Rapid Generation of Long Noncoding RNA Knockout Mice Using CRISPR/Cas9 Technology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
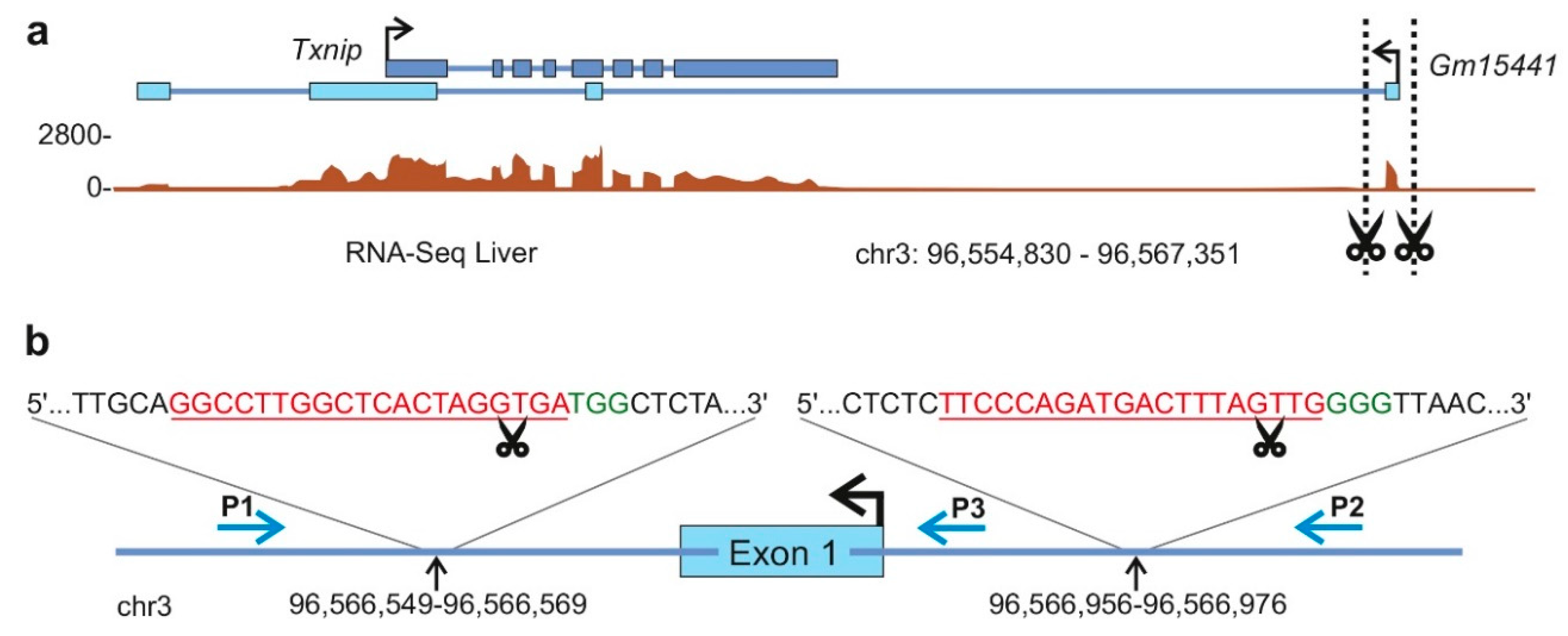
2. Results
2.1. Overview of the Protocol
2.1.1. General Considerations to Design Long Noncoding RNA Gene-Targeting Strategies
2.1.2. Long Noncoding RNA Target Selection (Step 1)
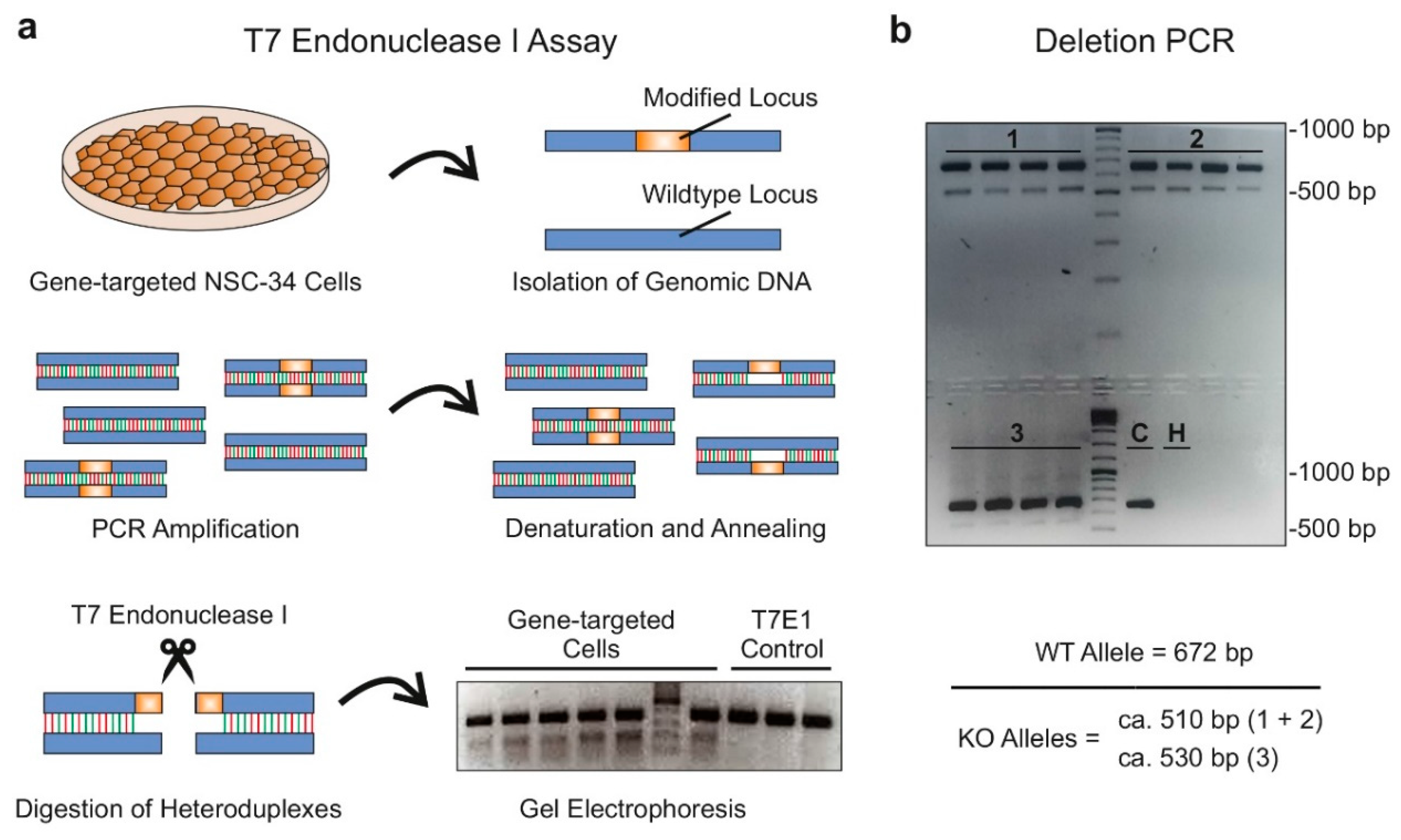
2.1.3. Validation of Single Guide RNA Activity In Vitro (Step 2):
2.1.4. Single Guide RNA–Cas9 Ribonucleoprotein Assembly and Pronuclear Microinjection (Step 3)
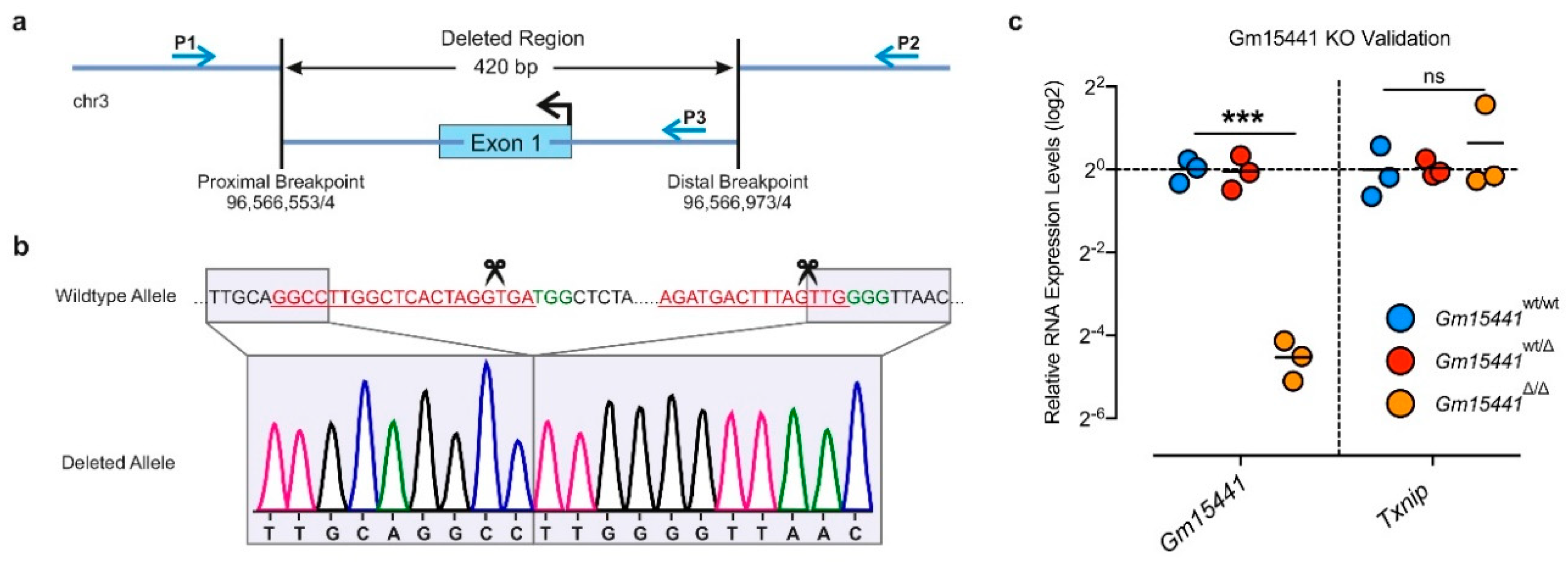
2.1.5. Identification and Validation of Long Noncoding RNA-Deficient Founder Animals (Step 4)
2.2. Experimental Design
2.2.1. Animal Use Authorization
2.2.2. Single-Guide RNA Design
2.2.3. Single-Guide RNA Synthesis
| T7 promotor | - | 5′-TTAATACGACTCACTATAGG-3′ |
| gRNA of interest | - | variable (18-20 bp of specific gRNA, see Figure S1) |
| pX330-5′tracrRNA | - | 5′-GTTTTAGAGCTAGAAATAGC-3′ |
2.2.4. In Vitro Single-Guide RNA Validation
| Primer for T7 Endonuclease I Assays | Sequence (5′->3′) | |
| Gm15441-5′-1 (20) forward | GCTCCTACTCAGACCCTTGTTC | |
| Gm15441-5′-1 (20) reverse | CTCCCTGAGTTGCTTTTGGTC | |
| Gm15441-5′-2 (20) forward | GAAGGGAGATAAAGCGCACG | |
| Gm15441-5′-2 (20) reverse | ATGGGGAGCAAGCCGATAAG | |
| Gm15441-3′-1 (20) forward | GACTAGTCTGATGGAGGCATC | |
| Gm15441-3′-1 (20) reverse | TGTGTGTGTGTGTGAGAGAGAG | |
| Gm15441-3′-2 (20) forward | TCAGCCTGCTTTCTTATATGGC | |
| Gm15441-3′-2 (20) reverse | TGCAAACACAGACATGCACAC | |
| Gm15441-5′-1 (18) forward | GCGCACGTTTAACTGACTCTC | |
| Gm15441-5′-1 (18) reverse | ATAAGCAGCACCCCTCCATG | |
