MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications

Abstract
1. Brain Immunity and Neuroinflammation
2. MicroRNAs
3. Key miRNAs Which Regulate Neuroinflammation
3.1. miR-155
3.2. miR-146a
3.3. miR-124
3.4. miR-21
3.5. Let-7
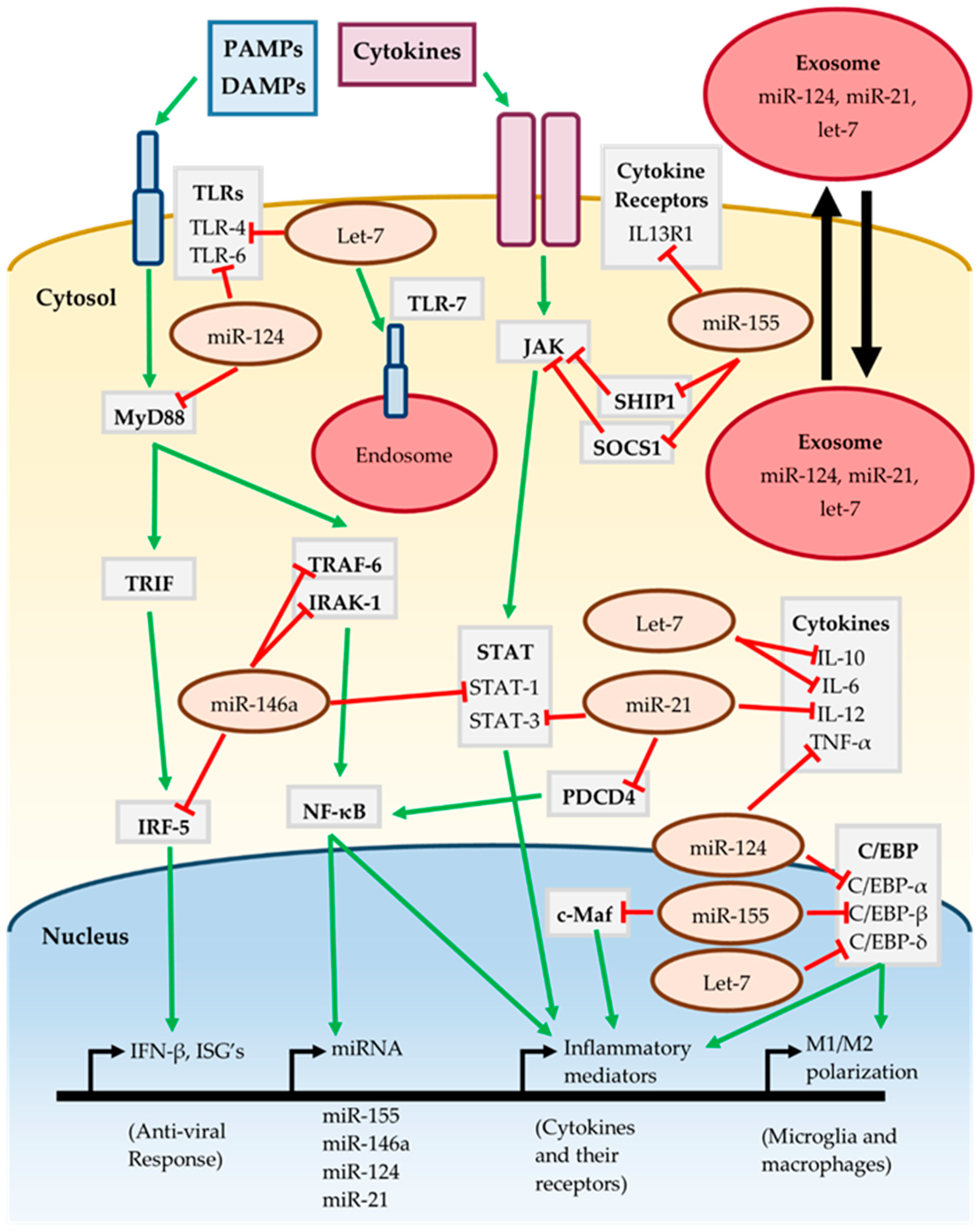
3.6. MiRNAs In Neuroinflammatory Signaling
4. MiRNAs in Disorders of Neuroinflammation
4.1. Neurodegenerative Diseases
4.1.1. Multiple Sclerosis
4.1.2. Alzheimer’s Disease
4.1.3. Parkinson’s Disease
4.1.4. Prion Diseases
4.2. CNS Viral Infection
4.2.1. Japanese Encephalitis Virus
4.2.2. Herpes Simplex Virus Encephalitis
4.3. CNS Injury
4.3.1. Ischemic Stroke
4.3.2. Traumatic Brain Injury
4.4. Notable miRNAs in Neuroinflammatory Disorders
5. MiRNAs as Biomarkers for Neuroinflammatory Diseases
6. Therapeutic Applications of miRNAs in Neuroinflammatory Disease
7. Conclusions/Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The Movers and Shapers in Immune Privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation Pathways: A General Review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Garber, C.; Howard, N. Infectious Immunity in the Central Nervous System and Brain Function. Nat. Immunol. 2017, 18, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Disabato, D.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2017, 139, 136–153. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [CrossRef] [PubMed]
- Wieghofer, P.; Prinz, M. Genetic Manipulation of Microglia during Brain Development and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 299–309. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during Development and Aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Buckwalter, M.S. Astrocytes: Integrative Regulators of Neuroinflammation in Stroke and Other Neurological Diseases. Neurotherapeutics 2016, 13, 685–701. [Google Scholar] [CrossRef]
- Vasile, F.; Dossi, E.; Rouach, N. Human Astrocytes: Structure and Functions in the Healthy Brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef]
- Dossi, E.; Vasile, F.; Rouach, N. Human Astrocytes in the Diseased Brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Barriers to Neurotoxic Inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Tohidpour, A.; Morgun, A.V.; Boitsova, E.B.; Malinovskaya, N.A.; Martynova, G.P.; Khilazheva, E.D.; Kopylevich, N.V.; Gertsog, G.E.; Salmina, A.B. Neuroinflammation and Infection: Molecular Mechanisms Associated with Dysfunction of Neurovascular Unit. Front. Cell. Infect. Microbiol. 2017, 7, 276. [Google Scholar] [CrossRef]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The Neurovascular Unit—Concept Review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Presta, I.; Vismara, M.; Novellino, F.; Donato, A.; Zaffino, P.; Scali, E.; Pirrone, K.C.; Spadea, M.F.; Malara, N.; Donato, G. Innate Immunity Cells and the Neurovascular Unit. Int. J. Mol. Sci. 2018, 19, 3856. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of MiRNAs and SiRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA Polymerase III Transcribes Human MicroRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Macias, S.; Cordiner, R.A.; Cáceres, J.F. Cellular Functions of the Microprocessor. Biochem. Soc. Trans. 2013, 41, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-S.; Rossi, J.J. Molecular Mechanisms of Dicer: Endonuclease and Enzymatic Activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular Mechanisms of RNA Interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Meijer, H.A.; Smith, E.M.; Bushell, M. Regulation of MiRNA Strand Selection: Follow the Leader? Biochem. Soc. Trans. 2014, 42, 1135–1140. [Google Scholar] [CrossRef]
- Gorski, S.A.; Vogel, J.; Doudna, J.A. RNA-Based Recognition and Targeting: Sowing the Seeds of Specificity. Nat. Rev. Mol. Cell Biol. 2017, 18, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shin, C. MicroRNA-Directed Cleavage of Targets: Mechanism and Experimental Approaches. BMB Rep. 2014, 47, 417–423. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of MRNA Translation and Stability by MicroRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human MiRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Bayraktar, R.; Van Roosbroeck, K.; Calin, G.A. Cell-to-Cell Communication: MicroRNAs as Hormones. Mol. Oncol. 2017, 11, 1673–1686. [Google Scholar] [CrossRef]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. In Methods in Molecular Biology; Springer: Berlin, Germany, 2017; Volume 1509, pp. 1–10. [Google Scholar] [CrossRef]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible MicroRNA-155 Feedback Promotes Type I IFN Signaling in Antiviral Innate Immunity by Targeting Suppressor of Cytokine Signaling 1. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-Regulation of MicroRNA-155 in Macrophages Contributes to Increased Tumor Necrosis Factor α (TNFα) Production via Increased MRNA Half-Life in Alcoholic Liver Disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Guedes, J.R.; Pereira de Almeida, L.; Pedroso de Lima, M.C. MiR-155 Modulates Microglia-Mediated Immune Response by down-Regulating SOCS-1 and Promoting Cytokine and Nitric Oxide Production. Immunology 2012, 135, 73–88. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 Is Induced during the Macrophage Inflammatory Response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol Phosphatase SHIP1 Is a Primary Target of MiR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef]
