du, Q.; Zou, D.; Xiao, J.; Zhang, Z.; Ma, L. LncExpDB: An expression database of human long non-coding RNAs. Nucleic Acids Res. 2021, 49, D962–D968. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Gao, Y.; Guo, Q.; Wang, Y.; Fang, Y.; Ma, X.; Zhi, H.; Zhou, D.; Shen, W.; et al. LncACTdb 2.0: An updated database of experimentally supported ceRNA interactions curated from low- and high-throughput experiments. Nucleic Acids Res. 2019, 47, D121–D127. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Lu, J. The roles of long noncoding RNAs in breast cancer metastasis. Cell Death Dis. 2020, 11, 749. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Chen, S.; Li, W.; Chen, W.; Gu, W. LncRNA MACC1-AS1 sponges multiple miRNAs and RNA-binding protein PTBP1. Oncogenesis 2019, 8, 73. [Google Scholar] [CrossRef]
- Mili, S.; Steitz, J.A. Evidence for reassociation of RNA-binding proteins after cell lysis: Implications for the interpretation of immunoprecipitation analyses. RNA 2004, 10, 1692–1694. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.J.; Yario, T.A.; Steitz, J.A. Association of Argonaute proteins and microRNAs can occur after cell lysis. RNA 2012, 18, 1581–1585. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Maeda, S.; Liu, C.; Karin, M.; Edgington, T.S. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 2007, 26, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Stamm, S. Regulation of Alternative Splicing by Reversible Protein Phosphorylation. J. Biol. Chem. 2008, 283, 1223–1227. [Google Scholar] [CrossRef]
- Calarco, J.A.; Superina, S.; O’Hanlon, D.; Gabut, M.; Raj, B.; Pan, Q.; Skalska, U.; Clarke, L.; Gelinas, D.; van der Kooy, D.; et al. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell 2009, 138, 898–910. [Google Scholar] [CrossRef]
- Bourgeois, C.F.; Lejeune, F.; Stévenin, J. Broad Specificity of SR (Serine/Arginine) Proteins in the Regulation of Alternative Splicing of Pre-Messenger RNA. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2004; Volume 78, pp. 37–88. [Google Scholar]
- Wang, D.; Ding, L.; Wang, L.; Zhao, Y.; Sun, Z.; Karnes, R.J.; Zhang, J.; Huang, H. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget 2015, 6, 41045–41055. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Bryant, R.J.; Cross, N.A.; Eaton, C.L.; Hamdy, F.C.; Cunliffe, V.T. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate 2007, 67, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Saramäki, O.R.; Tammela, T.L.J.; Martikainen, P.M.; Vessella, R.L.; Visakorpi, T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer 2006, 45, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Haberman, N.; Huppertz, I.; Attig, J.; König, J.; Wang, Z.; Hauer, C.; Hentze, M.W.; Kulozik, A.E.; Le Hir, H.; Curk, T.; et al. Insights into the design and interpretation of iCLIP experiments. Genome Biol. 2017, 18, 7. [Google Scholar] [CrossRef]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Vigilante, A.; Darbo, E.; Zirra, A.; Militti, C.; D’Ambrogio, A.; Luscombe, N.M.; Ule, J. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 2015, 519, 491–494. [Google Scholar] [CrossRef]
- Urlaub, H.; Hartmuth, K.; Lührmann, R. A two-tracked approach to analyze RNA-protein crosslinking sites in native, nonlabeled small nuclear ribonucleoprotein particles. Methods 2002, 26, 170–181. [Google Scholar] [CrossRef]
