Long Non-Coding RNAs in Multiple Myeloma

 , and
, and
Abstract
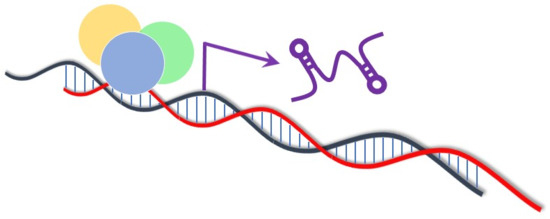
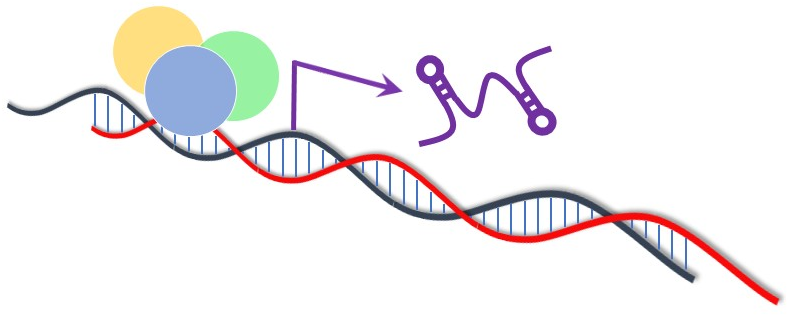
:
1. Introduction
2. Definition of Long Non-Coding RNAs
3. Comparison to Protein-Coding Genes
4. Biogenesis of Long Non-Coding RNAs
5. Localization of Long Non-Coding RNAs
6. Function of Long Non-Coding RNAs
7. Involvement of Long Non-Coding RNAs at the Onset of Various Pathologies
8. Long Non-Coding RNAs in Multiple Myeloma
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, 538–548. [Google Scholar] [CrossRef]
- Faiman, B. Myeloma Genetics and Genomics: Practice Implications and Future Directions. Clin. Lymphoma Myeloma Leuk. 2014, 14, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Fatima, R.; Akhade, V.S.; Pal, D.; MR Rao, S. Long noncoding RNAs in development and cancer: Potential biomarkers and therapeutic targets. Mol. Cell 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med. 2012, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, K.; Shi, Z.; Zhang, J.; Zhu, J.; Zhu, S.; Zhang, A.; Jia, Z.; Wang, G.; Yu, S.; et al. LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int. J. Oncol. 2012, 40, 2004–2012. [Google Scholar] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstien, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Lloret-Llinares, M.; Mapendano, C.K.; Martley, L.H.; Lykke-Andersen, S.; Jensen, T.H. Relationship between PROMPT and gene expression. RNA Biol. 2016, 13, 6–14. [Google Scholar] [CrossRef]
- Clark, M.B.; Johnston, R.L.; Inostroza-Pointa, M.; Fox, A.H.; Fortini, E.; Moscato, P.; Dinger, M.E.; Mattick, J.S. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012, 22, 885–898. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Ortutay, C.; Vihinen, M. PseudoGeneQuest—Service for identification of different pseudogene types in the human genome. BMC Bioinformat. 2008, 9, 299. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Ayers, D. Long non-coding RNAs: Novel emergent biomarkers for cancer diagnostics. J. Cancer Res. Treat. 2013, 1, 31–35. [Google Scholar]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature revers over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Rasmussen, M.; Pandey, G.K.; Isaksson, A.; Kanduri, C. Characterization of the RNA content of chromatin. Genome Res. 2010, 20, 899–907. [Google Scholar] [CrossRef]