| Gm15441-5′-2 (18) forward | CACAGAAGGGAGATAAAGCGC | |
| Gm15441-5′-2 (18) reverse | TTGCCTTCCCTCACTGATGG | |
| Gm15441-3′-1 (18) forward | ATCAGTGAGGGAAGGCAAGG | |
| Gm15441-3′-1 (18) reverse | AGCAAGCCAGTATCACATGC | |
| Gm15441-3′-2 (18) forward | ATGGAGGGGTGCTGCTTATC | |
| Gm15441-3′-2 (18) reverse | GCAGGAAGGCTAACAGGAGG |
2.2.5. Single-Guide RNA–Cas9 Ribonucleoprotein Assembly and Pronuclear Microinjection
2.2.6. Genotyping and Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yuan, J.; Chen, R. NONCODEv4: Annotation of Noncoding RNAs with Emphasis on Long Noncoding RNAs. Methods Mol. Biol. 2016, 1402, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Tichon, A.; Gil, N.; Lubelsky, Y.; Havkin Solomon, T.; Lemze, D.; Itzkovitz, S.; Stern-Ginossar, N.; Ulitsky, I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 2016, 7, 12209. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; Dhaouadi, I.; Gaziano, I.; Oliverio, M.; Klemm, P.; Awazawa, M.; Mitterer, G.; Fernandez-Rebollo, E.; Pradas-Juni, M.; Wagner, W.; et al. LincRNA H19 protects from dietary obesity by constraining expression of monoallelic genes in brown fat. Nat. Commun. 2018, 9, 3622. [Google Scholar] [CrossRef] [PubMed]
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 2018, 174, 350.e17–362.e17. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013, 9, e1003368. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014, 9, 1956–1968. [Google Scholar] [CrossRef] [PubMed]