- König, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef]
- Zheng, Z.-Q.; Li, Z.-X.; Zhou, G.-Q.; Lin, L.; Zhang, L.-L.; Lv, J.-W.; Huang, X.-D.; Liu, R.-Q.; Chen, F.; He, X.-J.; et al. Long Noncoding RNA FAM225A Promotes Nasopharyngeal Carcinoma Tumorigenesis and Metastasis by Acting as ceRNA to Sponge miR-590-3p/miR-1275 and Upregulate ITGB3. Cancer Res. 2019, 79, 4612–4626. [Google Scholar] [CrossRef]
- Petri, R.; Jakobsson, J. Identifying miRNA Targets Using AGO-RIPseq. In mRNA Decay: Methods and Protocols; Lamandé, S.R., Ed.; Springer: New York, NY, USA, 2018; pp. 131–140. ISBN 9781493975402. [Google Scholar]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.P.; Rajapakshe, K.I.; Bader, D.A.; Cerne, J.Z.; Smith, E.A.; Coarfa, C.; Hartig, S.M.; McGuire, S.E. The Landscape of microRNA Targeting in Prostate Cancer Defined by AGO-PAR-CLIP. Neoplasia 2016, 18, 356–370. [Google Scholar] [CrossRef]
- Orom, U.A.; Lund, A.H. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 2007, 43, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.-J.; Yang, H.-J.; Tsai, H.-J. Labeled microRNA pull-down assay system: An experimental approach for high-throughput identification of microRNA-target mRNAs. Nucleic Acids Res. 2009, 37, e77. [Google Scholar] [CrossRef] [PubMed]
- Baigude, H.; Li, Z.; Zhou, Y.; Rana, T.M. miR-TRAP: A benchtop chemical biology strategy to identify microRNA targets. Angew. Chem. Int. Ed. 2012, 51, 5880–5883. [Google Scholar] [CrossRef]
- Su, Z.; Ganbold, T.; Baigude, H. Analysis and Identification of Tumorigenic Targets of MicroRNA in Cancer Cells by Photoreactive Chemical Probes. Int. J. Mol. Sci. 2020, 21, 1545. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, L.; Xiao, X.; Chen, Y.; Wang, X.; Zhou, Z.; Zhang, C.; Zhang, Y. Photoclickable MicroRNA for the Intracellular Target Identification of MicroRNAs. J. Am. Chem. Soc. 2016, 138, 15943–15949. [Google Scholar] [CrossRef]
- Zhang, P.; Fu, H.; Du, S.; Wang, F.; Yang, J.; Cai, W.; Liu, D. Click RNA for Rapid Capture and Identification of Intracellular MicroRNA Targets. Anal. Chem. 2019, 91, 15740–15747. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Y.; Li, J.; Zhang, Y. A photoactivatable microRNA probe for identification of microRNA targets and light-controlled suppression of microRNA target expression. Chem. Commun. 2020, 56, 627–630. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Y.; Li, D.; Liu, Q.; Xuan, Z.; Li, W.-H. TargetLink, a new method for identifying the endogenous target set of a specific microRNA in intact living cells. RNA Biol. 2017, 14, 259–274. [Google Scholar] [CrossRef]
- Vencken, S.; Hassan, T.; McElvaney, N.G.; Smith, S.G.J.; Greene, C.M. miR-CATCH: MicroRNA Capture Affinity Technology. In RNA Interference: Challenges and Therapeutic Opportunities; Sioud, M., Ed.; Springer: New York, NY, USA, 2015; pp. 365–373. ISBN 9781493915385. [Google Scholar]
- Hassan, T.; Smith, S.G.J.; Gaughan, K.; Oglesby, I.K.; O’Neill, S.; McElvaney, N.G.; Greene, C.M. Isolation and identification of cell-specific microRNAs targeting a messenger RNA using a biotinylated anti-sense oligonucleotide capture affinity technique. Nucleic Acids Res. 2013, 41, e71. [Google Scholar] [CrossRef]
- Imig, J.; Brunschweiger, A.; Brümmer, A.; Guennewig, B.; Mittal, N.; Kishore, S.; Tsikrika, P.; Gerber, A.P.; Zavolan, M.; Hall, J. miR-CLIP capture of a miRNA targetome uncovers a lincRNA H19-miR-106a interaction. Nat. Chem. Biol. 2015, 11, 107–114. [Google Scholar] [CrossRef]
