Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives



Abstract
1. Introduction
2. Summary of Deep Learning Techniques that Are Applied in lncRNAome-Related Research Problems
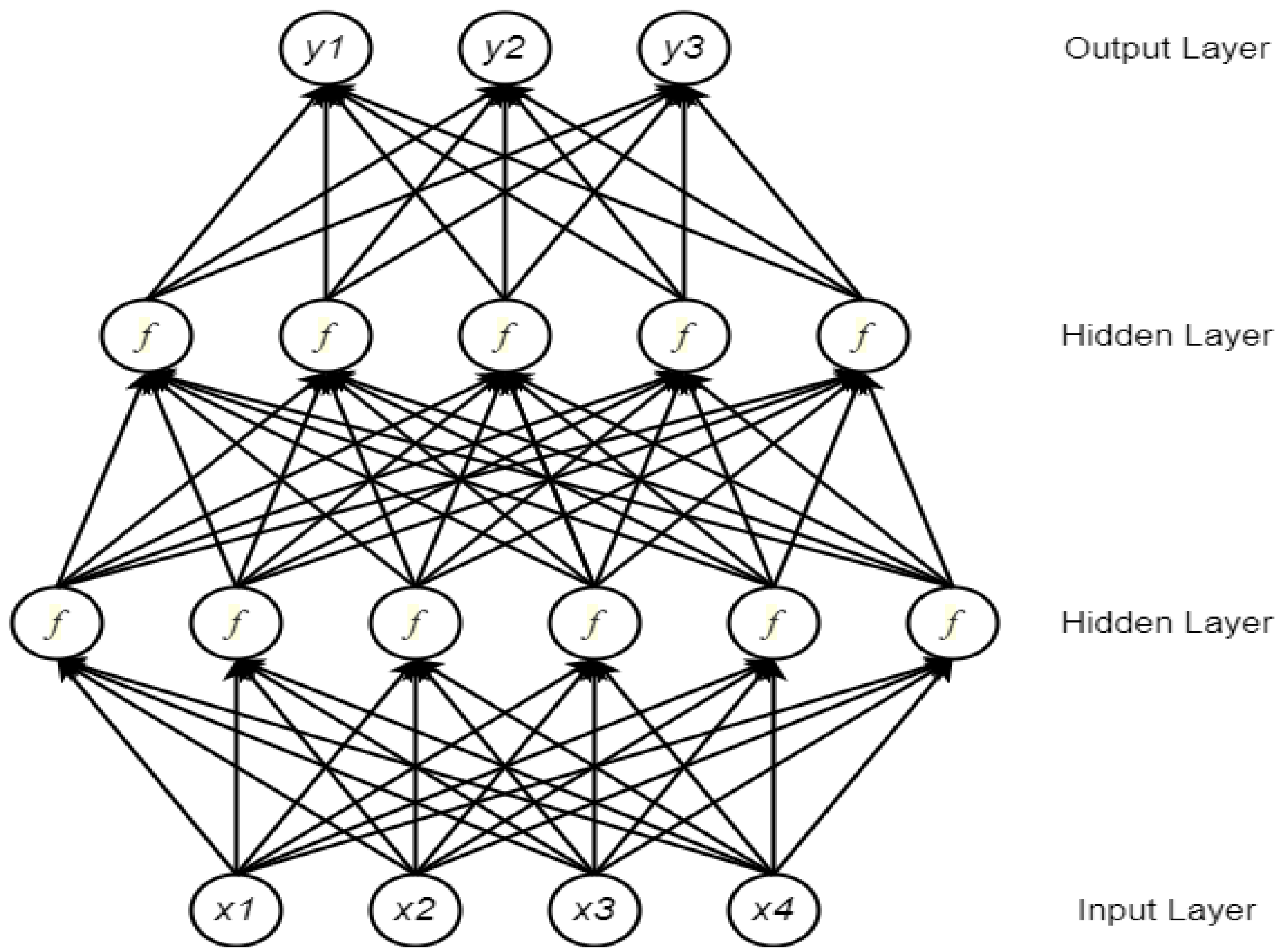
2.1. Neural Network
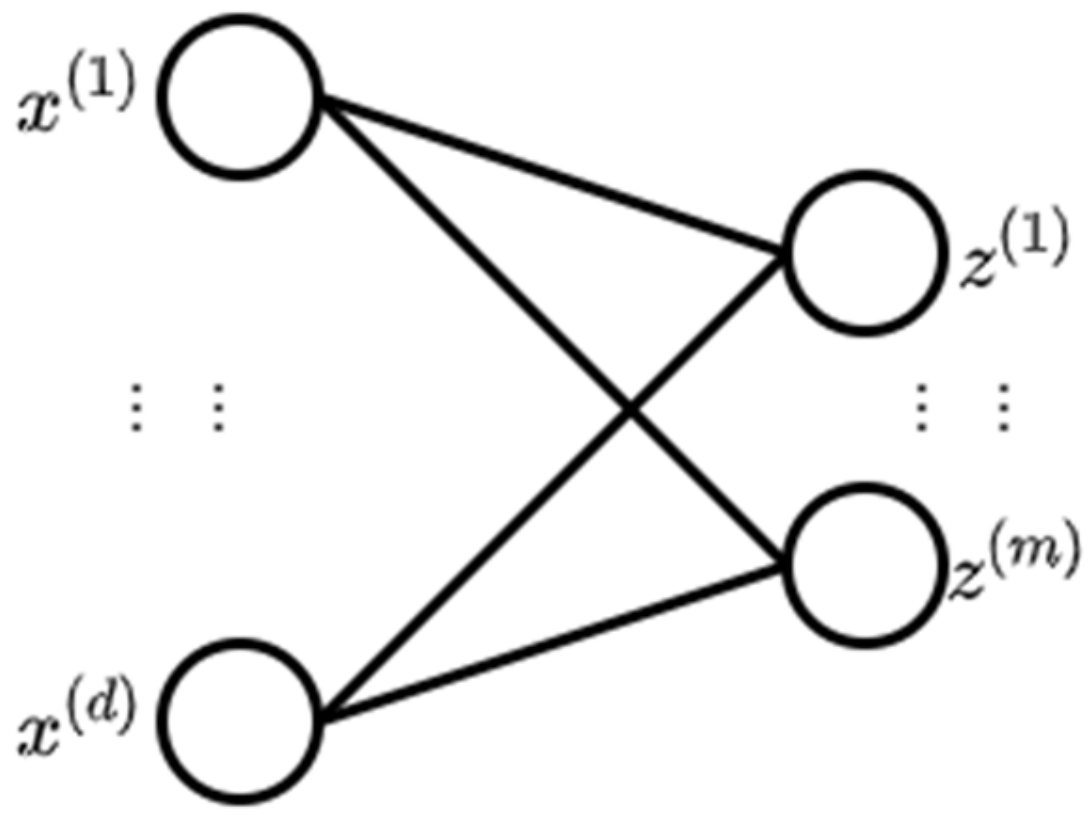
2.2. Deep Neural Network
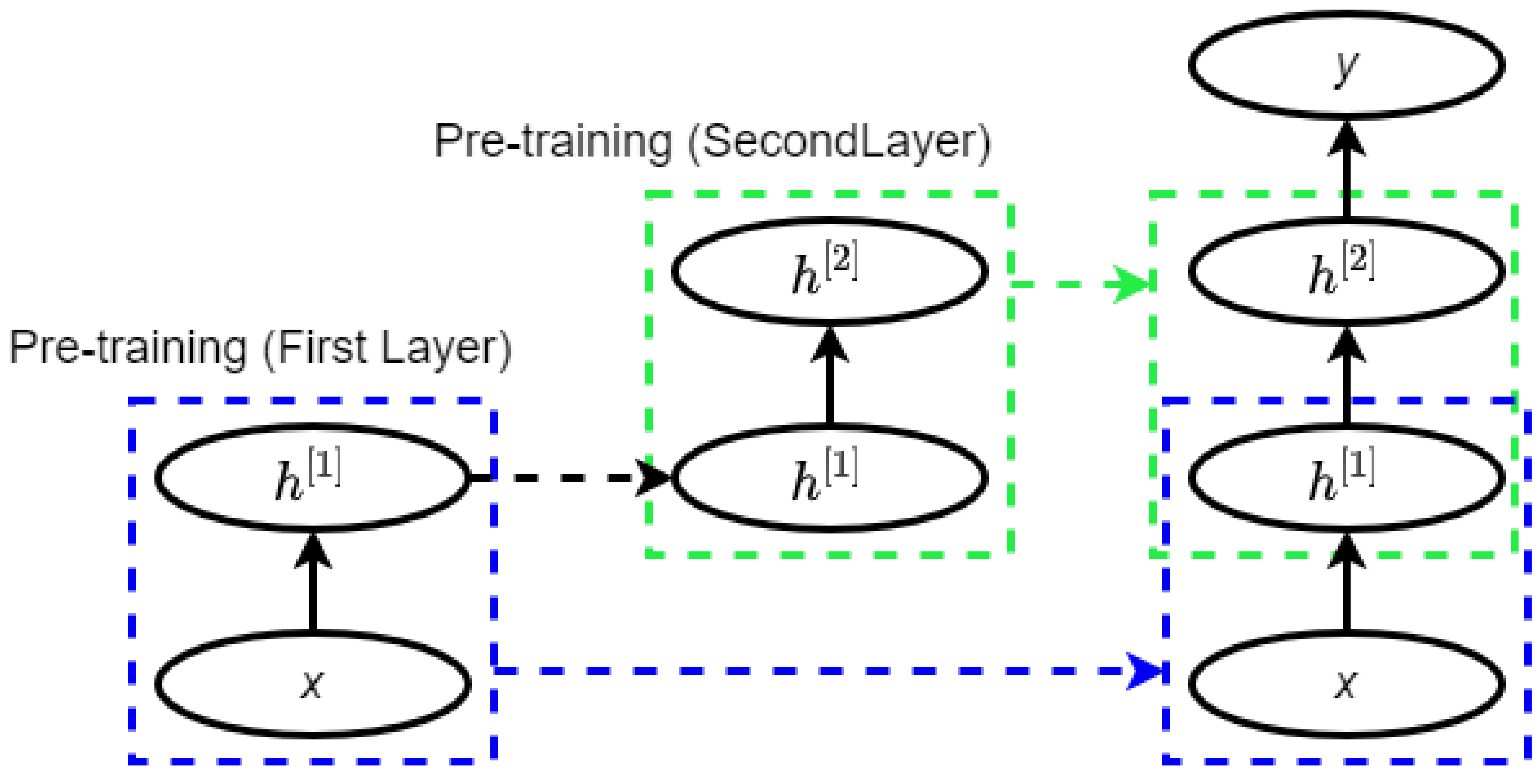
2.3. Deep Belief Network
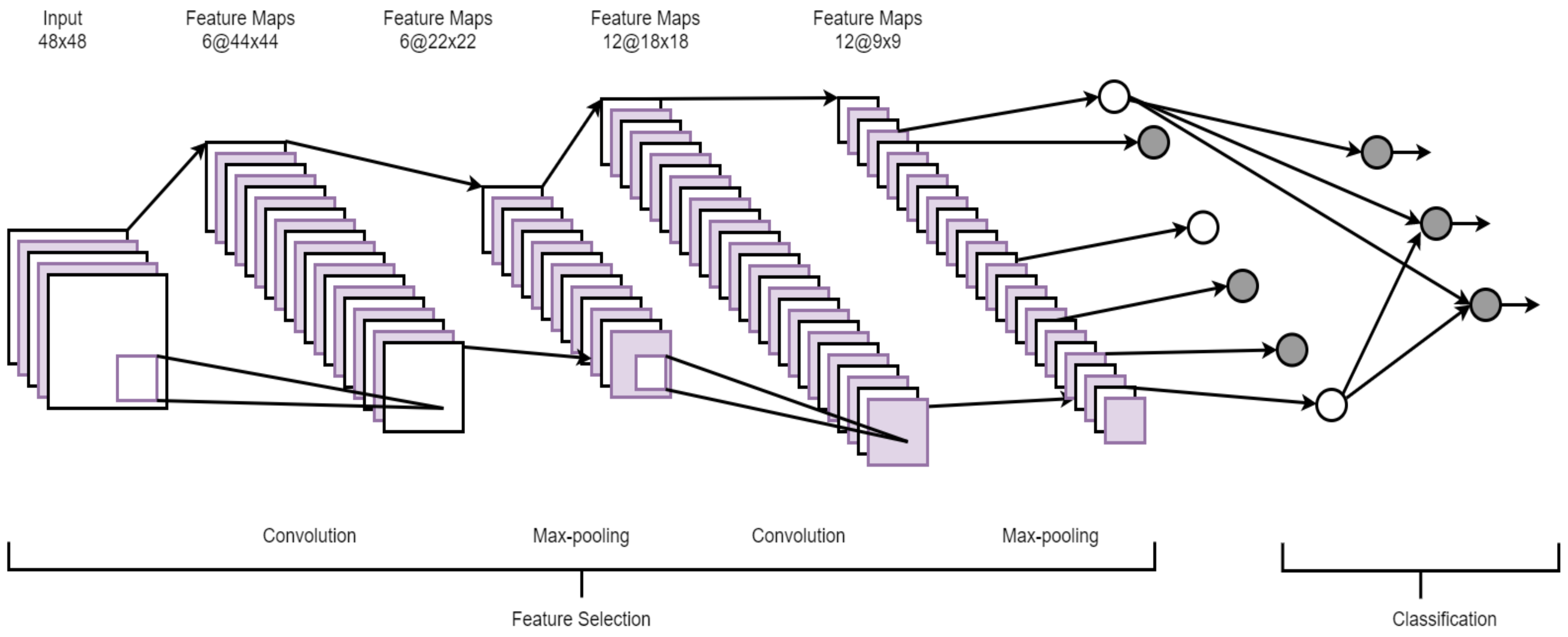
2.4. Convolutional Neural Network
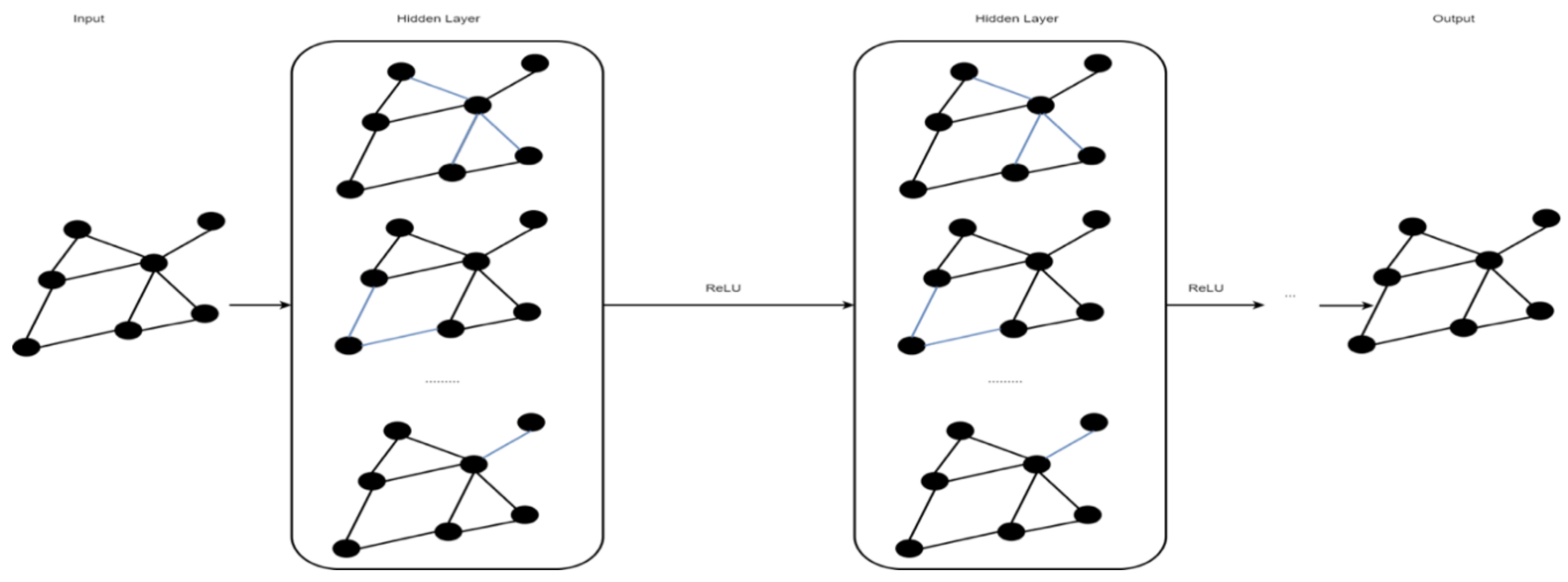
2.5. Graph Convolutional Network
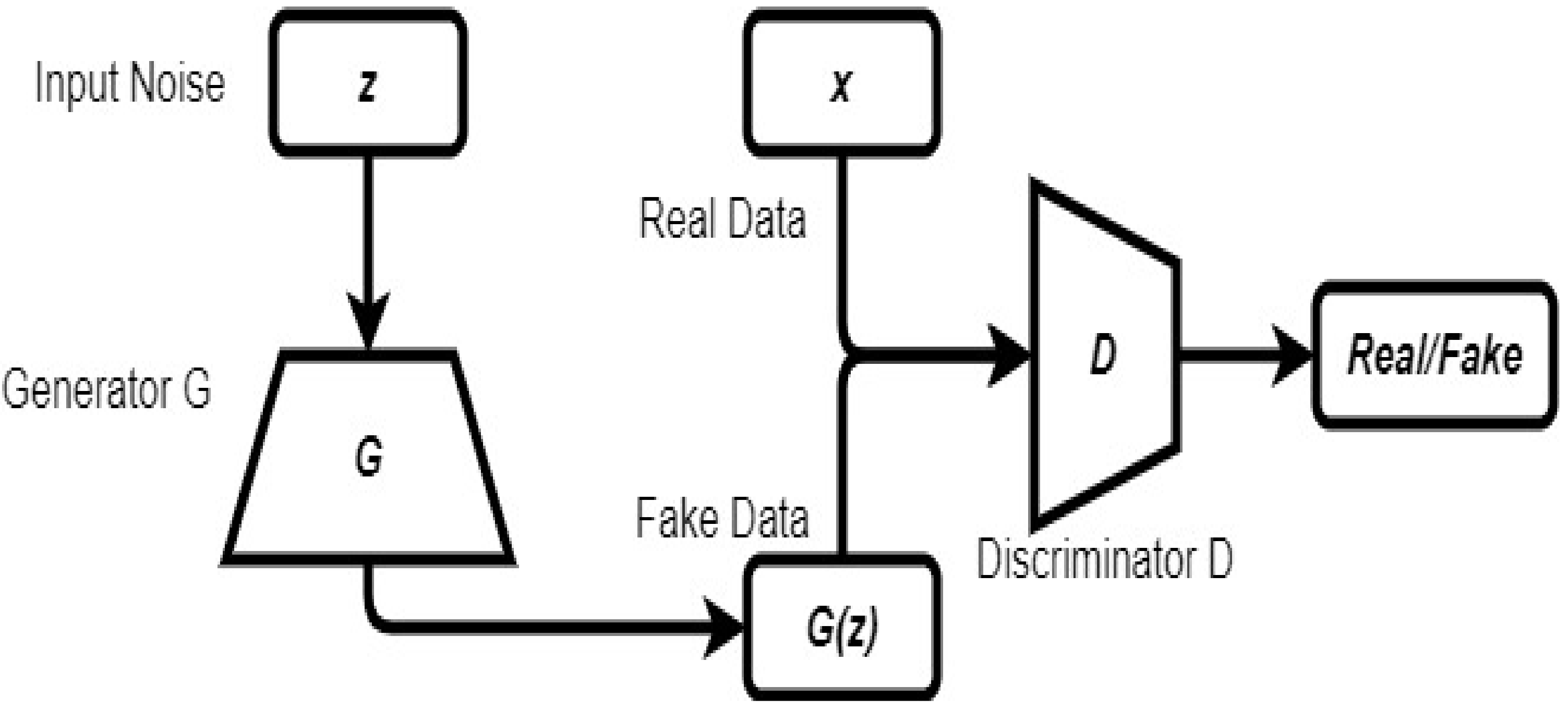
2.6. Generative Adversarial Network
2.7. Autoencoder
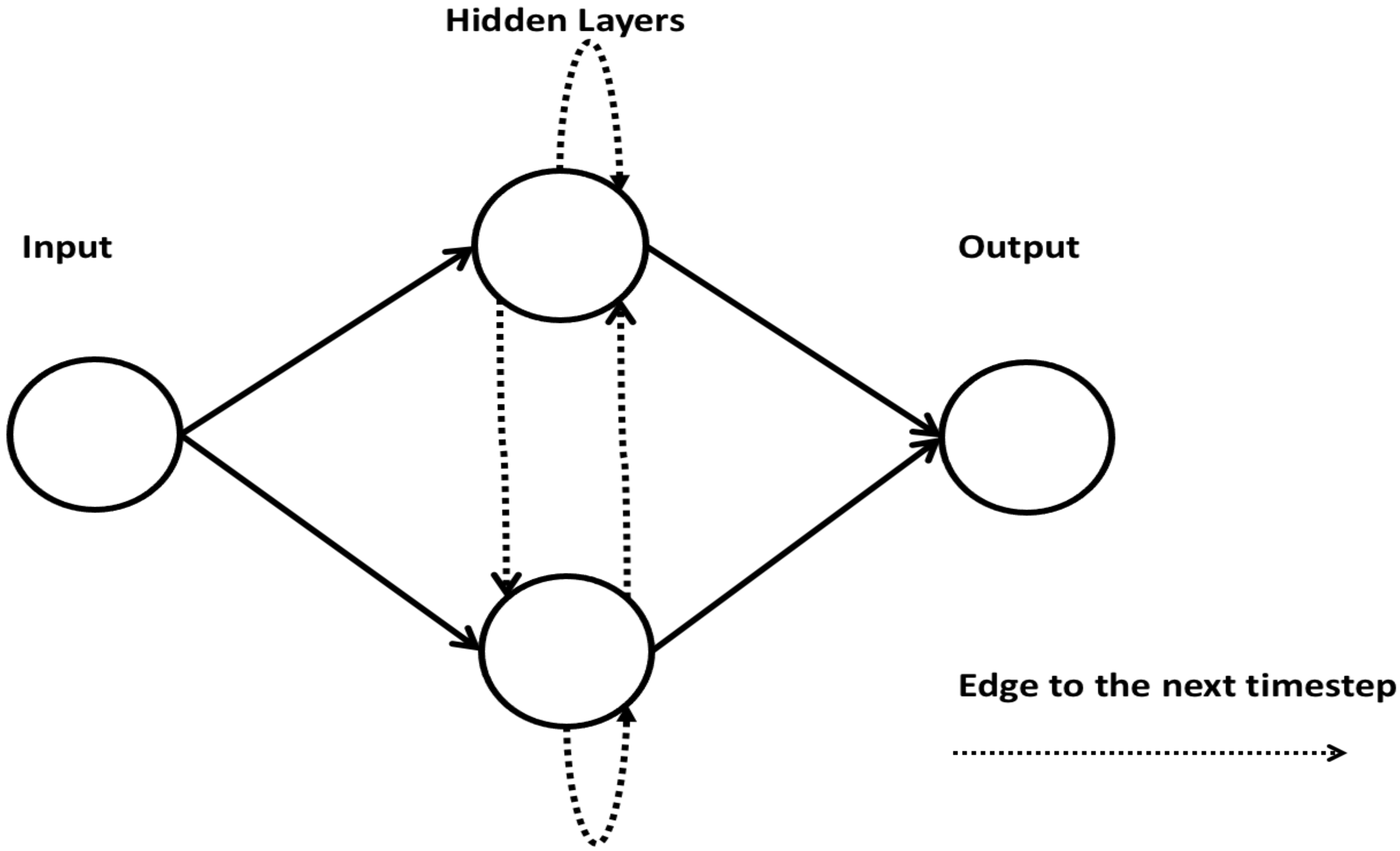
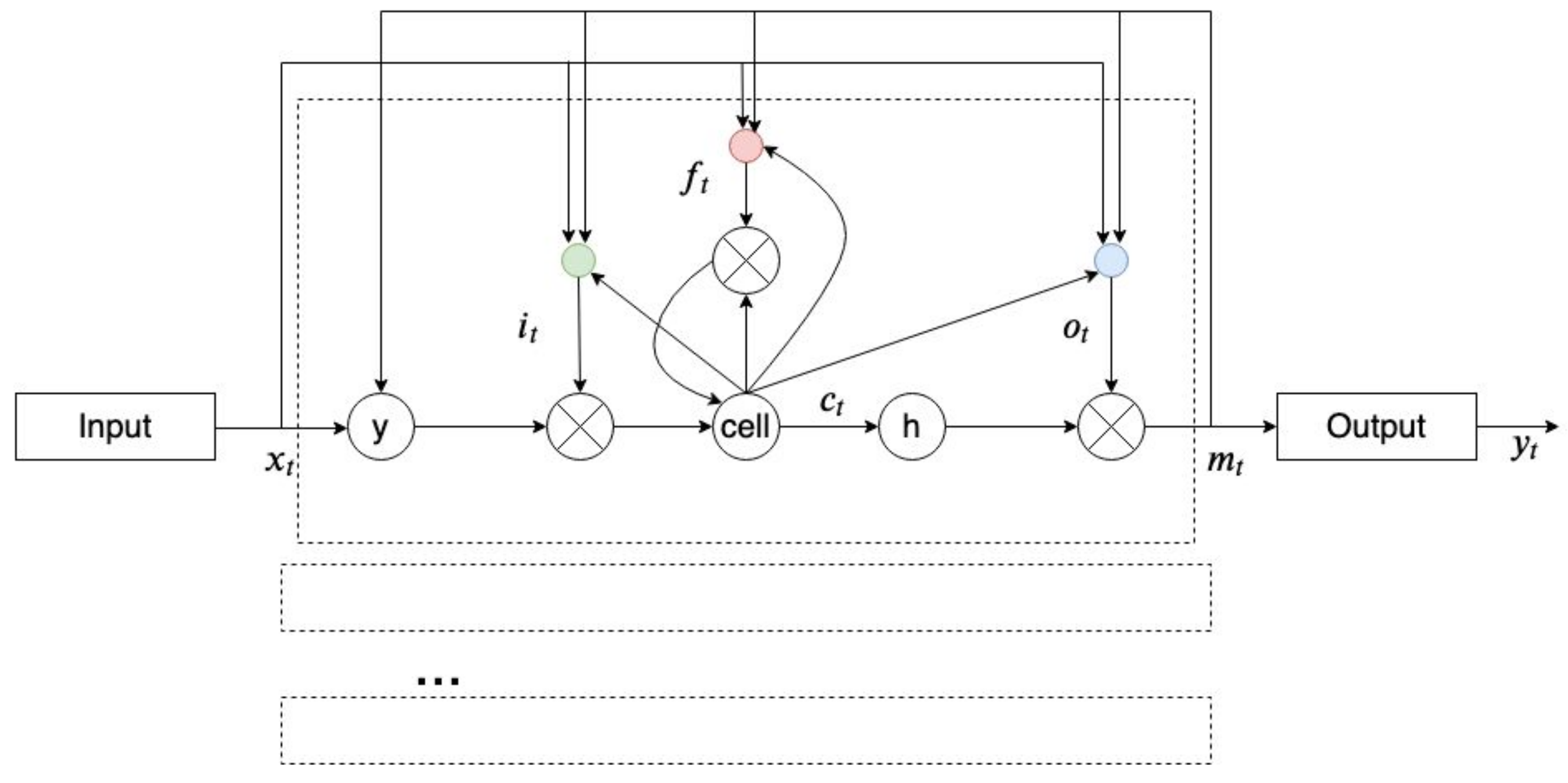
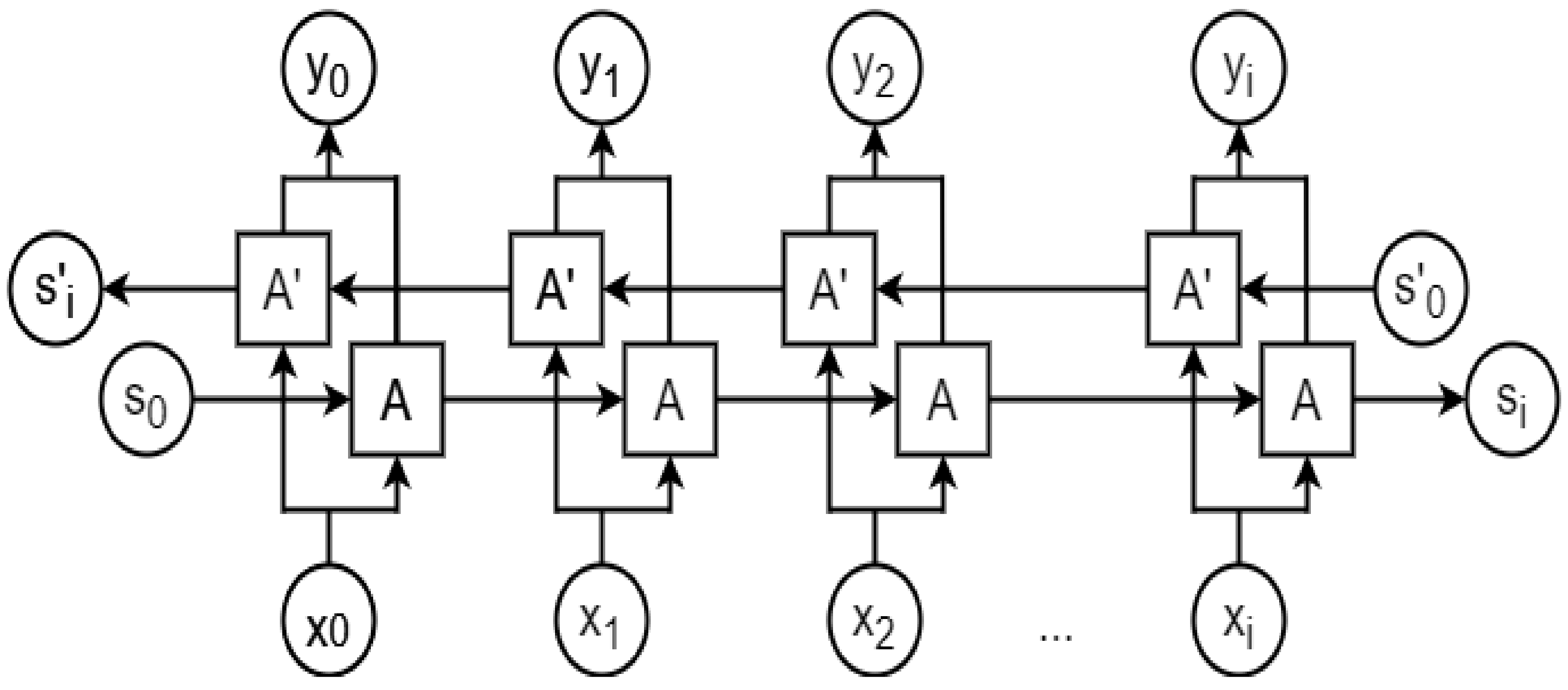
2.8. Recurrent Neural Network
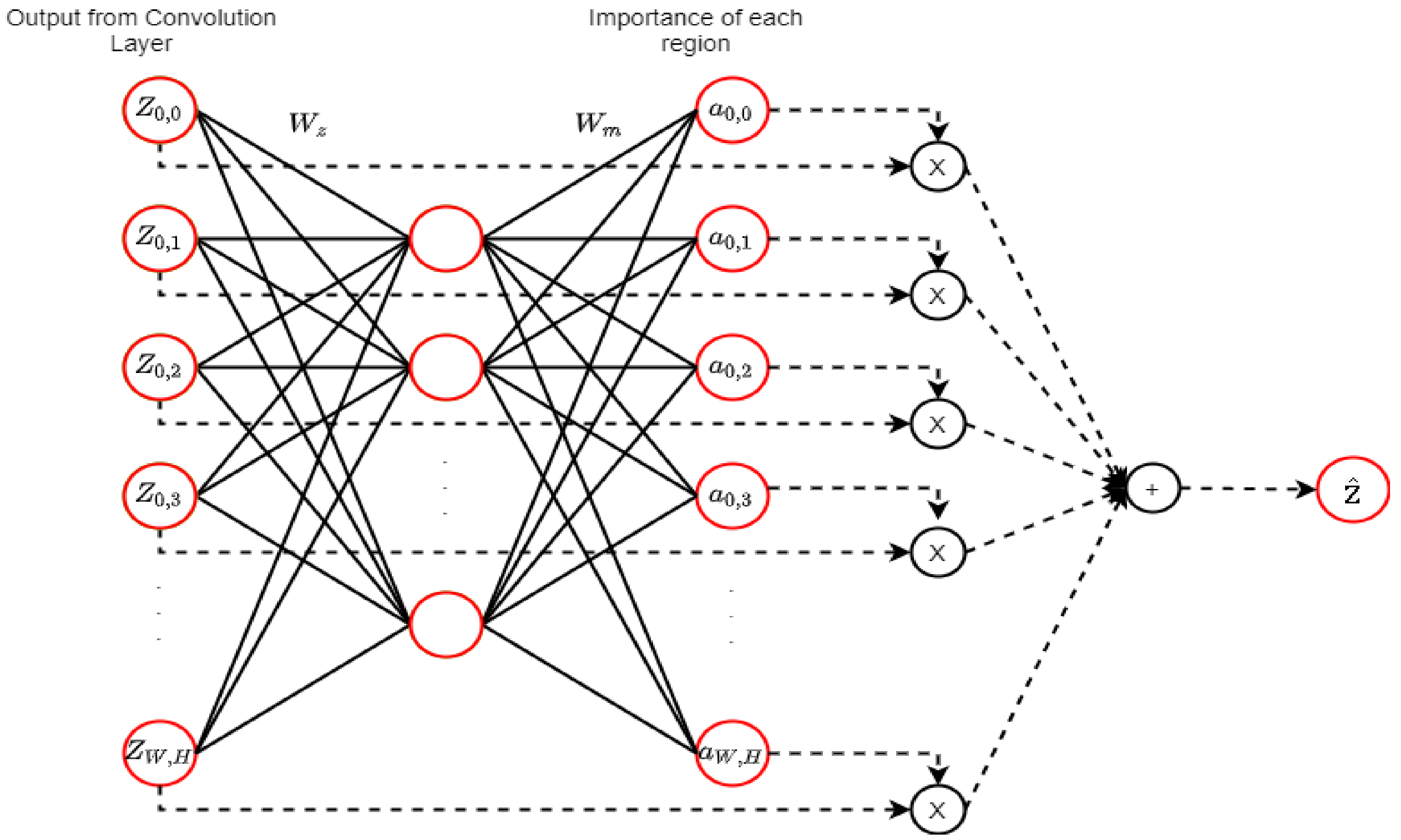
2.9. Attention Mechanism (AM)
3. Summary of the lncRNAome Research Domains Where Deep Learning-Based Techniques Have Made Significant Contributions
3.1. LncRNA Identification
3.2. Transcriptional Regulation of lncRNAs
3.3. Functional Annotation of lncRNAs
3.4. Predicting lncRNA Subcellular Localization
3.5. Predicting lncRNA–Protein Interactions
3.6. Predicting lncRNA–miRNA Interactions
3.7. Predicting lncRNA–DNA Binding
3.8. Predicting lncRNA-Disease Associations
3.9. Cancer Lassification
4. Challenges for Deep Learning in lncRNA Research
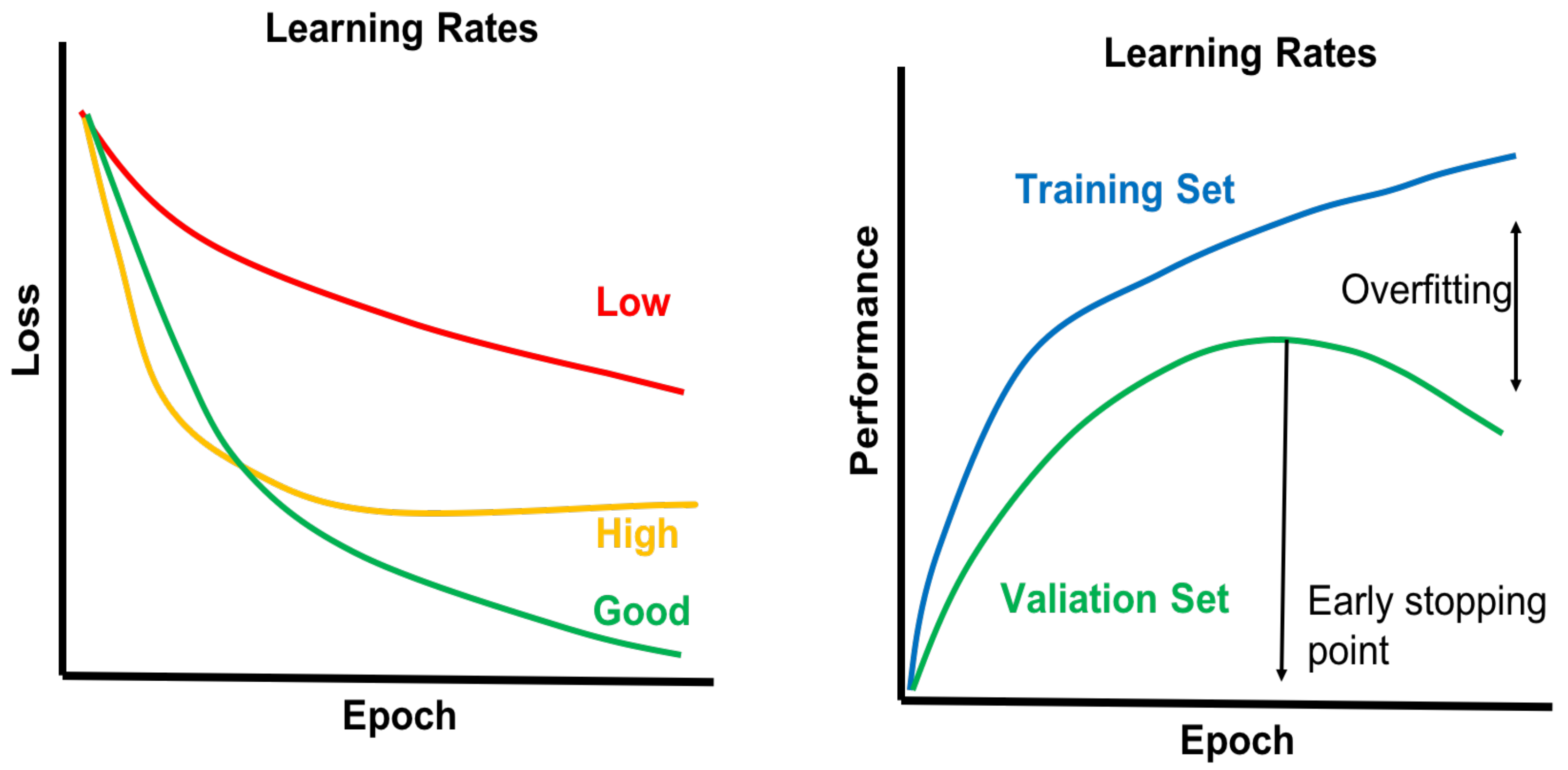
4.1. Required Data Set Sizes
4.2. Imbalanced Datasets