- Troder, S.E.; Ebert, L.K.; Butt, L.; Assenmacher, S.; Schermer, B.; Zevnik, B. An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS ONE 2018, 13, e0196891. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Baas, M.; Diederichs, S. Noncoding RNA gene silencing through genomic integration of RNA destabilizing elements using zinc finger nucleases. Genome Res. 2011, 21, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Paralkar, V.R.; Taborda, C.C.; Huang, P.; Yao, Y.; Kossenkov, A.V.; Prasad, R.; Luan, J.; Davies, J.O.; Hughes, J.R.; Hardison, R.C.; et al. Unlinking an lncRNA from Its Associated cis Element. Mol. Cell 2016, 62, 104–110. [Google Scholar] [CrossRef]
- Salsman, J.; Dellaire, G. Precision genome editing in the CRISPR era. Biochem. Cell Biol. 2017, 95, 187–201. [Google Scholar] [CrossRef]
- Brandl, C.; Ortiz, O.; Rottig, B.; Wefers, B.; Wurst, W.; Kuhn, R. Creation of targeted genomic deletions using TALEN or CRISPR/Cas nuclease pairs in one-cell mouse embryos. FEBS Open Bio 2015, 5, 26–35. [Google Scholar] [CrossRef]
- Aparicio-Prat, E.; Arnan, C.; Sala, I.; Bosch, N.; Guigo, R.; Johnson, R. DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genom. 2015, 16, 846. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Chen, L.; Shen, B.; Zhou, J.; Hu, B.; Du, Y.; Tate, P.H.; Huang, X.; Zhang, W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014, 11, 829–835. [Google Scholar] [CrossRef]
- Szafranski, P.; Karolak, J.A.; Lanza, D.; Gajecka, M.; Heaney, J.; Stankiewicz, P. CRISPR/Cas9-mediated deletion of lncRNA Gm26878 in the distant Foxf1 enhancer region. Mamm. Genome 2017, 28, 275–282. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem. 2008, 283, 2397–2406. [Google Scholar] [CrossRef]