- De Santi, C.; Vencken, S.; Blake, J.; Haase, B.; Benes, V.; Gemignani, F.; Landi, S.; Greene, C.M. Identification of MiR-21-5p as a Functional Regulator of Mesothelin Expression Using MicroRNA Capture Affinity Coupled with Next Generation Sequencing. PLoS ONE 2017, 12, e0170999. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Ueno, Y. Diazirine-containing RNA photo-cross-linking probes for capturing microRNA targets. J. Org. Chem. 2014, 79, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Minami, K.; Akao, Y.; Ueno, Y. Labeling of target mRNAs using a photo-reactive microRNA probe. Chem. Commun. 2016, 52, 6720–6722. [Google Scholar] [CrossRef]
- Nakamoto, K.; Akao, Y.; Ueno, Y. Diazirine-containing tag-free RNA probes for efficient RISC-loading and photoaffinity labeling of microRNA targets. Bioorg. Med. Chem. Lett. 2018, 28, 2906–2909. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, B.; Pollum, M.; Crespo-Hernández, C.E. Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases. Photochem. Photobiol. 2019, 95, 33–58. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, D.; Hoffmann, J.-E.; Hentze, M.W.; Schultz, C. A Genetically Encoded Diazirine Analogue for RNA-Protein Photo-crosslinking. Chembiochem 2020, 21, 88–93. [Google Scholar] [CrossRef]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Panda, A.C.; Kang, M.-J.; Guo, R.; Kim, J.; Grammatikakis, I.; Yoon, J.-H.; Dudekula, D.B.; Noh, J.H.; Yang, X.; et al. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res. 2014, 42, 10099–10111. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef]
- Helwak, A.; Tollervey, D. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH). Nat. Protoc. 2014, 9, 711–728. [Google Scholar] [CrossRef]
- Nguyen, T.C.; Cao, X.; Yu, P.; Xiao, S.; Lu, J.; Biase, F.H.; Sridhar, B.; Huang, N.; Zhang, K.; Zhong, S. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 2016, 7, 12023. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Lin, L.; Huang, Q.; He, W.; Zhang, S.; Dong, S.; Wen, Z.; Rao, J.; Liao, W.; et al. The lncRNA MACC1-AS1 promotes gastric cancer cell metabolic plasticity via AMPK/Lin28 mediated mRNA stability of MACC1. Mol. Cancer 2018, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Damas, N.D.; Marcatti, M.; Côme, C.; Christensen, L.L.; Nielsen, M.M.; Baumgartner, R.; Gylling, H.M.; Maglieri, G.; Rundsten, C.F.; Seemann, S.E.; et al. SNHG5 promotes colorectal cancer cell survival by counteracting STAU1-mediated mRNA destabilization. Nat. Commun. 2016, 7, 13875. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Gong, J.; Zhang, Q.C. PARIS: Psoralen Analysis of RNA Interactions and Structures with High Throughput and Resolution. Methods Mol. Biol. 2018, 1649, 59–84. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Sharma, E.; Sterne-Weiler, T.; O’Hanlon, D.; Blencowe, B.J. Global Mapping of Human RNA-RNA Interactions. Mol. Cell 2016, 62, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Calvet, J.P.; Pederson, T. Photochemical cross-linking of secondary structure in HeLa cell heterogeneous nuclear RNA in situ 1. Nucleic Acids Res. 1979, 6, 1993–2001. [Google Scholar] [CrossRef][Green Version]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.N.; Boon, K.-L.; Tapsin, S.; Chan, Y.-S.; Tan, C.-P.; Sim, A.Y.L.; et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature 2020, 582, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Szostak, J.W. A simple method for 3’-labeling of RNA. Nucleic Acids Res. 1996, 24, 4360–4361. [Google Scholar] [CrossRef] [PubMed]