4.3. Interpreting and Visualizing Convolutional Networks
4.4. Model Selection and Model Building
4.5. Confidence Score of the Prediction
4.6. Catastrophic Forgetting
5. Future Perspectives for Deep Learning in lncRNAome Research
6. Conclusions
Funding
Conflicts of Interest
References
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [PubMed]
- Mourtada-Maarabouni, M.; Pickard, M.R.; Hedge, V.L.; Farzaneh, F.; Williams, G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 2009, 28, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, M.; Liu, H.; Yao, Y.; Song, Y. Long non-coding RNAs: A new frontier in the study of human diseases. Cancer Lett. 2013, 339, 159–166. [Google Scholar] [CrossRef]
- Huang, Y.; Regazzi, R.; Cho, W. Emerging Roles of Long Noncoding RNAs in Neurological Diseases and Metabolic Disorders; Frontiers Media SAP: Lausanne, Switzerland, 2015; ISBN 9782889195718. [Google Scholar]
- Lin, N.; Rana, T.M. Dysregulation of Long Non-coding RNAs in Human Disease. In Molecular Biology of Long Non-Coding RNAs; Springer: Berlin/Heidelberg, Germany, 2013; pp. 115–136. [Google Scholar]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. Adv. Exp. Med. Biol. 2017, 1008, 1–46. [Google Scholar]
- Brannan, C.I.; Dees, E.C.; Ingram, R.S.; Tilghman, S.M. The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 1990, 10, 28–36. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; Cooper, P.; Smith, S.; McCabe, V.M.; Norris, D.P.; Penny, G.D.; Patel, D.; Rastan, S. