Herpes Simplex Virus 1 Deregulation of Host MicroRNAs


Abstract
:1. Introduction
2. Herpes Simplex Virus 1 and miRNAs
2.1. HSV-1 miRNAs
2.2. HSV-1 Deregulation and Interaction with Host miRNAs
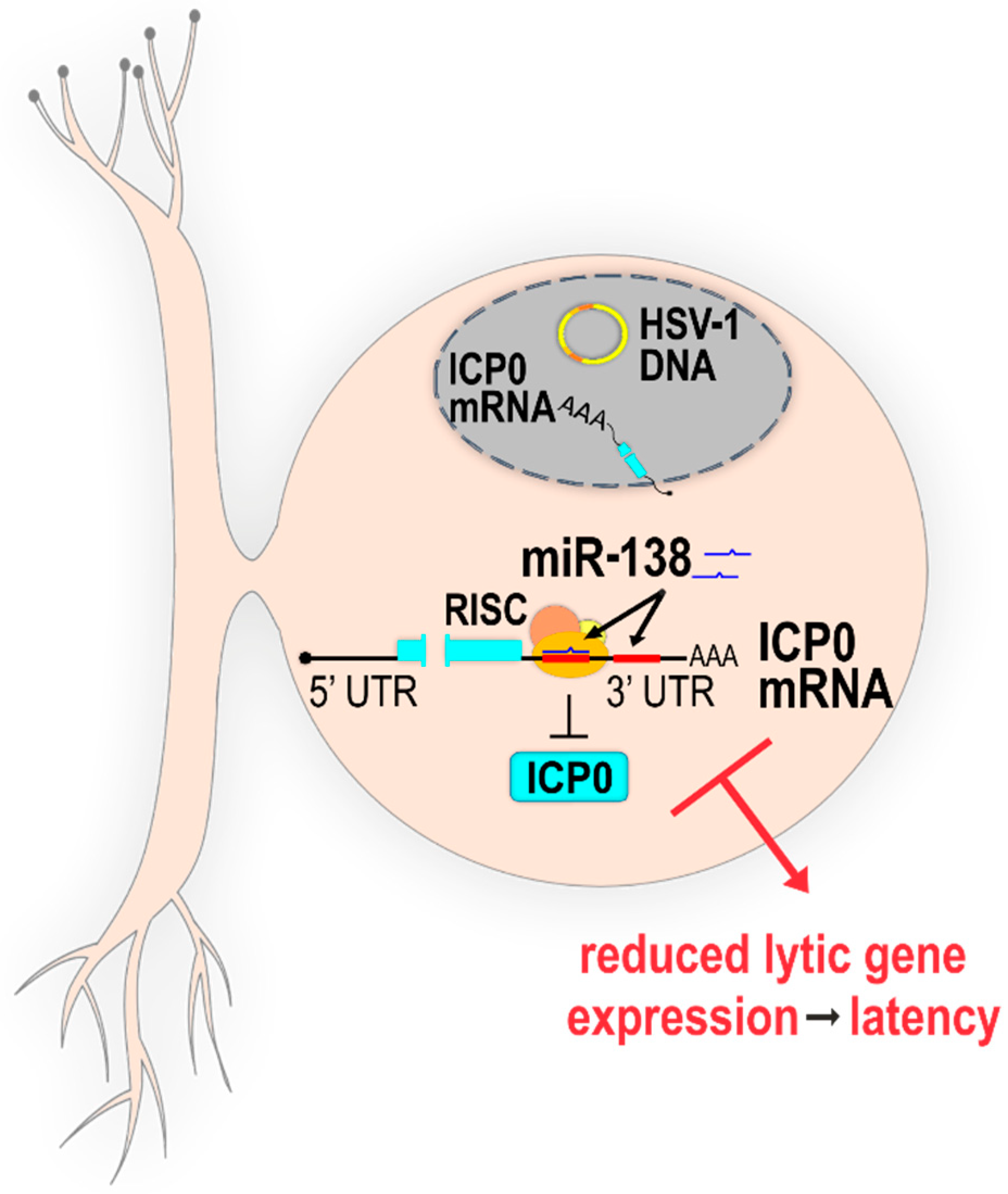
2.2.1. Direct Targeting of Host miRNAs to HSV-1 Transcripts
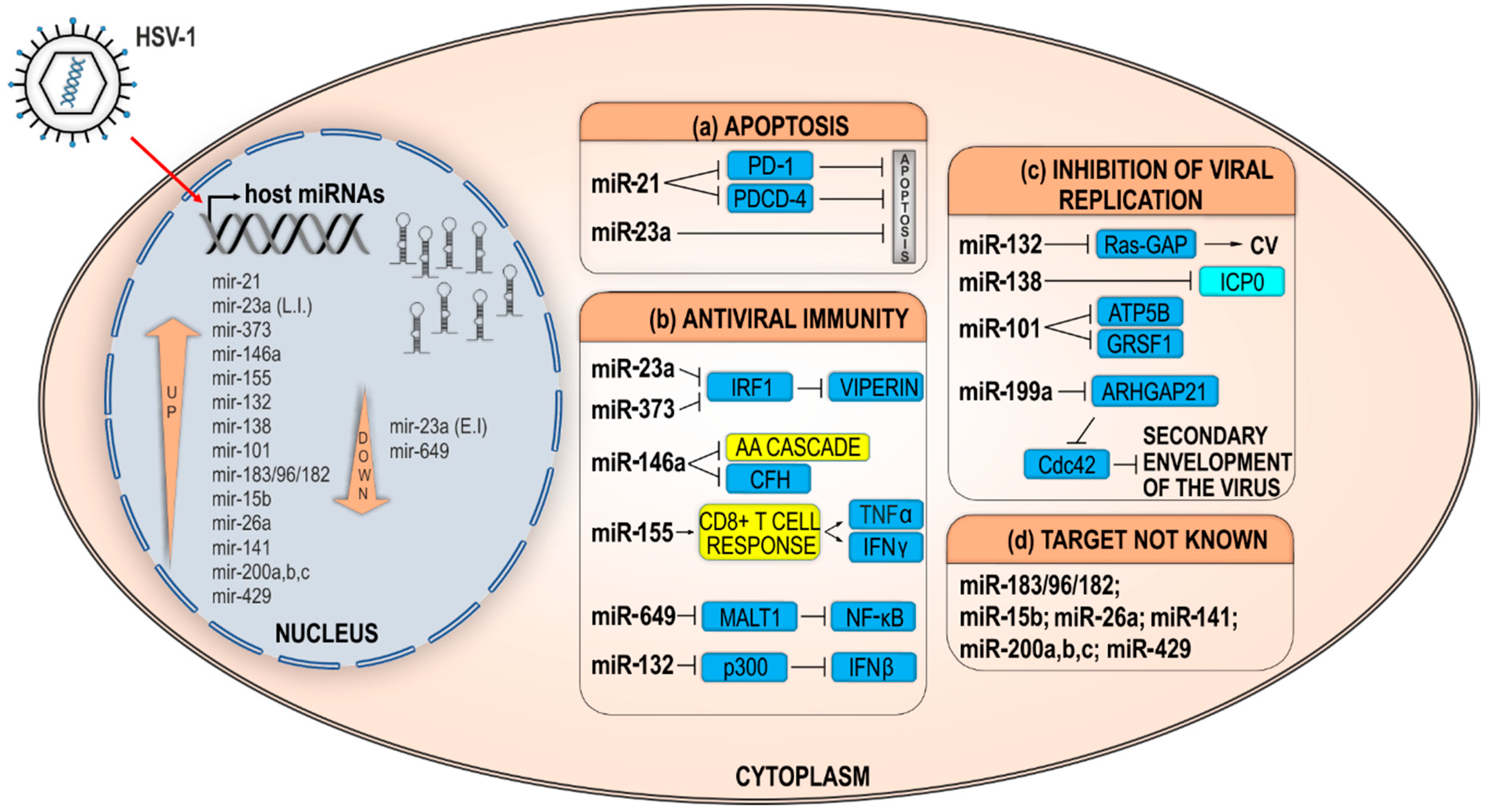
2.2.2. Indirect Effects of Host miRNAs on HSV-1 Infection
Host miRNAs Prolonging Cell Survival and/or Weakening Host Defense Mechanisms
Host miRNAs Modulate Host Factors Required for Efficient HSV-1 Replication
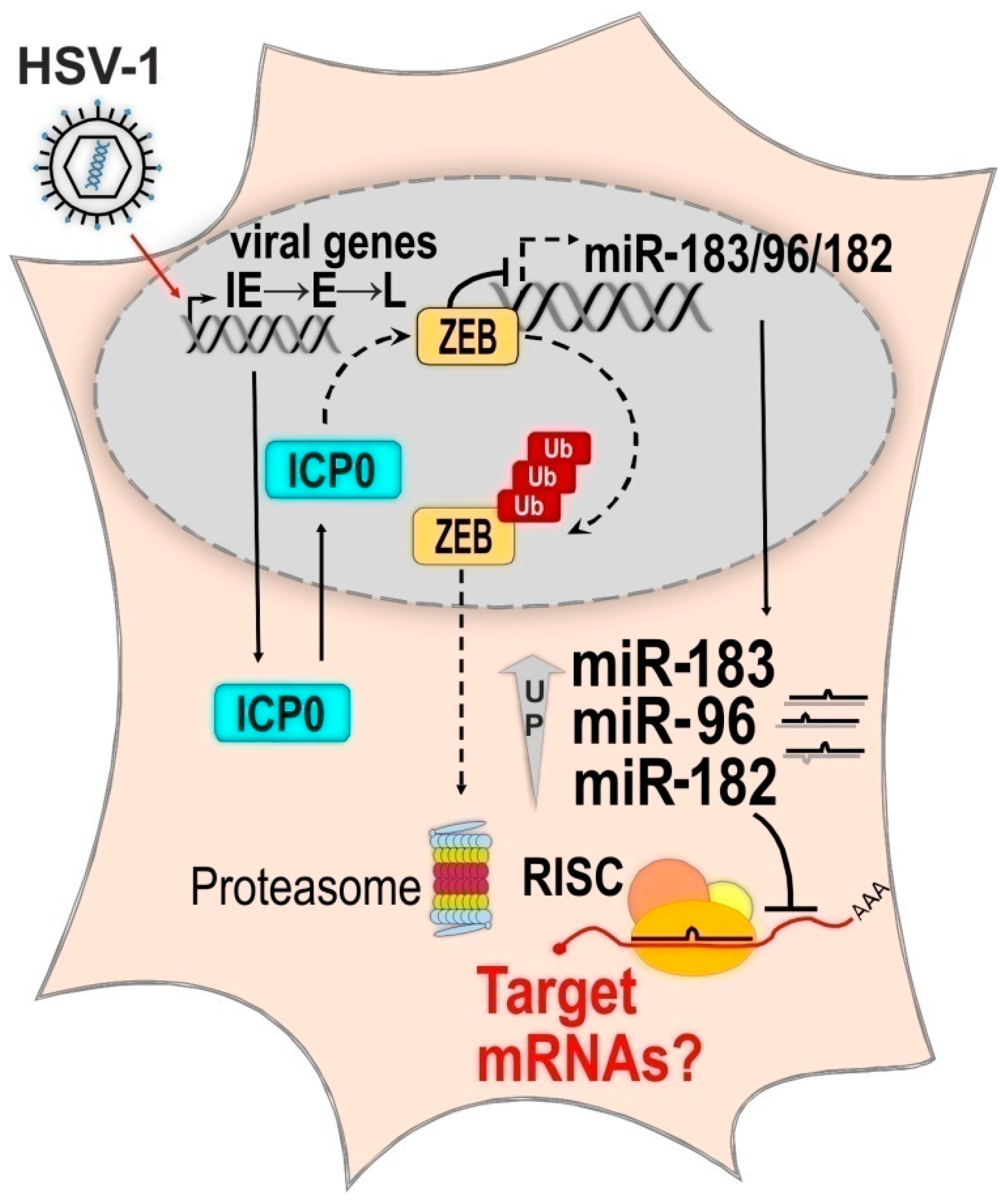
2.2.3. The Molecular Mechanism of HSV-1 Induced Upregulation of the miR-183/96/182 Cluster
3. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Moran, Y.; Agron, M.; Praher, D.; Technau, U. The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 2017, 1, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Jo, M.H.; Choi, Y.G.; Park, J.; Kwon, S.C.; Hohng, S.; Kim, V.N.; Woo, J.S. Functional Anatomy of the Human Microprocessor. Cell 2015, 161, 1374–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Cullen, B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Grey, F. Role of microRNAs in herpesvirus latency and persistence. J. Gen. Virol. 2015, 96, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: Implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurak, I.; Griffiths, A.; Coen, D.M. Mammalian alphaherpesvirus miRNAs. BBA-Gene Regul. Mech. 2011, 1809, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Grey, F.; Meyers, H.; White, E.A.; Spector, D.H.; Nelson, J. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog. 2007, 3, e163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cokaric Brdovcak, M.; Zubkovic, A.; Ferencic, A.; Sosa, I.; Stemberga, V.; Cuculic, D.; Rokic, F.; Vugrek, O.; Hackenberg, M.; Jurak, I. Herpes simplex virus 1 miRNA sequence variations in latently infected human trigeminal ganglia. Virus Res. 2018, 256, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Flores, O.; Umbach, J.L.; Pesola, J.M.; Bentley, P.; Rosato, P.C.; Leib, D.A.; Cullen, B.R.; Coen, D.M. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 2014, 15, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Girardi, E.; Lopez, P.; Pfeffer, S. On the importance of Host MicroRNA during Viral Ihfection. Front. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008, 27, 3300–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 2011, 108, 3193–3198. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, B.L.; Maximova, O.A.; Thach, D.C.; Speicher, J.M.; Pletnev, A.G. MicroRNA targeting of neurotropic flavivirus: Effective control of virus escape and reversion to neurovirulent phenotype. J. Virol. 2012, 86, 5647–5659. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Klimstra, W.B. MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol. Med. 2017, 23, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcinowski, L.; Tanguy, M.; Krmpotic, A.; Radle, B.; Lisnic, V.J.; Tuddenham, L.; Chane-Woon-Ming, B.; Ruzsics, Z.; Erhard, F.; Benkartek, C.; et al. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 2012, 8, e1002510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, A.H.; Perot, J.; Chisholm, M.A.; Kumar, D.S.; Tuddenham, L.; Cognat, V.; Marcinowski, L.; Dolken, L.; Pfeffer, S. Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA 2010, 16, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-regulation of a host microRNA by a herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Farrell, M.J.; Dobson, A.T.; Feldman, L.T. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA 1991, 88, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [PubMed]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [PubMed]
- Garber, D.A.; Schaffer, P.A.; Knipe, D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 1997, 71, 5885–5893. [Google Scholar] [PubMed]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.L.; Sawtell, N.M. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J. Virol. 2001, 75, 6660–6675. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.J.; Hamrah, P.; Gate, D.; Mott, K.R.; Mantopoulos, D.; Zheng, L.; Town, T.; Jones, C.; von Andrian, U.H.; Freeman, G.J.; et al. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. J. Virol. 2011, 85, 4184–4197. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Bogard, C.L.; Kosz-Vnenchak, M.; Hicks, K.A.; Coen, D.M.; Knipe, D.M.; Schaffer, P.A. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 1989, 63, 2893–2900. [Google Scholar] [PubMed]