- Worm, J.; Stenvang, J.; Petri, A.; Frederiksen, K.S.; Obad, S.; Elmén, J.; Hedtjärn, M.; Straarup, E.M.; Hansen, J.B.; Kauppinen, S. Silencing of MicroRNA-155 in Mice during Acute Inflammatory Response Leads to Derepression of c/Ebp Beta and down-Regulation of G-CSF. Nucleic Acids Res. 2009, 37, 5784–5792. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Nunez, R.T.; Louafi, F.; Sanchez-Elsner, T. The Interleukin 13 (IL-13) Pathway in Human Macrophages Is Modulated by MicroRNA-155 via Direct Targeting of Interleukin 13 Receptor A1 (IL13Rα1). J. Biol. Chem. 2011, 286, 1786–1794. [Google Scholar] [CrossRef]
- Su, W.; Hopkins, S.; Nesser, N.K.; Sopher, B.; Silvestroni, A.; Ammanuel, S.; Jayadev, S.; Moller, T.; Weinstein, J.; Garden, G.A. The P53 Transcription Factor Modulates Microglia Behavior through MicroRNA-Dependent Regulation of c-Maf. J. Immunol. 2014, 192, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-B-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Cui, J.G.; Li, Y.Y.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. Differential Regulation of Interleukin-1 Receptor-Associated Kinase-1 (IRAK-1) and IRAK-2 by MicroRNA-146a and NF-ΚB in Stressed Human Astroglial Cells and in Alzheimer Disease. J. Biol. Chem. 2010, 285, 38951–38960. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Cui, J.G.; Dua, P.; Pogue, A.I.; Bhattacharjee, S.; Lukiw, W.J. Differential Expression of MiRNA-146a-Regulated Inflammatory Genes in Human Primary Neural, Astroglial and Microglial Cells. Neurosci. Lett. 2011, 499, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cerutti, C.; Lopez-Ramirez, M.A.; Pryce, G.; King-Robson, J.; Simpson, J.E.; Van Der Pol, S.M.A.; Hirst, M.C.; De Vries, H.E.; Sharrack, B.; et al. Brain Endothelial MiR-146a Negatively Modulates T-Cell Adhesion through Repressing Multiple Targets to Inhibit NF-ΚB Activation. J. Cereb. Blood Flow Metab. 2015, 35, 412–423. [Google Scholar] [CrossRef]
- He, X.; Tang, R.; Sun, Y.; Wang, Y.-G.; Zhen, K.-Y.; Zhang, D.-M.; Pan, W.-Q. MicroR-146 Blocks the Activation of M1 Macrophage by Targeting Signal Transducer and Activator of Transcription 1 in Hepatic Schistosomiasis. EBioMedicine 2016, 13, 339–347. [Google Scholar] [CrossRef]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; De Vries, N.; Tak, P.P.; et al. MicroRNA-146a Contributes to Abnormal Activation of the Type I Interferon Pathway in Human Lupus by Targeting the Key Signaling Proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Zhao, Y.; Jian, G.C. An NF-ΚB-Sensitive Micro RNA-146a-Mediated Inflammatory Circuit in Alzheimer Disease and in Stressed Human Brain Cells. J. Biol. Chem. 2008, 283, 31315–31322. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, X.; QunZhou; Xie, J.; Ma, T.; Meng, X.; Li, J. MiR-146a Modulates Macrophage Polarization by Inhibiting Notch1 Pathway in RAW264.7 Macrophages. Int. Immunopharmacol. 2016, 32, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The MicroRNA MiR-124 Promotes Neuronal Differentiation by Triggering Brain-Specific Alternative Pre-MRNA Splicing. Mol. Cell 2007, 27, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 Promotes Microglia Quiescence and Suppresses EAE by Deactivating Macrophages via the C/EBP-α-PU.1 Pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Veremeyko, T.; Siddiqui, S.; Sotnikov, I.; Yung, A.; Ponomarev, E.D. IL-4/IL-13-Dependent and Independent Expression of MiR-124 and Its Contribution to M2 Phenotype of Monocytic Cells in Normal Conditions and during Allergic Inflammation. PLoS ONE 2013, 8, e81774. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Li, Y.; Li, M.; Deng, G.; Wu, X.; Zeng, J.; Hao, X.; Wang, X.; Liu, J.; Cho, W.C.S.; et al. MicroRNA-124 Negatively Regulates TLR Signaling in Alveolar Macrophages in Response to Mycobacterial Infection. Mol. Immunol. 2014, 62, 150–158. [Google Scholar] [CrossRef]
- Louw, A.M.; Kolar, M.K.; Novikova, L.N.; Kingham, P.J.; Wiberg, M.; Kjems, J.; Novikov, L.N. Chitosan Polyplex Mediated Delivery of MiRNA-124 Reduces Activation of Microglial Cells in Vitro and in Rat Models of Spinal Cord Injury. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Freilich, R.W.; Woodbury, M.E.; Ikezu, T. Integrated Expression Profiles of MRNA and MiRNA in Polarized Primary Murine Microglia. PLoS ONE 2013, 8, e79416. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J. Turning 21: Induction of MiR-21 as a Key Switch in the Inflammatory Response. Front. Immunol. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, L.Y.; Li, Y.J.; Hong, Z.; Wei, W.S. MiR-21 Represses FasL in Microglia and Protects against Microglia-Mediated Neuronal Cell Death Following Hypoxia/Ischemia. Glia 2012, 60, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-J.; Pan, Y.-B.; Sun, Z.-L.; Sun, Y.-Y.; Yang, X.-T.; Feng, D.-F. Inhibition of MiR-21 Ameliorates Excessive Astrocyte Activation and Promotes Axon Regeneration Following Optic Nerve Crush. Neuropharmacology 2018, 137, 33–49. [Google Scholar] [CrossRef]
- Han, Z.; Chen, F.; Ge, X.; Tan, J.; Lei, P.; Zhang, J. MiR-21 Alleviated Apoptosis of Cortical Neurons through Promoting PTEN-Akt Signaling Pathway in Vitro after Experimental Traumatic Brain Injury. Brain Res. 2014, 1582, 12–20. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J.; Hall, K.O.; Bonner, H.; Roller, A.M.; Syed, M.; Park, C.J.; Ball, J.P.; Rothenberg, M.E.; Stockmeier, C.A.; Romero, D.G. MicroRNA-21: Expression in Oligodendrocytes and Correlation with Low Myelin MRNAs in Depression and Alcoholism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 503–514. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Palsson-Mcdermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A.J. Negative Regulation of TLR4 via Targeting of the Proinflammatory Tumor Suppressor PDCD4 by the MicroRNA MiR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef]