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 1991, 351, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L.E.; Crick, F.H. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef] [PubMed]
- The FANTOM Consortium and the RIKEN Genome Exploration Research Group Phase I & II Team. The Transcriptional Landscape of the Mammalian Genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- The FANTOM Consortium and the RIKEN Genome Exploration Research Group Phase I & II Team. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar] [CrossRef]
- Hon, C.-C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Alam, T.; Uludag, M.; Essack, M.; Salhi, A.; Ashoor, H.; Hanks, J.B.; Kapfer, C.; Mineta, K.; Gojobori, T.; Bajic, V.B. FARNA: Knowledgebase of inferred functions of non-coding RNA transcripts. Nucleic Acids Res. 2017, 45, 2838–2848. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Chen, L.-C.; Papandreou, G.; Kokkinos, I.; Murphy, K.; Yuille, A.L. DeepLab: Semantic Image Segmentation with Deep Convolutional Nets, Atrous Convolution, and Fully Connected CRFs. IEEE Trans. Pattern Anal. Mach. Intell. 2018, 40, 834–848. [Google Scholar] [CrossRef]
- Bengio, Y.; Courville, A.; Vincent, P. Representation learning: A review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 1798–1828. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Commun. ACM 2017, 60, 84–90. [Google Scholar] [CrossRef]