- Cimino, G.D.; Gamper, H.B.; Isaacs, S.T.; Hearst, J.E. Psoralens as photoactive probes of nucleic acid structure and function: Organic chemistry, photochemistry, and biochemistry. Annu. Rev. Biochem. 1985, 54, 1151–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhong, S.; Luo, Y.; Zou, D.; Li, M.; Li, Y.; Lu, Y.; Miao, S.; Wang, L.; Song, W. A long noncoding RNA binding to QKI-5 regulates germ cell apoptosis via p38 MAPK signaling pathway. Cell Death Dis. 2019, 10, 699. [Google Scholar] [CrossRef]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302–307. [Google Scholar] [CrossRef]
- Siprashvili, Z.; Webster, D.E.; Kretz, M.; Johnston, D.; Rinn, J.L.; Chang, H.Y.; Khavari, P.A. Identification of proteins binding coding and non-coding human RNAs using protein microarrays. BMC Genom. 2012, 13, 633. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Xu, Y.; He, X.; Li, Y. Silencing the long noncoding RNA, TINCR, a molecular sponge of miR-335, inhibits the malignant phenotype of epithelial ovarian cancer via FGF2 suppression. Int. J. Oncol. 2019, 55, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Tian, G.; Cheung, H.-H.; Wei, W.; Lee, T.-L. Gas5 is an essential lncRNA regulator for self-renewal and pluripotency of mouse embryonic stem cells and induced pluripotent stem cells. Stem Cell Res. Ther. 2018, 9, 71. [Google Scholar] [CrossRef]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.-J.; Ju, H.-Q.; Liu, G.-P.; Tian, T.; Ma, G.-L.; Lu, Y.-X.; Liu, Z.-X.; Pan, R.-L.; Li, R.-H.; Piao, H.-L.; et al. LncRNA CamK-A Regulates Ca2+-Signaling-Mediated Tumor Microenvironment Remodeling. Mol. Cell 2018, 72, 71–83. [Google Scholar] [CrossRef]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-L.; Chen, L.-Z.; Lu, Y.-X.; Zhang, D.-S.; Zeng, Z.-L.; Pan, Z.-Z.; Huang, P.; Wang, F.-H.; Li, Y.-H.; Ju, H.-Q.; et al. Long noncoding RNA XIST expedites metastasis and modulates epithelial-mesenchymal transition in colorectal cancer. Cell Death Dis. 2017, 8, e3011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-T.; Pan, S.-X.; Wang, A.-H.; Kong, Q.-Y.; Jiang, K.-T.; Yu, Z.-B. Long Non-Coding RNA (lncRNA) X-Inactive Specific Transcript (XIST) Plays a Critical Role in Predicting Clinical Prognosis and Progression of Colorectal Cancer. Med. Sci. Monit. 2019, 25, 6429–6435. [Google Scholar] [CrossRef]
- Liang, X.-H.; Fournier, M.J. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol. Cell. Biol. 2006, 26, 7437–7450. [Google Scholar] [CrossRef]
- Fayet-Lebaron, E.; Atzorn, V.; Henry, Y.; Kiss, T. 18S rRNA processing requires base pairings of snR30 H/ACA snoRNA to eukaryote-specific 18S sequences. EMBO J. 2009, 28, 1260–1270. [Google Scholar] [CrossRef]
- Bak, G.; Han, K.; Kim, K.-S.; Lee, Y. Electrophoretic mobility shift assay of RNA-RNA complexes. Methods Mol. Biol. 2015, 1240, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Blum, Y.; Verma, A.; Liu, Z.; Pramanik, K.; Leigh, N.R.; Chun, C.Z.; Samant, G.V.; Zhao, B.; Garnaas, M.K.; et al. A noncoding antisense RNA in tie-1 locus regulates tie-1 function in vivo. Blood 2010, 115, 133–139. [Google Scholar] [CrossRef]