- Barnett, R.E.; Conklin, D.J.; Ryan, L.; Keskey, R.C.; Ramjee, V.; Sepulveda, E.A.; Srivastava, S.; Bhatnagar, A.; Cheadle, W.G. Anti-Inflammatory Effects of MiR-21 in the Macrophage Response to Peritonitis. J. Leukoc. Biol. 2015, 99, 361–371. [Google Scholar] [CrossRef]
- Sen, C.K.; Das, A.; Khanna, S.; Roy, S.; Ganesh, K. Engulfment of Apoptotic Cells by Macrophages: A Role of MicroRNA-21 in the Resolution of Wound Inflammation. J. Immunol. 2014, 192, 1120–1129. [Google Scholar] [CrossRef]
- Wang, Z.; Brandt, S.; Medeiros, A.; Wang, S.; Wu, H.; Dent, A.; Serezani, C.H. MicroRNA 21 Is a Homeostatic Regulator of Macrophage Polarization and Prevents Prostaglandin E2 -Mediated M2 Generation. PLoS ONE 2015, 10, e0115855. [Google Scholar] [CrossRef] [PubMed]
- Gruner, K.; Sahni, V.; Pan, L.; McGuire, T.L.; Bhalala, O.G.; Kessler, J.A.; Tourtellotte, W.G. MicroRNA-21 Regulates Astrocytic Response Following Spinal Cord Injury. J. Neurosci. 2012, 32, 17935–17947. [Google Scholar] [CrossRef]
- Lee, H.; Han, S.; Kwon, C.S.; Lee, D. Biogenesis and Regulation of the Let-7 MiRNAs and Their Functional Implications. Protein Cell 2016, 7, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Yang, S.; Liu, G.; Xie, N.; Icyuz, M.; Abraham, E.; Cui, H.; Banerjee, S. MicroRNA Let-7c Regulates Macrophage Polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef]
- Cho, K.J.; Song, J.; Oh, Y.; Lee, J.E. MicroRNA-Let-7a Regulates the Function of Microglia in Inflammation. Mol. Cell. Neurosci. 2015, 68, 167–176. [Google Scholar] [CrossRef]
- Schulte, L.N.; Eulalio, A.; Mollenkopf, H.J.; Reinhardt, R.; Vogel, J. Analysis of the Host MicroRNA Response to Salmonella Uncovers the Control of Major Cytokines by the Let-7 Family. EMBO J. 2011, 30, 1977–1989. [Google Scholar] [CrossRef]
- Teng, G.G.; Wang, W.H.; Dai, Y.; Wang, S.J.; Chu, Y.X.; Li, J. Let-7b Is Involved in the Inflammation and Immune Responses Associated with Helicobacter Pylori Infection by Targeting Toll-Like Receptor 4. PLoS ONE 2013, 8, e56709. [Google Scholar] [CrossRef]
- Shenoy, A.; Danial, M.; Blelloch, R.H. Let-7 and MiR-125 Cooperate to Prime Progenitors for Astrogliogenesis. EMBO J. 2015, 34, 1180–1194. [Google Scholar] [CrossRef]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An Unconventional Role for MiRNA: Let-7 Activates Toll-like Receptor 7 and Causes Neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Coleman, L.G.; Zou, J.; Crews, F.T. Microglial-Derived MiRNA Let-7 and HMGB1 Contribute to Ethanol-Induced Neurotoxicity via TLR7. J. Neuroinflamm. 2017, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Morton, M.C.; Neckles, V.N.; Seluzicki, C.M.; Holmberg, J.C.; Feliciano, D.M. Neonatal Subventricular Zone Neural Stem Cells Release Extracellular Vesicles That Act as a Microglial Morphogen. Cell Rep. 2018, 23, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with HSOD1-G93A Are Enriched in MiR-124 and Drive Alterations in Microglia Phenotype. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Simeoli, R.; Montague, K.; Jones, H.R.; Castaldi, L.; Chambers, D.; Kelleher, J.H.; Vacca, V.; Pitcher, T.; Grist, J.; Al-Ahdal, H.; et al. Exosomal Cargo Including MicroRNA Regulates Sensory Neuron to Macrophage Communication after Nerve Trauma. Nat. Commun. 2017, 8, 1778. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Jordana, A.; Montalban, X. Multiple Sclerosis: Epidemiologic, Clinical, and Therapeutic Aspects. Neuroimaging Clin. N. Am. 2017, 27, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of Neurodegeneration and Axonal Dysfunction in Multiple Sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA Profiling of Multiple Sclerosis Lesions Identifies Modulators of the Regulatory Protein CD47. Brain 2009, 132, 3342–3352. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, M.; Simpson, S.; Lucas, R.M.; Charlesworth, J.C.; Blackburn, N.; van der Mei, I.; Ponsonby, A.-L.; Taylor, B.V. Common Genetic Variation within MiR-146a Predicts Disease Onset and Relapse in Multiple Sclerosis. Neurol. Sci. 2018, 39, 297–304. [Google Scholar] [CrossRef]
- Park, R.; Lee, W.J.; Ji, J.D. Association between the Three Functional MiR-146a Single-Nucleotide Polymorphisms, Rs2910164, Rs57095329, and Rs2431697, and Autoimmune Disease Susceptibility: A Meta-Analysis. Autoimmunity 2016, 49, 451–458. [Google Scholar] [CrossRef]
- Martin, N.A.; Molnar, V.; Szilagyi, G.T.; Elkjaer, M.L.; Nawrocki, A.; Okarmus, J.; Wlodarczyk, A.; Thygesen, E.K.; Palkovits, M.; Gallyas, F.; et al. Experimental Demyelination and Axonal Loss Are Reduced in MicroRNA-146a Deficient Mice. Front. Immunol. 2018, 9, 490. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.G.; Lu, M.; Zhang, Y.; Shang, X.; Chopp, M. MiR-146a Promotes Oligodendrocyte Progenitor Cell Differentiation and Enhances Remyelination in a Model of Experimental Autoimmune Encephalomyelitis. Neurobiol. Dis. 2019, 125, 154–162. [Google Scholar] [CrossRef]
- Goldmann, T.; Prinz, M. Role of Microglia in CNS Autoimmunity. Clin. Dev. Immunol. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ramirez, M.A.; Wu, D.; Pryce, G.; Simpson, J.E.; Reijerkerk, A.; King-Robson, J.; Kay, O.; De Vries, H.E.; Hirst, M.C.; Sharrack, B.; et al. MicroRNA-155 Negatively Affects Blood-Brain Barrier Function during Neuroinflammation. FASEB J. 2014, 28, 2551–2565. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek-Winiarek, D.J.; Kacperska, M.J.; Glabinski, A. MicroRNAs as Novel Regulators of Neuroinflammation. Med. Inflamm. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid Precursor Protein Processing and Bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and Neurodegenerative Disease: The Story so Far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Delay, C.; Calon, F.; Mathews, P.; Hébert, S.S. Alzheimer-Specific Variants in the 3′UTR of Amyloid Precursor Protein Affect MicroRNA Function. Mol. Neurodegener. 2011, 6, 70. [Google Scholar] [CrossRef]