- Wu, Y.; Schuster, M.; Chen, Z.; Le, Q.V.; Norouzi, M.; Macherey, W.; Krikun, M.; Cao, Y.; Gao, Q.; Macherey, K.; et al. Google’s Neural Machine Translation System: Bridging the Gap between Human and Machine Translation. arXiv 2016, arXiv:1609.08144. [Google Scholar]
- Amodei, D.; Ananthanarayanan, S.; Anubhai, R.; Bai, J.; Battenberg, E.; Case, C.; Casper, J.; Catanzaro, B.; Cheng, Q.; Chen, G.; et al. Deep Speech 2: End-to-End Speech Recognition in English and Mandarin. In Proceedings of the International Conference on Machine Learning, New York, NY, USA, 19–24 June 2016; pp. 173–182. [Google Scholar]
- Khan, S.H.; Hayat, M.; Porikli, F. Regularization of deep neural networks with spectral dropout. Neural Netw. 2019, 110, 82–90. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, K.; Zhang, H. A Vibrating Mechanism to Prevent Neural Networks from Overfitting. In Proceedings of the 15th International Wireless Communications & Mobile Computing Conference (IWCMC), Tangier, Morocco, 24–28 June 2019. [Google Scholar]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef]
- Hinton, G.E. How neural networks learn from experience. Sci. Am. 1992, 267, 144–151. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Goldberg, Y. Neural Network Methods for Natural Language Processing. Synth. Lect. Hum. Lang. Technol. 2017, 10, 1–309. [Google Scholar] [CrossRef]