- Gilman, M. Ribonuclease protection assay. Curr. Protoc. Mol. Biol. 2001, 4. [Google Scholar] [CrossRef] [PubMed]
- Melton, D.A.; Krieg, P.A.; Rebagliati, M.R.; Maniatis, T.; Zinn, K.; Green, M.R. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 1984, 12, 7035–7056. [Google Scholar] [CrossRef] [PubMed]
- Jadaliha, M.; Gholamalamdari, O.; Tang, W.; Zhang, Y.; Petracovici, A.; Hao, Q.; Tariq, A.; Kim, T.G.; Holton, S.E.; Singh, D.K.; et al. A natural antisense lncRNA controls breast cancer progression by promoting tumor suppressor gene mRNA stability. PLoS Genet. 2018, 14, e1007802. [Google Scholar] [CrossRef] [PubMed]
- Ideue, T.; Hino, K.; Kitao, S.; Yokoi, T.; Hirose, T. Efficient oligonucleotide-mediated degradation of nuclear noncoding RNAs in mammalian cultured cells. RNA 2009, 15, 1578–1587. [Google Scholar] [CrossRef][Green Version]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Dethoff, E.A.; Chugh, J.; Mustoe, A.M.; Al-Hashimi, H.M. Functional complexity and regulation through RNA dynamics. Nature 2012, 482, 322–330. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Carter, J.-M.; Emmett, W.; Mozos, I.R.; Kotter, A.; Helm, M.; Ule, J.; Hussain, S. FICC-Seq: A method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res. 2019, 47, e113. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Aw, J.G.A.; Lim, S.W.; Wang, J.X.; Lambert, F.R.P.; Tan, W.T.; Shen, Y.; Zhang, Y.; Kaewsapsak, P.; Li, C.; Ng, S.B.; et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [PubMed]

| Database/Version/Ref. | Link | Conservation | Mutations | Expression | Localisation | Associations |
|---|---|---|---|---|---|---|
| LNCiPedia v5 (2019) [89] | https://lncipedia.org/ (accessed on 8 March 2021) | H. sapiens, D. melanogaster, D. rerio, M. musculus, P. troglodytes | NA | NA | NA | Relevant references |
| lncATLAS (2017) [90] | https://lncatlas.crg.eu/ (accessed on 8 March 2021) | NA | NA | GENCODE | GENCODE | NA |
| NONCODE v6 (2020) [88] | http://www.noncode.org/ (accessed on 8 March 2021) | H. sapiens, M. musculus and 15 more | dbSNP | Human Body Map; NCBI GEO | NA | Gene Ontology |
| lncWiki/Book (2019) [91,92] | https://bigd.big.ac.cn/lncrnawiki/index.php/Main_Page (accessed on 8 March 2021) https://bigd.big.ac.cn/lncbook/index (accessed on 8 March 2021) | NA | ClinVar; COSMIC | HPA; GTEx; Methylation | NA | Gene Ontology; MeSH Ontology; miRNA Interaction Prediction; |
| Lnc2Cancer v3 (2020) [93] | http://bio-bigdata.hrbmu.edu.cn/lnc2cancer/ (accessed on 8 March 2021) | NA | NA | Literature Mining | lncATLAS | Expression Correlation; Survival; TF Motif; lncBook |
| LncRNADisease v2 (2019) [94] | http://www.rnanut.net/lncrnadisease/ (accessed on 8 March 2021) | NA | NA | NA | NA | Disease Ontology; MeSH Ontology; Predictive Associations |
| LncMAP v2 (2018) [95] | http://bio-bigdata.hrbmu.edu.cn/LncMAP/ (accessed on 8 March 2021) | NA | NA | NA | NA | Associations with: TF, Genes, Drugs, Survival |
| TANRIC v2 (2019) [96] | https://www.tanric.org (accessed on 8 March 2021) | NA | TCGA Somatic Mutations | TCGA | NA | TCGA/CCLE Correlations: Expression, Stage; Survival |
| MNDR v3.1 (2020) [97] | https://www.rna-society.org/mndr/ (accessed on 8 March 2021) | NA | NA | Mammalian | NA | Evidenced disease associations and Predictor |