- Smith, P.; Al Hashimi, A.; Girard, J.; Delay, C.; Hébert, S.S. In Vivo Regulation of Amyloid Precursor Protein Neuronal Splicing by MicroRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of MicroRNA Cluster MiR-29a/b-1 in Sporadic Alzheimer’s Disease Correlates with Increased BACE1/Beta-Secretase Expression. Proc. Natl. Acad. Sci. USA 2008, 205, 6415–6420. [Google Scholar] [CrossRef]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 Downregulates Alzheimer’s Disease Amyloid-β Production by Targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Wang, W.-X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The Expression of MicroRNA MiR-107 Decreases Early in Alzheimer’s Disease and May Accelerate Disease Progression through Regulation of -Site Amyloid Precursor Protein-Cleaving Enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Zhao, J.; Yue, D.; Zhou, Y.; Jia, L.; Wang, H.; Guo, M.; Xu, H.; Chen, C.; Zhang, J.; Xu, L. The Role of MicroRNAs in Aβ Deposition and Tau Phosphorylation in Alzheimer’s Disease. Front. Neurol. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Goedeke, L.; Fernández-Hernando, C. MicroRNAs: A Connection between Cholesterol Metabolism and Neurodegeneration. Neurobiol. Dis. 2014, 72, 48–53. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. Deficits in the MiRNA-34a-Regulated Endogenous TREM2 Phagocytosis Sensor-Receptor in Alzheimer’s Disease (AD); an Update. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Zhao, Y.; Dua, P.; Rogaev, E.I.; Lukiw, W.J. MicroRNA-34α-Mediated down-Regulation of the Microglial-Enriched Triggering Receptor and Phagocytosis-Sensor TREM2 in Age-Related Macular Degeneration. PLoS ONE 2016, 11, e0150211. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Dua, P.; Lukiw, W.J. Up-Regulation of MiRNA-146a in Progressive, Age-Related Inflammatory Neurodegenerative Disorders of the Human CNS. Front. Neurol. 2014, 5. [Google Scholar] [CrossRef]
- Guedes, J.R.; Custódia, C.M.; Silva, R.J.; de Almeida, L.P.; de Lima, M.C.P.; Cardoso, A.L. Early MiR-155 Upregulation Contributes to Neuroinflammation in Alzheimer’s Disease Triple Transgenic Mouse Model. Hum. Mol. Genet. 2014, 23, 6286–6301. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Alexandrov, P.N. Regulation of Complement Factor H (CFH) by Multiple MiRNAs in Alzheimer’s Disease (AD) Brain. Mol. Neurobiol. 2012, 46, 11–19. [Google Scholar] [CrossRef]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H.; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for MiR-181 Involvement in Neuroinflammatory Responses of Astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortiz, C.J.; Baglietto-Vargas, D.; Martinez-Coria, H.; LaFerla, F.M.; Kitazawa, M. Upregulation of MiR-181 Decreases c-Fos and SIRT-1 in the Hippocampus of 3xTg-AD Mice. J. Alzheimer’s Dis. 2014, 42, 1229–1238. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s Disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K. α-Synuclein and Lewy Pathology in Parkinson’s Disease. Curr. Opin. Neurol. 2015, 28, 375–381. [Google Scholar] [CrossRef]
- Surendranathan, A.; Rowe, J.B.; O’Brien, J.T. Neuroinflammation in Lewy Body Dementia. Park. Relat. Disord. 2015, 21, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Fragkouli, A.; Doxakis, E. MiR-7 and MiR-153 Protect Neurons against MPP+-Induced Cell Death via Upregulation of MTOR Pathway. Front. Cell. Neurosci. 2014, 8, 182. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of MiR-34b and MiR-34c Enhances α-Synuclein Expression in Parkinson’s Disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of Alpha-Synuclein Expression and Toxicity by MicroRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA Profiling of Parkinson’s Disease Brains Identifies Early Downregulation of MiR-34b/c Which Modulate Mitochondrial Function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef] [PubMed]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. MicroRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Ye, Y.; Mao, H.; Lu, F.; He, X.; Lu, G.; Zhang, S. MicroRNA-124 Regulates the Expression of MEKK3 in the Inflammatory Pathogenesis of Parkinson’s Disease. J. Neuroinflamm. 2018, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Whitechurch, B.C.; Welton, J.M.; Collins, S.J.; Lawson, V.A. Prion Diseases. In Neurodegenerative Diseases, Advances in Neurobiology; Springer: Cham, Switzerland, 2017; pp. 335–364. [Google Scholar] [CrossRef]
- Hughes, D.; Halliday, M. What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings? Pathogens 2017, 6, 63. [Google Scholar] [CrossRef]
- Mabbott, N. How Do PrPSc Prions Spread between Host Species, and within Hosts? Pathogens 2017, 6, 60. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Martí, E.; Kanata, E.; Dafou, D.; Díaz-Lucena, D.; Vivancos, A.; Shomroni, O.; Zafar, S.; Schmitz, M.; et al. Regional and Subtype-Dependent MiRNA Signatures in Sporadic Creutzfeldt-Jakob Disease Are Accompanied by Alterations in MiRNA Silencing Machinery and Biogenesis. PLoS Pathog. 2018, 14, e1006802. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Dua, P.; Pogue, A.I.; Eicken, C.; Hill, J.M. Upregulation of Micro RNA-146a (MiRNA-146a), A Marker for Inflammatory Neurodegeneration, in Sporadic Creutzfeldt–Jakob Disease (SCJD) and Gerstmann–Straussler–Scheinker (GSS) Syndrome. J. Toxicol. Environ. Heal. Part A 2011, 74, 1460–1468. [Google Scholar] [CrossRef]