- University of Colorado, Deptartment of Computer Science; Smolensky, P. Information Processing in Dynamical Systems: Foundations of Harmony Theory; MIT Press: Cambridge, MA, USA, 1986. [Google Scholar]
- Sugiyama, M. Statistical Machine Learning. In Introduction to Statistical Machine Learning; Elsevier: Amsterdam, The Netherlands, 2016; pp. 3–8. [Google Scholar]
- Hinton, G. Deep belief networks. Scholarpedia 2009, 4, 5947. [Google Scholar] [CrossRef]
- Karhunen, J.; Raiko, T.; Cho, K. Unsupervised deep learning. In Advances in Independent Component Analysis and Learning Machines; Elsevier: Amsterdam, The Netherlands, 2015; pp. 125–142. [Google Scholar]
- Albawi, S.; Mohammed, T.A.; Al-Zawi, S. Understanding of a convolutional neural network. In Proceedings of the International Conference on Engineering and Technology (ICET), Antalya, Turkey, 21–23 August 2017. [Google Scholar]
- Lin, M.; Chen, Q.; Yan, S. Network in Network. arXiv 2013, arXiv:1312.4400. [Google Scholar]
- Alom, M.Z.; Taha, T.M.; Yakopcic, C.; Westberg, S.; Sidike, P.; Nasrin, M.S.; Hasan, M.; van Essen, B.C.; Awwal, A.A.S.; Asari, V.K. A State-of-the-Art Survey on Deep Learning Theory and Architectures. Electronics 2019, 8, 292. [Google Scholar] [CrossRef]
- Zhang, S.; Tong, H.; Xu, J.; Maciejewski, R. Graph convolutional networks: A comprehensive review. Comput. Soc. Netw. 2019, 6, 11. [Google Scholar] [CrossRef]
- Graph Convolutional Networks. Available online: https://tkipf.github.io/graph-convolutional-networks/ (accessed on 24 July 2020).