- Bhartiya, D.; Pal, K.; Ghosh, S.; Kapoor, S.; Jalali, S.; Panwar, B.; Jain, S.; Sati, S.; Sengupta, S.; Sachidanandan, C.; et al. LncRNome: A comprehensive knowledgebase of human long noncoding RNAs. Database 2013, 2013. [Google Scholar] [CrossRef]
- Fang, Y.; Fullwood, M.J. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef]
- Wilusz, J.E. Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim. Biophys. Acta 2016, 1859, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Trelles, F.; Tarrío, R.; Ayala, F.J. Origins and evolution of spliceosomal introns. Annu. Rev. Genet. 2006, 40, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long noncoding RNAs with snoRNA ends. Mol. Cell 2012, 48, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, 30733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Proudfoot, N.J.; Jopling, C.L. Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat. Struct. Mol Biol. 2015, 22, 319–327. [Google Scholar] [CrossRef]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef]
- Pontier, D.B.; Gribnau, J. Xist regulation and function explored. Hum. Genet. 2011, 130, 223–236. [Google Scholar] [CrossRef]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.Y.; Nakagawa, S. Long non-coding RNAs in nuclear bodies. Dev. Growth Differ. 2012, 54, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest-and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Cabili, M.N.; Rinn, J.; Raj, A. Linc-HOXA1 is noncoding RNA that repress Hoxa1 transcription in cis. Genes Dev. 2013, 27, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Ge, X.; Yang, S.; Huang, M.; Zhuang, W.; Chen, P.; Zhang, X.; Fu, J.; Qu, J.; Li, B. Upregulation of lncRNA MEG3 Promotes Osteogenic Differentiation of Mesenchymal Stem Cells from Multiple Myeloma Patients by Targeting BMP4 Transcription. Stem Cells 2015, 33, 1985–1997. [Google Scholar] [CrossRef]
- Ronchetti, D.; Agnelli, L.; Taiana, E.; Galletti, S.; Manzoni, M.; Todoerti, K.; Musto, P.; Strozzi, F.; Neri, A. Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget 2016, 7, 14814–14830. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Mohammad, F.; Mondal, T.; Kanduri, C. Epigenetics of imprinted long noncoding RNAs. Epigenetics 2009, 4, 277–286. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and IGF1R. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Rico, D.; Herrera, L.A. Regulated expression of the lncRNA TERRA and its impact on telomere biology. Mech. Ageing Dev. 2017, 167, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Carracedo, A.; Salmena, L.; Song, S.J.; Egia, A.; Malumbres, M.; Pandolfi, P.P. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011, 144, 187–199. [Google Scholar] [CrossRef]
- Kumar, M.M.; Goyal, R. LncRNAs as Therapeutic Target for Angiogenesis. Curr. Top. Med. Chem. 2017, 17, 1750–1757. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Brockdorff, N.; Kawano, S.; Tsutui, K.; Tsutui, K.; Nakagawa, S. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell 2010, 19, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, X.; Wang, Y.; Zhao, L.; Chen, W. Long non-coding RNA UCA1 regulated cell cycle distribution via CREB through PI3-K dependent pathway in bladder carcinoma cells. Gene 2012, 496, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef]
- Smola, M.J.; Christy, T.W.; Inoue, K.; Nicholson, C.O.; Friedersdorf, M.; Keene, D.J.; Lee, D.M.; Calabrese, J.M.; Wekks, K.M. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci. USA 2016, 113, 10322–10327. [Google Scholar] [CrossRef]
- Xue, Z.; Hennelly, S.; Doyle, B.; Gulati, A.A.; Novikova, I.V.; Sanbonmatsu, K.Y.; Boyer, L.A. A G-Rich Motif in the lncRNA Braveheart Interacts with a Zinc-Finger Transcription Factor to Specify the Cardiovascular Lineage. Mol. Cell 2016, 64, 37–50. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.T.; Hu, N.; Tan, L. Non-coding RNAs in Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R. Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol. Dis. 2012, 46, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Harvey, R.P.; Mattick, J.S. Long noncoding RNAs in cardiac development and pathophysiology. Circ. Res. 2012, 111, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Sui, G. Noncoding RNA in oncogenesis: A new era of identifying key players. Int. J. Mol. Sci. 2013, 14, 18319–18349. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Di Gesualdo, F.; Capaccioli, S.; Lulli, M. A pathophysiological view of the long non-coding RNA world. Oncotarget 2014, 5, 10976. [Google Scholar] [CrossRef]
- Ling, H.; Vincent, K.; Pichler, M.; Fodde, R.; Berindan-Neagoe, I.; Slack, F.J.; Calin, G.A. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015, 34, 5003–5011. [Google Scholar] [CrossRef]