- Gao, C.; Wei, J.; Zhang, B.Y.; Shi, Q.; Chen, C.; Wang, J.; Shi, Q.; Dong, X.P. MiRNA Expression Profiles in the Brains of Mice Infected with Scrapie Agents 139A, ME7 and S15. Emerg. Microbes Infect. 2016, 5, e115. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Goodman, C.D.; Huzarewich, R.L.C.H.; Robertson, C.; Booth, S.A. A MiRNA Signature of Prion Induced Neurodegeneration. PLoS ONE 2008, 3, e3652. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; Medina, S.J.; Niu, Y.; Abrenica, B.; Manguiat, K.J.; Frost, K.L.; Philipson, C.S.; Sorensen, D.L.; Booth, S.A. Early Mechanisms of Pathobiology Are Revealed by Transcriptional Temporal Dynamics in Hippocampal CA1 Neurons of Prion Infected Mice. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Boese, A.S.; Saba, R.; Campbell, K.; Majer, A.; Medina, S.; Burton, L.; Booth, T.F.; Chong, P.; Westmacott, G.; Dutta, S.M.; et al. MicroRNA Abundance Is Altered in Synaptoneurosomes during Prion Disease. Mol. Cell. Neurosci. 2016, 71, 13–24. [Google Scholar] [CrossRef]
- Saba, R.; Gushue, S.; Huzarewich, R.L.C.H.; Manguiat, K.; Medina, S.; Robertson, C.; Booth, S.A. MicroRNA 146a (MiR-146a) Is Over-Expressed during Prion Disease and Modulates the Innate Immune Response and the Microglial Activation State. PLoS ONE 2012, 7, e30832. [Google Scholar] [CrossRef]
- Misra, U.K.; Kalita, J. Overview: Japanese Encephalitis. Prog. Neurobiol. 2010, 91, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.L.; Hills, S.L.; Fischer, M.; Jacobson, J.A.; Hoke, C.H.; Hombach, J.M.; Marfin, A.A.; Solomon, T.; Tsai, T.F.; Tsu, V.D.; et al. Estimated Global Incidence of Japanese Encephalitis: A Systematic Review. Bull. World Health Organ. 2011, 89, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Lannes, N.; Summerfield, A.; Filgueira, L. Regulation of Inflammation in Japanese Encephalitis. J. Neuroinflamm. 2017, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Unni, S.K.; Růžek, D.; Chhatbar, C.; Mishra, R.; Johri, M.K.; Singh, S.K. Japanese Encephalitis Virus: From Genome to Infectome. Microbes Infect. 2011, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Thounaojam, M.C.; Kundu, K.; Kaushik, D.K.; Swaroop, S.; Mahadevan, A.; Shankar, S.K.; Basu, A. MicroRNA 155 Regulates Japanese Encephalitis Virus-Induced Inflammatory Response by Targeting Src Homology 2-Containing Inositol Phosphatase 1. J. Virol. 2014, 88, 4798–4810. [Google Scholar] [CrossRef] [PubMed]
- Pareek, S.; Roy, S.; Kumari, B.; Jain, P.; Banerjee, A.; Vrati, S. MiR-155 Induction in Microglial Cells Suppresses Japanese Encephalitis Virus Replication and Negatively Modulates Innate Immune Responses. J. Neuroinflamm. 2014, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Verma, R.; Kumawat, K.; Basu, A.; Singh, S.K. MiR-146a Suppresses Cellular Immune Response during Japanese Encephalitis Virus JaOArS982 Strain Infection in Human Microglial Cells. J. Neuroinflamm. 2015, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Du, G.; Zhao, J.; Du, X. MiR-146a Negatively Regulates the Induction of Proinflammatory Cytokines in Response to Japanese Encephalitis Virus Infection in Microglial Cells. Arch. Virol. 2017, 162, 1495–1505. [Google Scholar] [CrossRef]
- Smith, J.L.; Jeng, S.; McWeeney, S.K.; Hirsch, A.J. A MicroRNA Screen Identifies the Wnt Signaling Pathway as a Regulator of the Interferon Response during Flavivirus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Kumawat, K.L.; Rastogi, M.; Basu, A.; Singh, S.K. Japanese Encephalitis Virus Exploits the MicroRNA-432 to Regulate the Expression of Suppressor of Cytokine Signaling (SOCS) 5. Sci. Rep. 2016, 6, 27685. [Google Scholar] [CrossRef] [PubMed]
- Hazra, B.; Kumawat, K.L.; Basu, A. The Host MicroRNA MiR-301a Blocks the IRF1-Mediated Neuronal Innate Immune Response to Japanese Encephalitis Virus Infection. Sci. Signal. 2017, 10, eaaf5185. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Kaushik, D.K.; Kundu, K.; Basu, A. MicroRNA-29b Modulates Japanese Encephalitis Virus-Induced Microglia Activation by Targeting Tumor Necrosis Factor Alpha-Induced Protein 3. J. Neurochem. 2014, 129, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Ashraf, U.; Ye, J.; Duan, X.; Zohaib, A.; Wang, W.; Chen, Z.; Zhu, B.; Li, Y.; Chen, H.; et al. MicroRNA-22 Negatively Regulates Poly(I:C)-Triggered Type I Interferon and Inflammatory Cytokine Production via Targeting Mitochondrial Antiviral Signaling Protein (MAVS). Oncotarget 2016, 7, 76667–76683. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Li, W.; Cheng, P.; Nie, S. MiR-370 Mimic Inhibits Replication of Japanese Encephalitis Virus in Glioblastoma Cells. Neuropsychiatr. Dis. Treat. 2016, 12, 2411–2417. [Google Scholar] [CrossRef]
- Ashraf, U.; Zhu, B.; Ye, J.; Wan, S.; Nie, Y.; Chen, Z.; Cui, M.; Wang, C.; Duan, X.; Zhang, H.; et al. MicroRNA-19b-3p Modulates Japanese Encephalitis Virus-Mediated Inflammation via Targeting RNF11. J. Virol. 2016, 90, 4780–4795. [Google Scholar] [CrossRef]
- Zhu, B.; Ye, J.; Nie, Y.; Ashraf, U.; Zohaib, A.; Duan, X.; Fu, Z.F.; Song, Y.; Chen, H.; Cao, S. MicroRNA-15b Modulates Japanese Encephalitis Virus−Mediated Inflammation via Targeting RNF125. J. Immunol. 2015, 195, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Rechenchoski, D.Z.; Faccin-Galhardi, L.C.; Linhares, R.E.C.; Nozawa, C. Herpesvirus: An Underestimated Virus. Folia Microbiol. 2017, 62, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Wilson, A.C. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J. Virol. 2017, 91, 1–6. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef]