- Zhu, L.; Chen, Y.; Ghamisi, P.; Benediktsson, J.A. Generative Adversarial Networks for Hyperspectral Image Classification. IEEE Trans. Geosci. Remote Sens. 2018, 56, 5046–5063. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Nets. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 8–13 December 2014; pp. 2672–2680. [Google Scholar]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of Generative Autoencoder in De Novo Molecular Design. Mol. Inform. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, X.; Wang, Z.; Liang, J. Discriminant autoencoder for feature extraction in fault diagnosis. Chemom. Intell. Lab. Syst. 2019, 192, 103814. [Google Scholar] [CrossRef]
- Schuster, M.; Paliwal, K.K. Bidirectional recurrent neural networks. IEEE Trans. Signal Process. 1997, 45, 2673–2681. [Google Scholar] [CrossRef]
- Sterzing, V.; Schurmann, B. Recurrent neural networks for temporal learning of time series. In Proceedings of the IEEE International Conference on Neural Networks, San Francisco, CA, USA, 28 March–1 April 1993; pp. 843–850. [Google Scholar]
- Greff, K.; Srivastava, R.K.; Koutnik, J.; Steunebrink, B.R.; Schmidhuber, J. LSTM: A Search Space Odyssey. IEEE Trans. Neural Netw. Learn. Syst. 2017, 28, 2222–2232. [Google Scholar] [CrossRef] [PubMed]
- Azzouni, A.; Pujolle, G. A Long Short-Term Memory Recurrent Neural Network Framework for Network Traffic Matrix Prediction. arXiv 2017, arXiv:1705.05690. [Google Scholar]
- Bidirectional Recurrent Neural Networks—IEEE Journals & Magazine. Available online: https://ieeexplore.ieee.org/document/650093 (accessed on 18 September 2020).
- Yakura, H.; Shinozaki, S.; Nishimura, R.; Oyama, Y.; Sakuma, J. Neural malware analysis with attention mechanism. Comput. Secur. 2019, 87, 101592. [Google Scholar] [CrossRef]
- Baek, J.; Lee, B.; Kwon, S.; Yoon, S. LncRNAnet: Long non-coding RNA identification using deep learning. Bioinformatics 2018, 34, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, L.; Zhou, M.; Xie, H.; Zhang, C.; Wang, M.D.; Zhu, H. LncADeep: An ab initio lncRNA identification and functional annotation tool based on deep learning. Bioinformatics 2018, 34, 3825–3834. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Q.; Li, B.-X.; Zeng, G.-R.; Liu, Q.-Y.; Ai, D.-M. Prediction of Long Non-Coding RNAs Based on Deep Learning. Genes 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Patel, S.; Kumari, V.; Chakraborty, P.; Varadwaj, P.K. DeepLNC, a long non-coding RNA prediction tool using deep neural network. Netw. Model. Anal. Health Inform. Bioinform. 2016, 5, 21. [Google Scholar] [CrossRef]
- Alam, T.; Islam, M.T.; Househ, M.; Belhaouari, S.B.; Kawsar, F.A. DeepCNPP: Deep Learning Architecture to Distinguish the Promoter of Human Long Non-Coding RNA Genes and Protein-Coding Genes. Stud. Health Technol. Inform. 2019, 262, 232–235. [Google Scholar]
- Alam, T.; Islam, M.T.; Schmeier, S.; Househ, M.; Al-Thani, D.A. DeePEL: Deep learning architecture to recognize p-lncRNA and e-lncRNA promoters. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019. [Google Scholar]
- Gudenas, B.L.; Wang, L. Prediction of LncRNA Subcellular Localization with Deep Learning from Sequence Features. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Pan, X.; Fan, Y.-X.; Yan, J.; Shen, H.-B. IPMiner: Hidden ncRNA-protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genom. 2016, 17, 582. [Google Scholar] [CrossRef]
- Yi, H.-C.; You, Z.-H.; Huang, D.-S.; Li, X.; Jiang, T.-H.; Li, L.-P. A Deep Learning Framework for Robust and Accurate Prediction of ncRNA-Protein Interactions Using Evolutionary Information. Mol. Ther. Nucleic Acids 2018, 11, 337–344. [Google Scholar] [CrossRef]
- Zhan, Z.-H.; Jia, L.-N.; Zhou, Y.; Li, L.-P.; Yi, H.-C. BGFE: A Deep Learning Model for ncRNA-Protein Interaction Predictions Based on Improved Sequence Information. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Peng, C.; Han, S.; Zhang, H.; Li, Y. RPITER: A Hierarchical Deep Learning Framework for ncRNA–Protein Interaction Prediction. Int. J. Mol. Sci. 2019, 20, 1070. [Google Scholar] [CrossRef]
- Huang, Y.-A.; Huang, Z.-A.; You, Z.-H.; Zhu, Z.; Huang, W.-Z.; Guo, J.-X.; Yu, C.-Q. Predicting lncRNA-miRNA Interaction via Graph Convolution Auto-Encoder. Front. Genet. 2019, 10, 758. [Google Scholar] [CrossRef]
- Wang, F.; Chainani, P.; White, T.; Yang, J.; Liu, Y.; Soibam, B. Deep learning identifies genome-wide DNA binding sites of long noncoding RNAs. RNA Biol. 2018, 15, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Pan, S.; Zhang, T.; Liu, Y.; Sun, H. Graph Convolutional Network and Convolutional Neural Network Based Method for Predicting lncRNA-Disease Associations. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Cao, Y.; Zhang, T.; Kong, R.; Zhang, Z. Dual Convolutional Neural Networks with Attention Mechanisms Based Method for Predicting Disease-Related lncRNA Genes. Front. Genet. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Hu, J. Deep learning enables accurate prediction of interplay between lncRNA and disease. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Mamun, A.A.; Al Mamun, A.; Mondal, A.M. Long Non-coding RNA Based Cancer Classification using Deep Neural Networks. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics—BCB ’19, Niagara Falls, NY, USA, 7–10 September 2019. [Google Scholar]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Guo, J.-C.; Fang, S.-S.; Wu, Y.; Zhang, J.-H.; Chen, Y.; Liu, J.; Wu, B.; Wu, J.-R.; Li, E.-M.; Xu, L.-Y.; et al. CNIT: A fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res. 2019, 47, W516–W522. [Google Scholar] [CrossRef]
- Amin, N.; McGrath, A.; Chen, Y.-P.P. Evaluation of deep learning in non-coding RNA classification. Nat. Mach. Intell. 2019, 1, 246–256. [Google Scholar] [CrossRef]
- Han, S.; Liang, Y.; Ma, Q.; Xu, Y.; Zhang, Y.; Du, W.; Wang, C.; Li, Y. LncFinder: An integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief. Bioinform. 2018, 20, 2009–2027. [Google Scholar] [CrossRef]
- Lin, J.; Wen, Y.; He, S.; Yang, X.; Zhang, H.; Zhu, H. Pipelines for cross-species and genome-wide prediction of long noncoding RNA binding. Nat. Protoc. 2019, 14, 795–818. [Google Scholar] [CrossRef]
- Alam, T.; Medvedeva, Y.A.; Jia, H.; Brown, J.B.; Lipovich, L.; Bajic, V.B. Promoter analysis reveals globally differential regulation of human long non-coding RNA and protein-coding genes. PLoS ONE 2014, 9, e109443. [Google Scholar] [CrossRef] [PubMed]
- Schmeier, S.; Alam, T.; Essack, M.; Bajic, V.B. TcoF-DB v2: Update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017, 45, D145–D150. [Google Scholar] [CrossRef] [PubMed]