- Bloom, D.C.; Devi-Rao, G.B.; Hill, J.M.; Stevens, J.G.; Wagner, E.K. Molecular analysis of herpes simplex virus type 1 during epinephrine-induced reactivation of latently infected rabbits in vivo. J. Virol. 1994, 68, 1283–1292. [Google Scholar] [PubMed]
- Thompson, R.L.; Sawtell, N.M. The herpes simplex virus type 1 latency associated transcript locus is required for the maintenance of reactivation competent latent infections. J. Neurovirol. 2011, 17, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, C.; Griffiths, A.; Li, G.; Silva, L.M.; Kramer, M.F.; Gaasterland, T.; Wang, X.J.; Coen, D.M. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J. Virol. 2006, 80, 5499–5508. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Kramer, M.F.; Mellor, J.C.; van Lint, A.L.; Roth, F.P.; Knipe, D.M.; Coen, D.M. Numerous Conserved and Divergent MicroRNAs Expressed by Herpes Simplex Viruses 1 and 2. J. Virol. 2010, 84, 4659–4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Bertke, A.S.; Patel, A.; Wang, K.; Cohen, J.I.; Krause, P.R. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc. Natl. Acad. Sci. USA 2008, 105, 10931–10936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Patel, A.; Krause, P.R. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J. Virol. 2009, 83, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Wang, K.; Tang, S.; Krause, P.R.; Mont, E.K.; Cohen, J.I.; Cullen, B.R. Identification of viral microRNAs expressed in human sacral ganglia latently infected with herpes simplex virus 2. J. Virol. 2010, 84, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Munson, D.J.; Burch, A.D. A novel miRNA produced during lytic HSV-1 infection is important for efficient replication in tissue culture. Arch. Virol. 