- Rabinstein, A.A. Herpes Virus Encephalitis in Adults. Neurol. Clin. 2017, 35, 695–705. [Google Scholar] [CrossRef]
- Mancini, M.; Vidal, S.M. Insights into the Pathogenesis of Herpes Simplex Encephalitis from Mouse Models. Mamm. Genome 2018, 29, 425–445. [Google Scholar] [CrossRef]
- Sun, L.; Li, Q. The MiRNAs of Herpes Simplex Virus (HSV). Virol. Sin. 2012, 27, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Flores, O.; Umbach, J.L.; Pesola, J.M.; Bentley, P.; Rosato, P.C.; Leib, D.A.; Cullen, B.R.; Coen, D.M. A Neuron-Specific Host MicroRNA Targets Herpes Simplex Virus-1 ICP0 Expression and Promotes Latency. Cell Host Microbe 2014, 15, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 Infection of Human Brain Cells Induces MiRNA-146a and Alzheimer-Type Inflammatory Signaling. Neuroreport 2009, 20, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Bhela, S.; Mulik, S.; Gimenez, F.; Reddy, P.B.J.; Richardson, R.L.; Varanasi, S.K.; Jaggi, U.; Xu, J.; Lu, P.Y.; Rouse, B.T. Role of MiR-155 in the Pathogenesis of Herpetic Stromal Keratitis. Am. J. Pathol. 2015, 185, 1073–1084. [Google Scholar] [CrossRef]
- Majer, A.; Caligiuri, K.A.; Gale, K.K.; Niu, Y.; Phillipson, C.S.; Booth, T.F.; Booth, S.A. Induction of Multiple MiR-200/182 Members in the Brains of Mice Are Associated with Acute Herpes Simplex Virus 1 Encephalitis. PLoS ONE 2017, 12, e0169081. [Google Scholar] [CrossRef]
- Bhela, S.; Reddy, P.B.J.; Richardson, R.L.; Rajasagi, N.K.; Osmand, A.P.; Rouse, B.T.; Veiga-Parga, T.; Gimenez, F.; Mulik, S. Critical Role of MicroRNA-155 in Herpes Simplex Encephalitis. J. Immunol. 2014, 192, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.; Sun, H.; Fan, H.; Wang, C.; Li, Y.; Liu, M.; Tang, H. MiR-23a Facilitates the Replication of HSV-1 through the Suppression of Interferon Regulatory Factor 1. PLoS ONE 2014, 9, e114021. [Google Scholar] [CrossRef]
- Xie, Y.; He, S.; Wang, J. MicroRNA-373 Facilitates HSV-1 Replication through Suppression of Type I IFN Response by Targeting IRF1. Biomed. Pharmacother. 2018, 97, 1409–1416. [Google Scholar] [CrossRef]
- Meschia, J.F.; Brott, T. Ischaemic Stroke. Eur. J. Neurol. 2018, 25, 35–40. [Google Scholar] [CrossRef]
- Patel, R.A.G.; McMullen, P.W. Neuroprotection in the Treatment of Acute Ischemic Stroke. Prog. Cardiovasc. Dis. 2017, 59, 542–548. [Google Scholar] [CrossRef]
- Puig, B.; Brenna, S.; Magnus, T. Molecular Communication of a Dying Neuron in Stroke. Int. J. Mol. Sci. 2018, 19, 2834. [Google Scholar] [CrossRef]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of Inflammation and Its Mediators in Acute Ischemic Stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef]
- Li, G.; Morris-Blanco, K.C.; Lopez, M.S.; Yang, T.; Zhao, H.; Vemuganti, R.; Luo, Y. Impact of MicroRNAs on Ischemic Stroke: From Pre- to Post-Disease. Prog. Neurobiol. 2018, 163–164, 59–78. [Google Scholar] [CrossRef]
- Huang, L.; Ma, Q.; Li, Y.; Li, B.; Zhang, L. Inhibition of MicroRNA-210 Suppresses pro-Inflammatory Response and Reduces Acute Brain Injury of Ischemic Stroke in Mice. Exp. Neurol. 2018, 300, 41–50. [Google Scholar] [CrossRef]
- Ouyang, Y.B.; Lu, Y.; Yue, S.; Xu, L.J.; Xiong, X.X.; White, R.E.; Sun, X.; Giffard, R.G. MiR-181 Regulates GRP78 and Influences Outcome from Cerebral Ischemia in Vitro and in Vivo. Neurobiol. Dis. 2012, 45, 555–563. [Google Scholar] [CrossRef]
- Ma, Q.; Zhao, H.; Tao, Z.; Wang, R.; Liu, P.; Han, Z.; Ma, S.; Luo, Y.; Jia, J. MicroRNA-181c Exacerbates Brain Injury in Acute Ischemic Stroke. Aging Dis. 2016, 7, 705–714. [Google Scholar] [CrossRef]
- Xu, L.-J.; Ouyang, Y.-B.; Xiong, X.; Stary, C.M.; Giffard, R.G. Post-Stroke Treatment with MiR-181 Antagomir Reduces Injury and Improves Long-Term Behavioral Recovery in Mice after Focal Cerebral Ischemia. Exp. Neurol. 2015, 264, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Z.; Tian, Y.; Ander, B.P.; Xu, H.; Stamova, B.S.; Zhan, X.; Turner, R.J.; Jickling, G.; Sharp, F.R. Brain and Blood MicroRNA Expression Profiling of Ischemic Stroke, Intracerebral Hemorrhage, and Kainate Seizures. J. Cereb. Blood Flow Metab. 2010, 30, 92–101. [Google Scholar] [CrossRef]
- Caballero-Garrido, E.; Pena-Philippides, J.C.; Lordkipanidze, T.; Bragin, D.; Yang, Y.; Erhardt, E.B.; Roitbak, T. In Vivo Inhibition of Mir-155 Promotes Recovery after Experimental Mouse Stroke. J. Neurosci. 2015, 35, 12446–12464. [Google Scholar] [CrossRef] [PubMed]
- Pena-Philippides, J.C.; Caballero-Garrido, E.; Lordkipanidze, T.; Roitbak, T. In Vivo Inhibition of MiR-155 Significantly Alters Post-Stroke Inflammatory Response. J. Neuroinflamm. 2016, 13, 287. [Google Scholar] [CrossRef]
- Hamzei Taj, S.; Kho, W.; Aswendt, M.; Collmann, F.M.; Green, C.; Adamczak, J.; Tennstaedt, A.; Hoehn, M. Dynamic Modulation of Microglia/Macrophage Polarization by MiR-124 after Focal Cerebral Ischemia. J. Neuroimmune Pharmacol. 2016, 11, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Hamzei Taj, S.; Kho, W.; Riou, A.; Wiedermann, D.; Hoehn, M. MiRNA-124 Induces Neuroprotection and Functional Improvement after Focal Cerebral Ischemia. Biomaterials 2016, 91, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Gao, L.; Wang, R.; Liu, X.; Gao, Z.; Tao, Z.; Xu, C.; Song, J.; Ji, X.; et al. MiRNA-424 Protects against Permanent Focal Cerebral Ischemia Injury in Mice Involving Suppressing Microglia Activation. Stroke 2013, 44, 1706–1713. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, H.; Wang, R.; Wang, P.; Tao, Z.; Gao, L.; Yan, F.; Liu, X.; Yu, S.; Ji, X.; et al. MicroRNA-424 Protects against Focal Cerebral Ischemia and Reperfusion Injury in Mice by Suppressing Oxidative Stress. Stroke 2015, 46, 513–519. [Google Scholar] [CrossRef]