- Noboli, C.; Lionetti, M.; Neri, A. Long non-coding RNAs in normal and malignant hematopoiesis. Oncotarget 2016, 7, 50666–50681. [Google Scholar] [CrossRef]
- Nobili, L.; Ronchetti, D.; Taiana, E.; Neri, A. Long non-coding RNAs in B-cell malignancies: A comprehensive overview. Oncotarget 2017, 8, 60605–60623. [Google Scholar] [CrossRef]
- Maluskova, D.; Svobodova, I.; Kucerova, M.; Brozova, L.; Muzik, J.; Jarkovsky, J.; Hajek, R.; Maisnar, V.; Dusek, L. Epidemiology of Multiple Myeloma in the Czech Republic. Klin Onkol. 2017, 30 (Suppl. 2), 35–42. [Google Scholar] [CrossRef]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.D.; Wall, C.; Patel, A.; Lim, J. Multiple myeloma: A review. Orthop. Trauma 2014, 28, 187–193. [Google Scholar] [CrossRef]
- Nagoshi, H.; Taki, T.; Hanamura, I.; Nitta, M.; Otsuki, T.; Nishida, K.; Okuda, K.; Sakamoto, N.; Kobayashi, S.; Yamamoto-Sugitani, M.; et al. Frequent PVT1 rearrangement and novel chimeric genes PVT1-NBEA and PVT1-WWOX occur in multiple myeloma with 8q24 abnormality. Cancer Res. 2012, 72, 4954–4962. [Google Scholar] [CrossRef] [PubMed]
- Pour, L.; Ševčíková, S.; Greslíková, H.; Kupska, R.; Majkova, P.; Zahradova, L.; Sandecka, V.; Adam, Z.; Krejci, M.; Kuglik, P.; et al. Soft-tissue extramedullary multiple myeloma prognosis is significantly worse in comparison to bone-related extramedullary relapse. Haematologica 2014, 99, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Mahindra, A.; Hideshima, T.; Anderson, K.C. Multiple myeloma: Biology of the disease. Blood Rev. 2010, 24, 5–11. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Zahid, S.; Basit Ashraf, M.A.; Qazi, M.H.; Asif, M.; Zaheer, A.; Arshad, M.; Raza, A.; Jamal, M.S. Non-coding RNAs in cancer diagnosis and therapy. Non-Coding RNA Res. 2016, 1, 69–76. [Google Scholar] [CrossRef]
- Gutierréz, N.C.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; De Las Rivas, J.; Ticona, F.V.; Ferminan, E.; Martin-Jimenez, P.; Chillon, C.; Riueuno, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef]
- Lionetti, M.; Biasiolo, M.; Angelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, 20–26. [Google Scholar] [CrossRef]