- Salhi, A.; Essack, M.; Alam, T.; Bajic, V.P.; Ma, L.; Radovanovic, A.; Marchand, B.; Schmeier, S.; Zhang, Z.; Bajic, V.B. DES-ncRNA: A knowledgebase for exploring information about human micro and long noncoding RNAs based on literature-mining. RNA Biol. 2017, 14, 963–971. [Google Scholar] [CrossRef]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Cao, Z.; Pan, X.; Yang, Y.; Huang, Y.; Shen, H.-B. The lncLocator: A subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics 2018, 34, 2185–2194. [Google Scholar] [CrossRef]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef]
- Kim, M.Y.; Hur, J.; Jeong, S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009, 42, 125–130. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Pan, X.; Shen, H.-B. Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 2018, 34, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Ren, S.; Lu, M.; Zhang, Y.; Zhu, D.; Zhang, X.; Li, T. Computational prediction of associations between long non-coding RNAs and proteins. BMC Genom. 2013, 14, 651. [Google Scholar] [CrossRef] [PubMed]
- Suresh, V.; Liu, L.; Adjeroh, D.; Zhou, X. RPI-Pred: Predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 2015, 43, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Muppirala, U.K.; Honavar, V.G.; Dobbs, D. Predicting RNA-protein interactions using only sequence information. BMC Bioinform. 2011, 12, 489. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.A.; Walia, R.R.; Terribilini, M.; Ferguson, J.; Zheng, C.; Honavar, V.; Dobbs, D. PRIDB: A Protein-RNA interface database. Nucleic Acids Res. 2011, 39, D277–D282. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-W.; Fan, X.-N. Computational Methods for Predicting ncRNA-protein Interactions. Med. Chem. 2017, 13, 515–525. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, T.; Xu, Z.; Wai, C.M.; Chen, K.; Zhang, X.; Wang, S.; Ji, B.; Ming, R.; Sunkar, R. Identification of microRNAs, phasiRNAs and Their Targets in Pineapple. Trop. Plant Biol. 2016, 9, 176–186. [Google Scholar] [CrossRef]
- Jalali, S.; Bhartiya, D.; Lalwani, M.K.; Sivasubbu, S.; Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS ONE 2013, 8, e53823. [Google Scholar] [CrossRef]
- Huang, Z.-A.; Huang, Y.-A.; You, Z.-H.; Zhu, Z.; Sun, Y. Novel link prediction for large-scale miRNA-lncRNA interaction network in a bipartite graph. BMC Med. Genom. 2018, 11, 17–27. [Google Scholar] [CrossRef]
- Antonov, I.V.; Mazurov, E.; Borodovsky, M.; Medvedeva, Y.A. Prediction of lncRNAs and their interactions with nucleic acids: Benchmarking bioinformatics tools. Brief. Bioinform. 2019, 20, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Hon, J.; Martínek, T.; Rajdl, K.; Lexa, M. Triplex: An R/Bioconductor package for identification and visualization of potential intramolecular triplex patterns in DNA sequences. Bioinformatics 2013, 29, 1900–1901. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Kuo, C.-C.; Kalwa, M.; Wagner, W.; Costa, I.G. Triplex Domain Finder: Detection of Triple Helix Binding Domains in Long Non-Coding RNAs. bioRxiv 2015. [Google Scholar] [CrossRef]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplexator: Detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012, 22, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplex-Inspector: An analysis tool for triplex-mediated targeting of genomic loci. Bioinformatics 2013, 29, 1895–1897. [Google Scholar] [CrossRef]
- He, S.; Zhang, H.; Liu, H.; Zhu, H. LongTarget: A tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics 2015, 31, 178–186. [Google Scholar] [CrossRef]
- Ping, P.; Wang, L.; Kuang, L.; Ye, S.; Iqbal, M.F.B.; Pei, T. A Novel Method for LncRNA-Disease Association Prediction Based on an lncRNA-Disease Association Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 688–693. [Google Scholar] [CrossRef]
- Lan, W.; Li, M.; Zhao, K.; Liu, J.; Wu, F.-X.; Pan, Y.; Wang, J. LDAP: A web server for lncRNA-disease association prediction. Bioinformatics 2017, 33, 458–460. [Google Scholar] [CrossRef]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.-X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef]
- Fu, G.; Wang, J.; Domeniconi, C.; Yu, G. Matrix factorization-based data fusion for the prediction of lncRNA-disease associations. Bioinformatics 2018, 34, 1529–1537. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Luo, C.; Ji, W.; Zhang, Y.; Dai, Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 2015, 5, 11338. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y. Practical Recommendations for Gradient-Based Training of Deep Architectures. Lect. Notes Comput. Sci. 2012, 7700, 437–478. [Google Scholar]
- Alam, T.; Alazmi, M.; Naser, R.; Huser, F.; Momin, A.A.; Walkiewicz, K.W.; Canlas, C.G.; Huser, R.G.; Ali, A.J.; Merzaban, J.; et al. Proteome-level assessment of origin, prevalence and function of Leucine-Aspartic Acid (LD) motifs. Bioinformatics 2020, 36, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.; Islam, M.T.; Househ, M.; Bouzerdoum, A.; Kawsar, F.A. DeepDSSR: Deep Learning Structure for Human Donor Splice Sites Recognition. Stud. Health Technol. Inform. 2019, 262, 236–239. [Google Scholar] [PubMed]
- Kalkatawi, M.; Magana-Mora, A.; Jankovic, B.; Bajic, V.B. DeepGSR: An optimized deep-learning structure for the recognition of genomic signals and regions. Bioinformatics 2019, 35, 1125–1132. [Google Scholar] [CrossRef]
- Greenside, P.G. Interpretable Machine Learning Methods for Regulatory and Disease Genomics; Stanford University: Stanford, CA, USA, 2018. [Google Scholar]
- Park, Y.; Kellis, M. Deep learning for regulatory genomics. Nat. Biotechnol. 2015, 33, 825–826. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Samek, W.; Binder, A.; Montavon, G.; Lapuschkin, S.; Muller, K.-R. Evaluating the Visualization of What a Deep Neural Network Has Learned. IEEE Trans Neural Netw. Learn Syst. 2017, 28, 2660–2673. [Google Scholar] [CrossRef]
- Shrikumar, A.; Greenside, P.; Kundaje, A. Learning important features through propagating activation differences. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; Volume 70, pp. 3145–3153. [Google Scholar]
- Lipton, Z.C. The mythos of model interpretability. Commun. ACM 2018, 61, 36–43. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.-M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Dauphin, Y.; Pascanu, R.; Gulcehre, C.; Cho, K.; Ganguli, S.; Bengio, Y. Identifying and attacking the saddle point problem in high-dimensional non-convex optimization. Adv. Inf. Proces. Syst. 2014, 2014, 2933–2941. [Google Scholar]
- Leibig, C.; Allken, V.; Ayhan, M.S.; Berens, P.; Wahl, S. Leveraging uncertainty information from deep neural networks for disease detection. Sci. Rep. 2017, 7, 17816. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Pleiss, G.; Sun, Y.; Weinberger, K.Q. On Calibration of Modern Neural Networks. arXiv 2017, arXiv:1706.04599. [Google Scholar]
- Probabilistic Outputs for Support Vesctor Machines and Comparisons to Refularized Likelihood Methods. Available online: https://www.cs.colorado.edu/~mozer/Teaching/syllabi/6622/papers/Platt1999.pdf (accessed on 24 July 2020).