| lncRNASNP v2 (2018) [98] | http://bioinfo.life.hust.edu.cn/lncRNASNP/#!/ (accessed on 8 March 2021) | NA | TCGA and COSMIC SNVs | NA | NA | miRNA binding & SNP effects; GWAS LD; Mutation effects |
| lncRNAMAP (2014) [99] | https://lncrnamap.mbc.nctu.edu.tw (accessed on 8 March 2021) | NA | NA | NCBI GEO | NA | miRNA and endo-siRNA predictors |
| LncTarD (2020) [100] | http://bio-bigdata.hrbmu.edu.cn/LncTarD/ (accessed on 8 March 2021) | NA | NA | NA | NA | Disease-related Target Prediction |
| EVLncRNAs (2017) [101] | http://biophy.dzu.edu.cn/EVLncRNAs/ (accessed on 8 March 2021) | NA | NA | NA | NA | Manually curated disease association |
| LncSPA (2020) [102] | http://bio-bigdata.hrbmu.edu.cn/LncSpA/ (accessed on 8 March 2021) | NA | NA | GTEx, HPA, HBM2, FANTOM, TCGA, TARGET | NA | Expression in diseased tissues |
| Database/Version/Ref. | Link | Species | Data Sources | Integrations | Predictions |
|---|---|---|---|---|---|
| CircAtlas (2020) [103] | http://159.226.67.237:8080/new/index.php (accessed on 8 March 2021) | H. sapiens, M. mulatta, M. musculus, R norvegicus, S. scrofa and G gallus | 1070 RNA-seq samples across 6 species | Integrates circR2Disease and circRNADIsease for disease associations | Co-expression network; Functional inference from GO/KEGG; RBP and miRNA binding |
| circRNAdb (2016) [104] | http://reprod.njmu.edu.cn/cgi-bin/circrnadb/circRNADb.php (accessed on 8 March 2021) | H. sapiens | Literature and RNA-seq dataset | UniProt | Protein domains, post-translational modifications, half-lifes |
| CircFunBase (2019) [105] | http://bis.zju.edu.cn/CircFunBase/ (accessed on 8 March 2021) | H. sapiens, M. musculus + 13 more. | Literature search | CircInteractome (CLIP data), miRBase | miRNA-circRNA interactions |
| circBase (2017) [106] | http://www.circbase.org/ (accessed on 8 March 2021) | H. sapiens, C. elegans, D. melanogaster, M. musculus, L. chalumnae, L. menadoensis | Various publications [18,107,108,109,110,111] | doRiNA | NA |
| Circbank (2019) [112] | http://www.circbank.cn/ (accessed on 8 March 2021) | M. musculus, R. norvegicus, D. melanogaster | circBase, miRBase | m6A literature, COSMIC somatic mutations | IRES, circRNA-miRNA prediction |
| CIRCpedia v2 (2018) [113] | https://www.picb.ac.cn/rnomics/circpedia/ (accessed on 8 March 2021) | H. sapiens, M. musculus, R. norvegicus, D. rerio, D. melanogaster, C | 180 RNA-seq samples across 6 species | NA | Putative circRNAs |
| CircRNADisease (2018) [114] | http://cgga.org.cn:9091/circRNADisease/ (accessed on 8 March 2021) | H. sapiens | Manual curation of 800 publications | NA | Association to diseases |
| CircR2Disease (2018) [115] | http://bioinfo.snnu.edu.cn/CircR2Disease/ (accessed on 8 March 2021) | H. sapiens | Manual curation of literature | NA | Association to diseases |
| TSCD (2017) [116] | http://gb.whu.edu.cn/tscd/ (accessed on 8 March 2021) | H. sapiens, M. musculus | ENCODE + NCBI GEO RNA-seq | Starbase, Gene Ontology | MRE, Protein binding sites |
| circad (2020) [117] | http://clingen.igib.res.in/circad/ (accessed on 8 March 2021) | H. sapiens, M. musculus, R. rattus | Manual curation of literature | NA | Asssociation to diseases |
| circVAR (2020) [118] | http://soft.bioinfo-minzhao.org/circvar/ (accessed on 8 March 2021) | H. sapiens | circBase, circNet, circRNAdb | 1000 Genomes, ClinVAR, GWASCatalog, ClinVAR, COSMIC | Association to diseases/cancer |