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Sedlarikova, L.; Gromesová, B.; Kubaczkova, V.; Radova, L.; Filipova, J.; Jarkovsky, J.; Brozova, L.; Velichova, R.; Almasi, M.; Penka, M.; et al. Deregulated expression of long non-coding RNA UCA1 in multiple myeloma. Eur. J. Haematol. 2017, 99, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Ronchetti, D.; Favasuli, V.; Todoerti, K.; Manzoni, M.; Amodio, N.; Tassone, P.; Agnelli, L.; Neri, A. Long non-coding RNA NEAT1 shows high expression unrelated to molecular features and clinical outcome in multiple myeloma. Haematologica 2018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leuk 2018, 32, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Minvielle, S.; Gulla, A.; Fulciniti, M.; Cleynen, A.; Aktus Samur, A.; Szalat, R.; Shammas, M.; Magrangeas, F.; Tai, Y.T.; et al. Long intergenic non-coding RNAs have an independent impact on survival in multiple myeloma. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT 1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef]
- Amodio, N.; Stamato, M.A.; Gulla, A.M.; Morelli, E.; Romeo, E.; Raimondi, L.; Pitari, M.R.; Ferrandino, I.; Misso, G.; Caraglia, M.; et al. Therapeutic Targeting of miR-29b/HDAC4 Epigenetic Loop in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1364–1375. [Google Scholar] [CrossRef]
- Stamato, M.A.; Juli, G.; Romeo, E.; Ronchetti, D.; Arbitrio, M.; Caracciolo, D.; Neri, A.; Tagliaferri, P.; Tassone, P.; Amodio, N. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget 2017, 8, 106527–106537. [Google Scholar] [CrossRef]
- Calura, E.; Bisognin, A.; Manzoni, M.; Todoerti, K.; Taiana, E.; Sales, G.; Morgan, G.J.; Tonon, G.; Amodio, N.; Tassone, P.; et al. Disentangling the microRNA regulatory milieu in multiple myeloma: Integrative genomics analysis outlines mixed miRNA-TF circuits and pathway-derived networks modulated in t(4;14) patients. Oncotarget 2016, 7, 2367–2378. [Google Scholar] [CrossRef]
- Shen, X.; Bai, H.; Zhu, H.; Yan, Q.; Yang, Y.; Yu, W.; Shi, Q.; Wang, J.; Li, J.; Chen, L. Long Non-Coding RNA MEG3 Functions as a Competing Endogenous RNA to Regulate HOXA11 Expression by Sponging miR-181a in Multiple Myeloma. Cell Physiol. Biochem. 2018, 49, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yang, C.; Zhang, Z.; Cong, Y.; Xiao, M. Knockdown of Linc00515 Inhibits Multiple Myeloma Autophagy and Chemoresistance by Upregulating miR-140-5p and Downregulating ATG14. Cell Physiol. Biochem. 