- Yu, H.; Wu, M.; Zhao, P.; Huang, Y.; Wang, W.; Yin, W. Neuroprotective Effects of Viral Overexpression of MicroRNA-22 in Rat and Cell Models of Cerebral Ischemia-Reperfusion Injury. J. Cell. Biochem. 2015, 116, 233–241. [Google Scholar] [CrossRef]
- Corrigan, J.D.; Hammond, F.M. Traumatic Brain Injury as a Chronic Health Condition. Arch. Phys. Med. Rehabil. 2013, 94, 1199–1201. [Google Scholar] [CrossRef]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and Severe Traumatic Brain Injury in Adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2014, 32, 1834–1848. [Google Scholar] [CrossRef] [PubMed]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef]
- Ge, X.; Han, Z.; Chen, F.; Wang, H.; Zhang, B.; Jiang, R.; Lei, P.; Zhang, J. MiR-21 Alleviates Secondary Blood–Brain Barrier Damage after Traumatic Brain Injury in Rats. Brain Res. 2015, 1603, 150–157. [Google Scholar] [CrossRef]
- Harrison, E.B.; Hochfelder, C.G.; Lamberty, B.G.; Meays, B.M.; Morsey, B.M.; Kelso, M.L.; Fox, H.S.; Yelamanchili, S.V. Traumatic Brain Injury Increases Levels of MiR-21 in Extracellular Vesicles: Implications for Neuroinflammation. FEBS Open Bio. 2016, 6, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.-T.; Lei, P.; Wang, H.-C.; Zhang, A.-L.; Han, Z.-L.; Chen, X.; Li, S.-H.; Jiang, R.-C.; Kang, C.-S.; Zhang, J.-N. MiR-21 Improves the Neurological Outcome after Traumatic Brain Injury in Rats. Sci. Rep. 2015, 4, 6718. [Google Scholar] [CrossRef] [PubMed]
- Redell, J.B.; Liu, Y.; Dash, P.K. Traumatic Brain Injury Alters Expression of Hippocampal MicroRNAs: Potential Regulators of Multiple Pathophysiological Processes. J. Neurosci. Res. 2009, 87, 1435–1448. [Google Scholar] [CrossRef]
- Wang, W.X.; Huang, Q.; Hu, Y.; Stromberg, A.J.; Nelson, P.T. Patterns of MicroRNA Expression in Normal and Early Alzheimer’s Disease Human Temporal Cortex: White Matter versus Gray Matter. Acta Neuropathol. 2011, 121, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.X.; Wilfred, B.R.; Madathil, S.K.; Tang, G.; Hu, Y.; Dimayuga, J.; Stromberg, A.J.; Huang, Q.; Saatman, K.E.; Nelson, P.T. MiR-107 Regulates Granulin/Progranulin with Implications for Traumatic Brain Injury and Neurodegenerative Disease. Am. J. Pathol. 2010, 177, 334–345. [Google Scholar] [CrossRef]
- Cenik, B.; Sephton, C.F.; Cenik, B.K.; Herz, J.; Yu, G. Progranulin: A Proteolytically Processed Protein at the Crossroads of Inflammation and Neurodegeneration. J. Biol. Chem. 2012, 287, 32298–32306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, F.; Pan, C.; Lou, Y.; Zhang, P.; Guo, S.; Yin, J.; Deng, Z. Effects of Low Temperatures on Proliferation-Related Signaling Pathways in the Hippocampus after Traumatic Brain Injury. Exp. Biol. Med. 2012, 237, 1424–1432. [Google Scholar] [CrossRef]
- Truettner, J.S.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D. Therapeutic Hypothermia Alters MicroRNA Responses to Traumatic Brain Injury in Rats. J. Cereb. Blood Flow Metab. 2011, 31, 1897–1907. [Google Scholar] [CrossRef]
- Truettner, J.S.; Motti, D.; Dietrich, W.D. MicroRNA Overexpression Increases Cortical Neuronal Vulnerability to Injury. Brain Res. 2013, 1533, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-X.; Visavadiya, N.P.; Pandya, J.D.; Nelson, P.T.; Sullivan, P.G.; Springer, J.E. Mitochondria-Associated MicroRNAs in Rat Hippocampus Following Traumatic Brain Injury. Exp. Neurol. 2015, 265, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J.; Doran, S.J.; Barrett, J.P.; Meadows, V.E.; Sabirzhanov, B.; Stoica, B.A.; Loane, D.J.; Faden, A.I. Inhibition of MiR-155 Limits Neuroinflammation and Improves Functional Recovery After Experimental Traumatic Brain Injury in Mice. Neurotherapeutics 2019, 16, 216–230. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Guo, J.-S.; Li, S.-S.; Wu, X.-B.; Cao, D.-L.; Jiang, B.-C.; Jing, P.-B.; Bai, X.-Q.; Li, C.-H.; Wu, Z.-H.; et al. TLR8 and Its Endogenous Ligand MiR-21 Contribute to Neuropathic Pain in Murine DRG. J. Exp. Med. 2018, 215, 3019–3037. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.; Soares-dos-Reis, R.; Melo, P.N.; Relvas, J.B.; Guimarães, J.; Sá, M.J.; Cruz, A.P.; Mendes Pinto, I. Emerging Biosensing Technologies for Neuroinflammatory and Neurodegenerative Disease Diagnostics. Front. Mol. Neurosci. 2018, 11, 164. [Google Scholar] [CrossRef]
- Morand, P.; Lecuit, M.; Piroth, L.; Crabol, Y.; Honnorat, J.; Stahl, J.P.; Fillatre, P. Infectious Encephalitis: Management without Etiological Diagnosis 48 Hours after Onset. Méd. Mal. Infect. 2017, 47, 236–251. [Google Scholar] [CrossRef]
- Cook, G.A.; Hawley, J.S. A Review of Mild Traumatic Brain Injury Diagnostics: Current Perspectives, Limitations, and Emerging Technology. Mil. Med. 2014, 179, 1083–1089. [Google Scholar] [CrossRef]
- Backes, C.; Meese, E.; Keller, A. Specific MiRNA Disease Biomarkers in Blood, Serum and Plasma: Challenges and Prospects. Mol. Diagn. Ther. 2016, 20, 509–518. [Google Scholar] [CrossRef]
- Gandhi, R. MiRNA in Multiple Sclerosis: Search for Novel Biomarkers. Mult. Scler. J. 2015, 21, 1095–1103. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Shaik, M.M.; Tamargo, I.A.; Kamal, M.A. MiRNAs as Circulating Biomarkers for Alzheimer’s Disease and Parkinson’s Disease. Med. Chem. 2016, 12, 217–225. [Google Scholar] [CrossRef]