| CSCD (2018) [119] | http://gb.whu.edu.cn/cscd/ (accessed on 8 March 2021) | H. sapiens | 228 RNA-seq samples from ENCODE | Starbase | Cancer Association, MRE, RBP, ORFs |
| Circ2Traits (2013) [120] | http://gyanxet-beta.com/circdb/ (accessed on 8 March 2021) | H. sapiens | RNA-seq [107] | Starbase, TargetScan, miRCode, dbSNP, GWAS catalog, PAR-CLIP Data [121] | miRNA interactions |
| Circ2Disease (2018) [122] | http://bioinformatics.zju.edu.cn/Circ2Disease/index.html (accessed on 8 March 2021) | H. sapiens | Manual curation of literature | HMDD, OncomiRDB, miRTarBase, dbDEMC, miRecords | miRNA interactions |
| CircInteractome (2016) [123] | https://circinteractome.nia.nih.gov/ (accessed on 8 March 2021) | H. sapiens | circBase | Starbase, miRBase | IRES, RBP and miRNA binding sites |
| Method | Specifications | Limitations | Requirements (Time/Special Resources) |
|---|---|---|---|
| RIP/RIP-seq [182] (tagged/endogenous RBP mediated RNA co-occupancy) | Characterization of native RNA-protein complexes without crosslinking; antibody enrichment | Low specificity; dependent on antibody availability | 3–4 d/IP compatible antibody; Autoradiograph facilities |
| CLIP/CLIP-seq [187] (tagged/endogenous RBP mediated RNA co-occupancy) | RNA-protein interaction sites via RNA-Protein UV crosslinking; antibody enrichment | 5′ and 3′ sites of RNA tags affected by cleavage and ligation biases; dependent on antibody availability | 5–8 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| hiCLIP [190] (tagged/endogenous RBP mediated RNA co-occupancy and RNA-duplexes) | RNA-protein interaction sites and RNA duplexes via UV crosslinking; antibody enrichment | May only capture highly expressed RNA species; dependent on antibody availability | 5 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| iCLIP [192] (tagged/endogenous RBP mediated RNA co-occupancy) | RNA-protein interaction sites at nucleotide resolution via UV crosslinking; antibody enrichment | miRNA-target interaction strength; dependent on antibody availability | 5 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| PAR-CLIP [121] (tagged/endogenous RBP mediated RNA co-occupancy) | RNA-protein interaction sites at nucleotide resolution; enhanced UV cross-linking and analysis choices; antibody enrichment | cultured cells only; 4-SU can induce cellular stress; dependent on antibody availability | 5 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| Biotin-mimics/LAMP [199] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; Biotin enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence | 2 d/Streptavidin magnetic beads |
| miR-TRAP/PCP-seq [200,201] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions at nucleotide resolution; UVA crosslinking; Poly-A enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence | 2–3 d/UV Crosslinker |
| DBCO-tagged mimics [203] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; increased loading affinity; Click enrichment | Requires known miRNA sequence | 3 d/Azide-immobilized magnetic Beads |
| PA-miRNA [204] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; Photocleavable linker; Biotin enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence; linker is not easily acquired | 5 d/Solid phase synthesis; HPLC; Mass spectrometry; UV Crosslinker; Streptavidin magnetic beads |
| TargetLink [205] (tagged miRNA mimic probing RNA targets) | One or more miRNAs to many RNA targets; LNA+Biotin enrichment | Requires KO control; Requires known miRNA sequence | 6 d/UV Crosslinker; HPLC; Streptavidin magnetic beads |