2018, 48, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, L.; Hu, N.; Zhao, H. Long non-coding RNA FEZF1-AS1 promotes cell growth in multiple myeloma via miR-610/Akt3 axis. Biomed. Pharmacother. 2018, 103, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Li, Y.; Wang, Y.; Zhao, B.; Zou, Z.; Hou, S.; Jia, X.; Liu, X.; Yao, Y.; Wan, J.; et al. LncRNA PRAL is closely related to clinical prognosis of multiple myeloma and the bortezomib sensitivity. Exp. Cell Res. 2018, 370, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hu, N.; Wang, C.; Zhao, H.; Gu, Y. Long non-coding RNA CCAT1 promotes multiple myeloma progression by acting as a molecular sponge of miR-181a-5p to modulate HOXA1 expression. Cell Cycle 2017, 17, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chen, J.; Zhang, H.; Wang, X.; Yao, H.; Peng, Y.; Zhang, W. LncRNA OIP5-AS1 loss-induced microRNA-410 accumulation regulates cell proliferation and apoptosis by targeting KLF10 via activating PTEN/PI3K/AKT pathway in multiple myeloma. Cell Death Dis. 2017, 8, 2975. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.B.; He, X.; Huang, Y.F.; Wu, Q.N.; Zhou, Y.C.; Hao, D.J. Long Noncoding RNA CRNDE Promotes Multiple Myeloma Cell Growth by Suppressing miR-451. Oncol. Res. 2017, 25, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Pan, J.; Zhang, N.; Wei, W.; Yu, S.; Ai, L. Knockdown of long non-coding RNA H19 inhibits multiple myeloma cell growth via NF-κB pathway. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, Y.; Wu, X.; Guo, Y.; Shi, W.; Qi, J.; Cong, H.; Wang, X.; Wu, X.; Ju, S. Upregulated lncRNA-PCAT1 is closely related to clinical diagnosis of multiple myeloma as a predictive biomarker in serum. Cancer Biomark. 2017, 18, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sedlarikova, L.; Bollova, B.; Radova, L.; Brozova, L.; Jarkovsky, J.; Almasi, M.; Penka, M.; Kuglik, P.; Sandecka, V.; Stork, M.; et al. Circulating exosomal long noncoding RNA PRINS-First findings in monoclonal gammopathies. Hematol. Oncol. 2018, 36, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, H.; Shen, X.; Wang, X.; Ju, S.; Lu, M.; Cong, H. Serum level of long noncoding RNA H19 as a diagnostic biomarker of multiple myeloma. Clin. Chim. Acta 2018, 480, 199–205. [Google Scholar] [CrossRef] [PubMed]
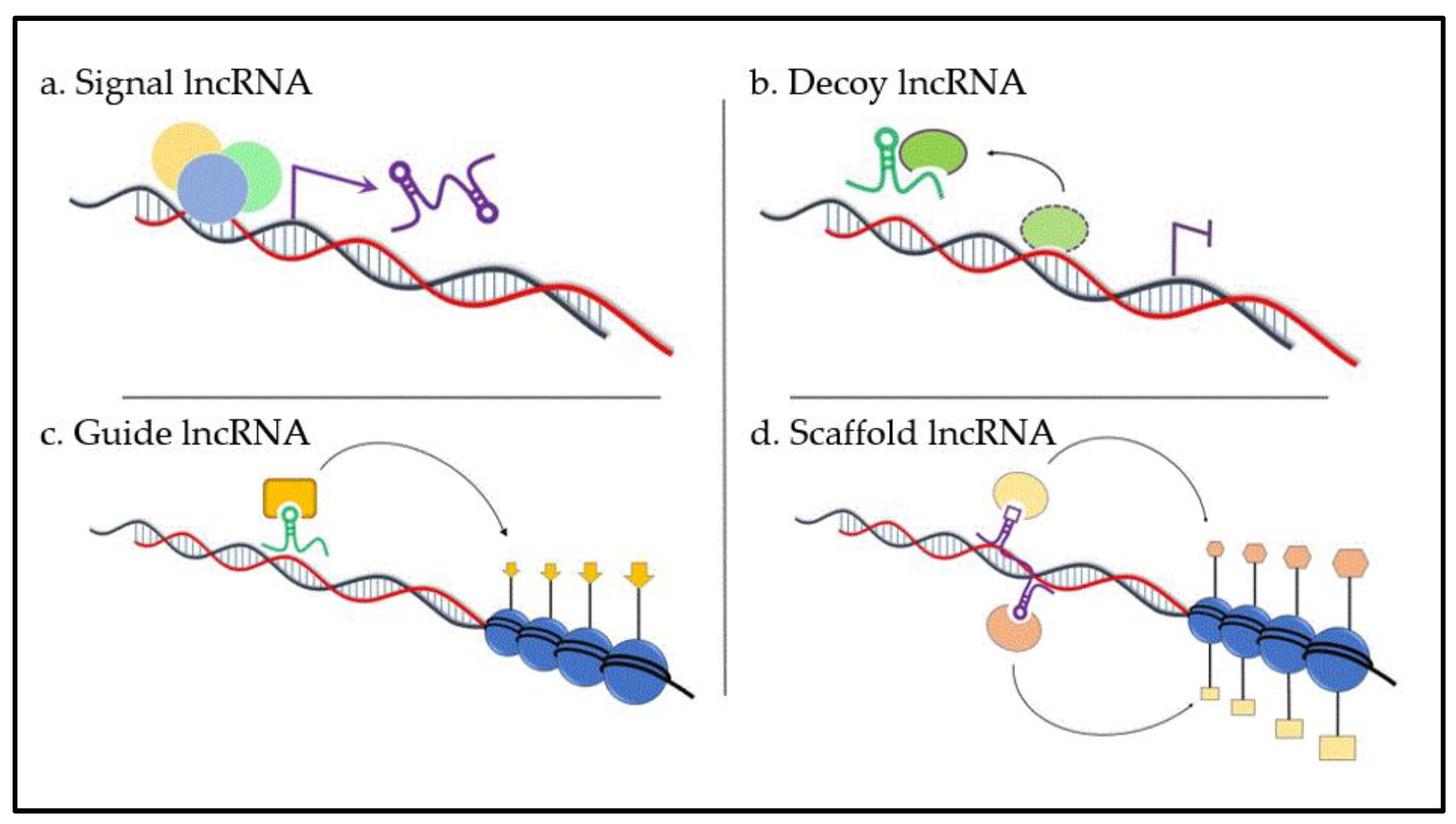

{kind=link}
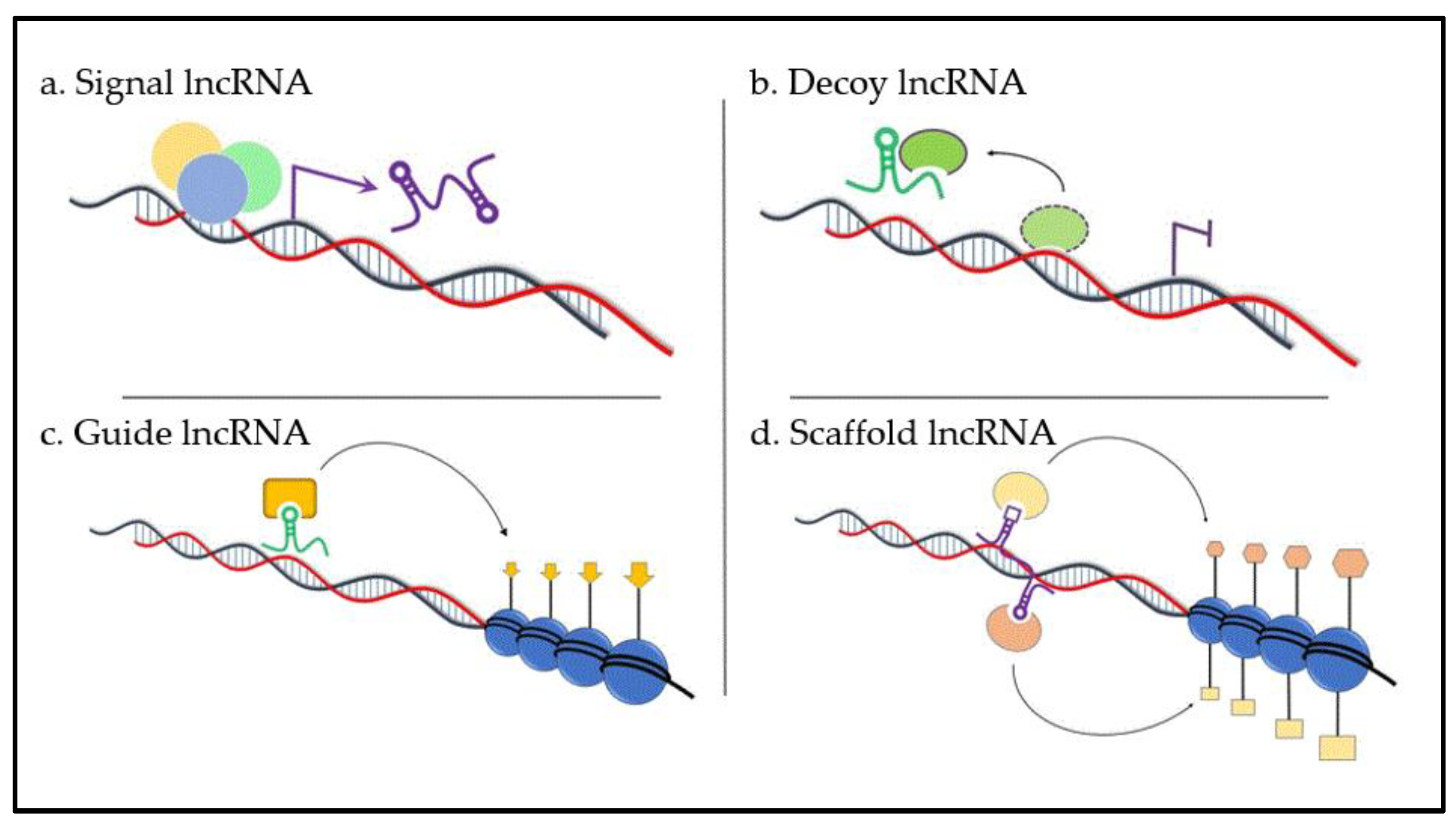{kind=link}