- Zadrozny, B.; Elkan, C. Transforming classifier scores into accurate multiclass probability estimates. In Proceedings of the Eighth ACM SIGKDD International Conference on Knowledge Discovery and Data Mining—KDD ’02, Montreal, QC, Canada, 23–26 July 2002. [Google Scholar]
- Kirkpatrick, J.; Pascanu, R.; Rabinowitz, N.; Veness, J.; Desjardins, G.; Rusu, A.A.; Milan, K.; Quan, J.; Ramalho, T.; Grabska-Barwinska, A.; et al. Overcoming catastrophic forgetting in neural networks. Proc. Natl. Acad. Sci. USA 2017, 114, 3521–3526. [Google Scholar] [CrossRef]
- Rebuffi, S.-A.; Kolesnikov, A.; Sperl, G.; Lampert, C.H. iCaRL: Incremental Classifier and Representation Learning. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 26 June–1 July 2016. [Google Scholar]
- Tieleman, T.; Hinton, G. Using fast weights to improve persistent contrastive divergence. In Proceedings of the 26th Annual International Conference on Machine Learning—ICML ’09, Montreal, QC, Canada, 14–18 June 2009. [Google Scholar]
- Di Lena, P.; Nagata, K.; Baldi, P. Deep architectures for protein contact map prediction. Bioinformatics 2012, 28, 2449–2457. [Google Scholar] [CrossRef]
- Lena, P.D.; Nagata, K.; Baldi, P.F. Deep Spatio-Temporal Architectures and Learning for Protein Structure Prediction. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, NV, USA, 3–6 December 2012; pp. 512–520. [Google Scholar]
- Baldi, P.; Pollastri, G. The Principled Design of Large-Scale Recursive Neural Network Architectures--DAG-RNNs and the Protein Structure Prediction Problem. J. Mach. Learn. Res. 2003, 4, 575–602. [Google Scholar]
- Graves, A.; Schmidhuber, J. Offline Handwriting Recognition with Multidimensional Recurrent Neural Networks. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 6–8 December 2009; pp. 545–552. [Google Scholar]
- Masci, J.; Meier, U.; Cireşan, D.; Schmidhuber, J. Stacked Convolutional Auto-Encoders for Hierarchical Feature Extraction. Lect. Notes Comput. Sci. 2011, 6791, 52–59. [Google Scholar]
- Gupta, A.; Zou, J. Feedback GAN for DNA optimizes protein functions. Nat. Mach. Intell. 2019, 1, 105–111. [Google Scholar] [CrossRef]
- Hinton, G.E.; Sabour, S.; Frosst, N. Matrix capsules with EM routing. In Proceedings of the International Conference on Learning Representations, Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]
- Afshar, P.; Mohammadi, A.; Plataniotis, K.N. Brain Tumor Type Classification via Capsule Networks. In Proceedings of the 25th IEEE International Conference on Image Processing (ICIP), Athens, Greece, 7–10 October 2018. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Area | Proposed DL Based Architecture | References |
|---|---|---|
| LncRNA Identification | CNN and RNN | LncRNAnet [52] |
| DBN | LncADeep [53] | |
| Embedding vector, BLSTM, CNN | Liu et al. [54] | |
| DNN | DeepLNC [55] | |
| Distinct transcription regulation of lncRNAs | CNN | DeepCNPP [56], DeePEL [57] |
| Functional annotation of lncRNAs | DNN | LncADeep [53] |
| Localization prediction | DNN | DeepLncRNA [58] |
| lncRNA–protein interaction | Stacked auto-encoder, Random forest | IPminer [59], RPI-SAN [60], BGFE [61] |
| Stacked auto-encoder, CNN | RPITER [62] | |
| LncRNA–miRNA interaction | GCN | GCLMI [63] |
| LncRNA–DNA interaction | GCN | [64] |
| LncRNA–disease association | GCN and AM | GCNLDA [65] |
| CNN and AM | CNNLDA [66] | |
| DNN | NNLDA [67] | |
| Cancer type classification | MLP, CNN, LSTM, DAE | [68] |
| LncRNAnet [52] | LncADeep [53] | Liu et al. [54] | DeepLNC [55] | |
|---|---|---|---|---|
| Publication Year | 2018 | 2018 | 2019 | 2016 |
| Species | Human and Mouse | Human and Mouse | Human and Mouse | Human |
| Data source used | GENCODE 25, Ensembl | GENCODE 24, Refseq | GENCODE 28, Refseq | LNCipedia 3.1, Refseq |
| Number of lncRNA considered for training | ~21k (~21k) lncRNA transcripts from human (mouse) | ~66k (~42k) full length lncRNA transcripts from human (mouse) | 28k (~17k) lncRNA transcripts from human (mouse) | ~80k lncRNA transcripts and ~100k mRNA transcripts |
| Performance metric | SN, SP, ACC, F1-Score, AUC | SN, SP, Hm | SN, SP, ACC, F1-Score, AUC | SN, SP, ACC, F1-Score, Precision |
| Metrics for comparison against traditional ML based model * | ACC:91.79 # | Hm: 97.7 # | ACC:96.4 # | ACC: 98.07 |
| Intriguing features from the proposed model | ORF length and ratio | ORF length and ratio, k-mer composition and hexamer score, position specific nucleotide frequency etc. | k-mer embedding | Solely based on k-mer patterns |
| Source code/Implementation | N/A | https://github.com/cyang235/LncADeep/ | N/A | http://bioserver.iiita.ac.in/deeplnc |
| DeepCNPP [56] | DeePEL [57] | |
|---|---|---|
| Publication Year | 2019 | 2019 |
| Species | Human | Human |
| Data source used | Dataset from [75] | FANTOM CAT [17] |
| Number of lncRNA transcripts or genes considered | ~19k lncRNA genes | ~7k (~3k) p-lncRNA (e-lncRNA) transcripts |
| Performance metric | SN, SP, ACC | SN, SP, MCC, AUC |
| Metrics for comparison against traditional ML based model * | ACC: 83.34 | Traditional ML model does not exist for this task |
| Intriguing features from the proposed model | k-mer embedding of promoter regions | k-mer embedding of promoter regions, transcription factor binding sites |
| IPminer [59] | RPI-SAN [60] | BGFE [61] | RPITER [62] | |
|---|---|---|---|---|
| Publication Year | 2016 | 2018 | 2019 | 2019 |
| Species | Multi-species | Multi-species | Multi-species | Multi-species |
| Benchmark Data source used | NPInter 2.0, RPI369, RPI488, RPI1807, RPI2241, RPI13254 | NPInter 2.0, RPI488, RPI1807, RPI2241 | RPI488, RPI1807, RPI2241 | NPInter 2.0, RPI369, RPI488, RPI1807, RPI2241 |
| Performance metric | SN, SP, ACC, Precision, AUC, MCC | SN, SP, ACC, Precision, AUC, MCC | SN, SP, ACC, Precision, AUC, MCC | SN, SP, ACC, Precision, AUC, MCC |
| Metrics for comparison against traditional ML based model for different dataset * | NPInter 2.0 (ACC: 95.7) #, RPI369 (ACC: 75.2), RPI488 (ACC: 89.1), RPI1807 (ACC: 98.6), RPI2241 (ACC: 82.4), RPI13254 (ACC: 94.5) | NPInter 2.0 (ACC: 99.33) #, RPI488 (ACC: 89.7), RPI1807 (ACC: 96.1), RPI2241 (ACC: 90.77) | RPI488 (ACC: 88.68), RPI1807 (ACC: 96.0), RPI2241 (ACC: 91.30) | NPInter 2.0 (ACC: 95.5) #, RPI369 (ACC: 72.8), RPI488 (ACC: 89.3), RPI1807 (ACC: 96.8), RPI2241 (ACC: 89.0) |
| Intriguing features from the proposed model | Sequence composition features, specifically 3-mer and 4-mer from protein and RNA sequences, respectively | k-mer sparse matrix from RNA sequences and PSSM from protein sequences | k-mer sparse matrix from RNA sequences and PSSM from protein sequences. Stacked auto-encoder was employed to get high accuracy | k-mer frequency of sequence and two types of structural information (bracket and dot) from RNA. k-mer frequency of sequence and three types of structural information (α-helix, β-sheet and coil) from protein |
| Source code/Implementation | https://github.com/xypan1232/IPMiner; http://www.csbio.sjtu.edu.cn/bioinf/IPMiner | N/A | N/A | https://github.com/Pengeace/RPITER |
| GCNLDA [65] | CNNLDA [66] | NNLDA [67] | |
|---|---|---|---|
| Publication Year | 2019 | 2019 | 2019 |
| Data source used | LncRNADisease, Lnc2cancer, GeneRIF | LncRNADisease, Lnc2cancer, GeneRIF | LncRNADisease |
| Number of lncRNA considered | 240 | 240 | 19166 |
| Number of diseases considered | 402 | 402 | 529 |
| Performance metric | AUC, AUPRC, Precision, Recall | AUC, AUPRC, Precision, Recall | HR(k): Probability for the predicted samples to appear in top-k ranked list |
| Metrics for comparison against traditional ML based models | AUC $: 0.959 AUPRC $: 0.223 | AUC $: 0.952 AUPRC $: 0.251 | HR(k); k = 1.10 |
| Intriguing features from the proposed model * | For ncRNA-lncRNA similarity Chen’s method was applied [105]. For disease-disease similarity Wang’s method was applied [106] | For ncRNA-lncRNA similarity Chen’s method was applied [105]. For disease-disease similarity Wang’s method was applied [106] | Matrix factorization method was modified in two aspects to fit into this model: (a) cross-entropy was used as a loss function; (b) only one batch data per round was used to minimize loss |
| Source code/Implementation | N/A | N/A | https://github.com/gao793583308/NNLDA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, T.; Al-Absi, H.R.H.; Schmeier, S. Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives. Non-Coding RNA 2020, 6, 47. https://doi.org/10.3390/ncrna6040047
Alam T, Al-Absi HRH, Schmeier S. Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives. Non-Coding RNA. 2020; 6(4):47. https://doi.org/10.3390/ncrna6040047
Chicago/Turabian StyleAlam, Tanvir, Hamada R. H. Al-Absi, and Sebastian Schmeier. 2020. "Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives" Non-Coding RNA 6, no. 4: 47. https://doi.org/10.3390/ncrna6040047
APA StyleAlam, T., Al-Absi, H. R. H., & Schmeier, S. (2020). Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives. Non-Coding RNA, 6(4), 47. https://doi.org/10.3390/ncrna6040047