| miR-CATCH [206] (tagged RNA mimic probing miRNA targetors) | One RNA to many miRNA interactions; RNA-RISC crosslinking by formaldehyde; Biotin enrichment | Delivered by transfection to cultured cells; Requires known RNA sequence | 3–4 d/Dynamag-2; FastPrep-24; Hybridization Oven; Streptavidin magnetic beads |
| miR-CLIP [208] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; RNA-RNA crosslinking by psoralen; Biotin enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence; Probe needs testing | 3–4 d/HPLC; UV Crosslinker; Streptavidin magnetic beads |
| CLASH [219] (tagged RBP mediated RNA-Protein/duplex capture) | RNA-protein interaction sites and RNA duplexes via UV crosslinking; IgG+Ni-NTA enrichment | Delivered by transfection to cultured cells; Tagged protein expression design may be challenging | 4–5 d/UV Crosslinker; Autoradiography facilities |
| MARIO [220] (endogenous RBP mediated RNA-duplex capture) | Global RNA-RNA interactions mediated by RBPs; RNA-Protein UV crosslinking; 2-step biotin enrichment; proximity ligation | Limited to RBP mediated interactions | 5 d/UV Crosslinker; Streptavidin magnetic beads |
| RIA-seq [215] (endogenous RNA-duplex capture) | One RNA to all RNA interactions; glutaraldehyde crosslinking; biotin enrichment | Limited to RBP mediated interactions; probe preparation may be challenging | 5 d/Streptavidin magnetic beads |
| PARIS [223] (endogenous RNA-duplex capture) | All to all RNA interactions; psoralen crosslinking of RNAs; 2D enrichment of crosslinked duplexes; proximity ligation | Possible AMT side effects; 2D gel setup may be challenging | 5 d/UV Crosslinker; SequaGel UreaGel System |
| LIGR-seq [225] (endogenous RNA-duplex capture) | All to all RNA interactions; psoralen crosslinking of RNAs; RNAseR enrichment of crosslinked duplexes; proximity ligation | Possible AMT side effects | 4 d/UV Crosslinker; RNAseR |
| SPLASH [227] (endogenous RNA-duplex capture) | All to all RNA interactions; psoralen crosslinking of RNAs; biotin enrichment of crosslinked duplexes; proximity ligation | Possible AMT side effects | 4 d/UV Crosslinker; Streptavidin magnetic beads |
| RIC-seq [228] (endogenous RBP mediated RNA-duplex capture) | Global RNA-RNA interactions mediated by RBPs; RNA-Protein formaldehyde crosslinking; biotin enrichment; in situ proximity ligation | Limited to RBP mediated interactions; cell permeabilization may need optimizing | 5 d/Streptavidin magnetic beads |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carter, J.-M.; Ang, D.A.; Sim, N.; Budiman, A.; Li, Y. Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer. Non-Coding RNA 2021, 7, 19. https://doi.org/10.3390/ncrna7010019
Carter J-M, Ang DA, Sim N, Budiman A, Li Y. Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer. Non-Coding RNA. 2021; 7(1):19. https://doi.org/10.3390/ncrna7010019
Chicago/Turabian StyleCarter, Jean-Michel, Daniel Aron Ang, Nicholas Sim, Andrea Budiman, and Yinghui Li. 2021. "Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer" Non-Coding RNA 7, no. 1: 19. https://doi.org/10.3390/ncrna7010019
APA StyleCarter, J.-M., Ang, D. A., Sim, N., Budiman, A., & Li, Y. (2021). Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer. Non-Coding RNA, 7(1), 19. https://doi.org/10.3390/ncrna7010019