| Specific lncRNA | Expression in MM vs. Normal | Results of Changed Expression in MM | Effect of Reversed Expression Profile | References |
|---|---|---|---|---|
| UCA1 | ↑ | Cell cycle positive regulation via CREB regulation | [82] | |
| NEAT1 | ↑ | Increased DEX resistance | Sensitivity to DEX | [83] |
| MALAT1 | ↑ | Increased proliferation | Decreased proliferation, proteasome inhibition | [37,58,84,85,86] |
| MEG3 | ↓ | Negative effect to osteogenesis of mesenchymal stem cells (MSC) | MM–MSC differentiation | [36] |
| linc00515 | ↑ | Chemoresistance | Inhibition of myeloma autophagy | [92] |
| FEZF1-AS1 | ↑ | Increased cell proliferation | Cell inhibition, cell arrest, and apoptosis induction | [93] |
| PRAL | ↓ | Increased tumor growth | Tumor growth inhibition, promotion of apoptosis, and sensitivity to bortezomib | [87] |
| CCAT1 | ↑ | Oncogenic role via positive regulation of HOXA1 expression | Cell proliferation inhibition, promotion of apoptosis, and tumor growth suppression in vivo | [94] |
| OIP5-AS1 | ↓ | Increased cell proliferation via miR-410 accumulation | [96] | |
| CRNDE | ↑ | Increased tumor progression via negative regulation of miR-451 | Cell proliferation inhibition, promotion of apoptosis | [97] |
| H19 | ↑ | Increased proliferation through NF-κB pathway | Inhibition of proliferation and viability | [98,101] |
| TUG1 | ↑ | Promoting of proliferation, migration, and invasion | Suppression of cell proliferation, invasion, and colony formation | [57] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butova, R.; Vychytilova-Faltejskova, P.; Souckova, A.; Sevcikova, S.; Hajek, R. Long Non-Coding RNAs in Multiple Myeloma. Non-Coding RNA 2019, 5, 13. https://doi.org/10.3390/ncrna5010013
Butova R, Vychytilova-Faltejskova P, Souckova A, Sevcikova S, Hajek R. Long Non-Coding RNAs in Multiple Myeloma. Non-Coding RNA. 2019; 5(1):13. https://doi.org/10.3390/ncrna5010013
Chicago/Turabian StyleButova, Romana, Petra Vychytilova-Faltejskova, Adela Souckova, Sabina Sevcikova, and Roman Hajek. 2019. "Long Non-Coding RNAs in Multiple Myeloma" Non-Coding RNA 5, no. 1: 13. https://doi.org/10.3390/ncrna5010013
APA StyleButova, R., Vychytilova-Faltejskova, P., Souckova, A., Sevcikova, S., & Hajek, R. (2019). Long Non-Coding RNAs in Multiple Myeloma. Non-Coding RNA, 5(1), 13. https://doi.org/10.3390/ncrna5010013