- Ferreira, N.E.; Omae, S.; Pereira, A.; Rodrigues, M.V.; Miyakawa, A.A.; Campos, L.C.; Santos, P.C.; Dallan, L.A.; Martinez, T.L.; Santos, R.D.; et al. Thioredoxin interacting protein genetic variation is associated with diabetes and hypertension in the Brazilian general population. Atherosclerosis 2012, 221, 131–136. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Haeusler, R.A.; Kaestner, K.H.; Accili, D. FoxOs function synergistically to promote glucose production. J. Biol. Chem. 2010, 285, 35245–35248. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Durham, H.D.; Dahrouge, S.; Cashman, N.R. Evaluation of the spinal cord neuron × neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. Neurotoxicology 1993, 14, 387–395. [Google Scholar]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansmeier, N.R.; Widdershooven, P.J.M.; Khani, S.; Kornfeld, J.-W. Rapid Generation of Long Noncoding RNA Knockout Mice Using CRISPR/Cas9 Technology. Non-Coding RNA 2019, 5, 12. https://doi.org/10.3390/ncrna5010012
Hansmeier NR, Widdershooven PJM, Khani S, Kornfeld J-W. Rapid Generation of Long Noncoding RNA Knockout Mice Using CRISPR/Cas9 Technology. Non-Coding RNA. 2019; 5(1):12. https://doi.org/10.3390/ncrna5010012
Chicago/Turabian StyleHansmeier, Nils R., Pia J. M. Widdershooven, Sajjad Khani, and Jan-Wilhelm Kornfeld. 2019. "Rapid Generation of Long Noncoding RNA Knockout Mice Using CRISPR/Cas9 Technology" Non-Coding RNA 5, no. 1: 12. https://doi.org/10.3390/ncrna5010012
APA StyleHansmeier, N. R., Widdershooven, P. J. M., Khani, S., & Kornfeld, J.-W. (2019). Rapid Generation of Long Noncoding RNA Knockout Mice Using CRISPR/Cas9 Technology. Non-Coding RNA, 5(1), 12. https://doi.org/10.3390/ncrna5010012