- Kanata, E.; Thüne, K.; Xanthopoulos, K.; Ferrer, I.; Dafou, D.; Zerr, I.; Sklaviadis, T.; Llorens, F. MicroRNA Alterations in the Brain and Body Fluids of Humans and Animal Prion Disease Models: Current Status and Perspectives. Front. Aging Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Banerjee, A.; Kumari, B.; Bandopadhyay, B.; Bhattacharya, N.; Basu, N.; Vrati, S.; Banerjee, A. Differential Expression and Significance of Circulating MicroRNAs in Cerebrospinal Fluid of Acute Encephalitis Patients Infected with Japanese Encephalitis Virus. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dewdney, B.; Trollope, A.; Moxon, J.; Thomas Manapurathe, D.; Biros, E.; Golledge, J. Circulating MicroRNAs as Biomarkers for Acute Ischemic Stroke: A Systematic Review. J. Stroke Cerebrovasc. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P. MicroRNAs as Diagnostic Markers and Therapeutic Targets for Traumatic Brain Injury. Neural Regen. Res. 2017, 12, 1749. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Sen, S. MicroRNA as Biomarkers and Diagnostics. J. Cell. Physiol. 2016, 231, 25–30. [Google Scholar] [CrossRef]
- Burgos, K.; Malenica, I.; Metpally, R.; Courtright, A.; Rakela, B.; Beach, T.; Shill, H.; Adler, C.; Sabbagh, M.; Villa, S.; et al. Profiles of Extracellular MiRNA in Cerebrospinal Fluid and Serum from Patients with Alzheimer’s and Parkinson’s Diseases Correlate with Disease Status and Features of Pathology. PLoS ONE 2014, 9, e94839. [Google Scholar] [CrossRef]
- Pereira, P.; Queiroz, J.A.; Figueiras, A.; Sousa, F. Current Progress on MicroRNAs-Based Therapeutics in Neurodegenerative Diseases. Wiley Interdiscip. Rev. RNA 2017, 8, e1409. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Van Rooij, E.; Kauppinen, S. Development of MicroRNA Therapeutics Is Coming of Age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. Therapeutic Targeting of MicroRNAs: Current Status and Future Challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J. Progress on RNAi-Based Molecular Medicines. Int. J. Nanomed. 2012, 7, 3971. [Google Scholar] [CrossRef][Green Version]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Aryal, M.; Arvanitis, C.D.; Alexander, P.M.; McDannold, N. Ultrasound-Mediated Blood–Brain Barrier Disruption for Targeted Drug Delivery in the Central Nervous System. Adv. Drug Deliv. Rev. 2014, 72, 94–109. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Hudry, E.; Maguire, C.A.; Sena-Esteves, M.; Breakefield, X.O.; Grandi, P. Viral Vectors for Therapy of Neurologic Diseases. Neuropharmacology 2017, 120, 63–80. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| miRNA | Disorder | Expression | Consequences | Reference |
|---|---|---|---|---|
| miR-155 | MS | Up | Increased activation of microglia, enhancement of phagocytosis and increased BBB permeability | [80,86] |
| AD | Up | Increased inflammation | [102] | |
| PD | Up | Microglial inflammatory response | [113] | |
| JEV | Up | Microglial activation and suppression of viral replication and innate immune signaling | [130,131] | |
| HSVE | Up | Protective inflammatory response and decreases viral replication | [150,151,152] | |
| Stroke | Down | Decreased inflammation and tissue damage | [164,165,166] | |
| TBI | Up | Increased inflammation and tissue damage | [186,187] | |
| miR-146a | MS | Up | Complex, influences inflammation and demyelination/remyelination | [80,83,84] |
| AD | Up | Attenuates inflammatory signaling | [43,103] | |
| PrD | Up | Dampens microglial inflammatory response | [125] | |
| JEV | Up | Decreased inflammation and ISG secretion, promotes JEV replication | [132,133] | |
| HSVE | Up | Decreased inflammation, promotes viral replication | [149,151] | |
| miR-124 | MS | Down | Release from microglial quiescence | [51] |
| PD | - | Attenuates inflammatory signaling in microglia | [114] | |
| PrD | Up then down | Release from microglial quiescence | [119,123] | |
| Stroke | - | M2 microglial polarization, decreased inflammation, protective effect | [167,168] | |
| miR-21 | MS | Up | CNS specific function unknown | [80] |
| TBI | Up | Increased BBB repair and angiogenesis, impaired apoptosis and inflammation | [176,177,178] | |
| Let-7 | AD | Up | Acts as a DAMP for TLR-7 | [72] |
| miR-181 | AD | Up | Neuronal dysfunction | [105] |
| Stroke | Up | Promotes neuronal death and inflammatory signaling | [161,162,163] | |
| miR-34 | MS | Up | Enhanced macrophage phagocytosis | [80] |
| AD | Up | Impaired Aβ42 clearance by microglia | [99,100] | |
| PD | Down | Increased α-synuclein expression and increased inflammation | [112] | |
| JEV | - | Induces type I interferon signaling, decreases viral replication | [134] | |
| TBI | - | Promotes release of pro-inflammatory and pro-apoptotic factors | [185] |
| Disorder | Plasma/Serum | Whole Blood | PBMCs | CSF | Reference |
|---|---|---|---|---|---|
| MS | let-7 | miR-146b | miR-146a, miR-155 | miR-181c | [193] |
| AD | let-7, miR-34, miR-181c, miR-21 | let-7, miR-103a, miR-107 | miR-34a, miR-181b | miR-146a, miR-155, miR-34a, miR-124, miR-181a | [194,200] |
| PD | miR-181c | - | - | let-7 | [194,200] |
| PrD | miR-21 | - | - | - | [195] |
| JEV | - | - | - | miR-21, let-7, miR-181a | [196] |
| Stroke | - | let-7, miR-21 | - | - | [197] |
| TBI | miR-21 | - | - | miR-181 | [198] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slota, J.A.; Booth, S.A. MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Non-Coding RNA 2019, 5, 35. https://doi.org/10.3390/ncrna5020035
Slota JA, Booth SA. MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Non-Coding RNA. 2019; 5(2):35. https://doi.org/10.3390/ncrna5020035
Chicago/Turabian StyleSlota, Jessy A., and Stephanie A. Booth. 2019. "MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications" Non-Coding RNA 5, no. 2: 35. https://doi.org/10.3390/ncrna5020035
APA StyleSlota, J. A., & Booth, S. A. (2019). MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Non-Coding RNA, 5(2), 35. https://doi.org/10.3390/ncrna5020035