2012, 157, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Liu, X.; Chen, X.; Zhou, X.; Du, T.; Roizman, B.; Zhou, G. miR-H28 and miR-H29 expressed late in productive infection are exported and restrict HSV-1 replication and spread in recipient cells. Proc. Natl. Acad. Sci. USA 2016, 113, E894–E901. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.F.; Jurak, I.; Pesola, J.M.; Boissel, S.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 2011, 417, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, O.; Nakayama, S.; Whisnant, A.W.; Javanbakht, H.; Cullen, B.R.; Bloom, D.C. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J. Virol. 2013, 87, 6589–6603. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pesola, J.M.; Li, G.; McCarron, S.; Coen, D.M. Mutations Inactivating Herpes Simplex Virus 1 MicroRNA miR-H2 Do Not Detectably Increase ICP0 Gene Expression in Infected Cultured Cells or Mouse Trigeminal Ganglia. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Brown, D.; Osorio, N.; Hsiang, C.; BenMohamed, L.; Wechsler, S.L. Increased neurovirulence and reactivation of the herpes simplex virus type 1 latency-associated transcript (LAT)-negative mutant dLAT2903 with a disrupted LAT miR-H2. J. Neurovirol. 2016, 22, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Brown, D.; Osorio, N.; Hsiang, C.; Li, L.; Chan, L.; BenMohamed, L.; Wechsler, S.L. A herpes simplex virus type 1 mutant disrupted for microRNA H2 with increased neurovirulence and rate of reactivation. J. Neurovirol. 2015, 21, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, Y.; Bosch-Marce, M.; Tang, S.; Patel, A.; Krause, P.R. Herpes Simplex Virus 2 Latency-Associated Transcript (LAT) Region Mutations Do Not Identify a Role for LAT-Associated MicroRNAs in Viral Reactivation in Guinea Pig Genital Models. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Liao, J.; Huang, Q.; Nie, Y.; Wu, K. HSV-1 miR-H6 inhibits HSV-1 replication and IL-6 expression in human corneal epithelial cells in vitro. Clin. Dev. Immunol. 2012, 2012, 192791. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C.; Wiel, C.; Rubin, G.M. Complementary miRNA pairs suggest a regulatory role for miRNA:miRNA duplexes. RNA 2004, 10, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurak, I.; Silverstein, L.B.; Sharma, M.; Coen, D.M. Herpes Simplex Virus Is Equipped with RNA- and Protein-Based Mechanisms To Repress Expression of ATRX, an Effector of Intrinsic Immunity. J. Virol. 2012, 86, 10093–10102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.; Liu, Q.; Wang, S.; Ren, Z.; Kitazato, K.; Yang, D.; Wang, Y. HSV-1-encoded microRNA miR-H1 targets Ubr1 to promote accumulation of neurodegeneration-associated protein. Virus Genes 2018, 54, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.R.; Shango, J.; Seal, A.; Shukla, D.; Nares, S. Viral miRNAs Alter Host Cell miRNA Profiles and Modulate Innate Immune Responses. Front. Immunol. 2018, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Levi, A.; Weisblum, Y.; Yamin, R.; Charpak-Amikam, Y.; Wolf, D.G.; Mandelboim, O. HSV1 MicroRNA Modulation of GPI Anchoring and Downstream Immune Evasion. Cell Rep. 2016, 17, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, H.H.; Wang, D.D.; Chen, D.; Liu, S.W.; Wang, Z.; Yan, D.L.; Dong, S.C.; Feng, J.F. miR-138: A promising therapeutic target for cancer. Tumour Biol. 2017, 39, 1010428317697575. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Hengartner, M.O. miRNAs and apoptosis: RNAs to die for. Oncogene 2006, 25, 6176–6187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.; Cheng, A.C.; Wang, M.S.; Jia, R.Y.; Sun, K.F.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; Liu, M.F.; et al. The suppression of apoptosis by alpha-herpesvirus. Cell Death Dis. 2017, 8, e2749. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.; Sun, H.; Fan, H.; Wang, C.; Li, Y.; Liu, M.; Tang, H. miR-23a facilitates the replication of HSV-1 through the suppression of interferon regulatory factor 1. PLoS ONE 2014, 9, e114021. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ru, J.; Zhang, J.; Zhu, L.H.; Liu, M.; Li, X.; Tang, H. miR-23a targets interferon regulatory factor 1 and modulates cellular proliferation and paclitaxel-induced apoptosis in gastric adenocarcinoma cells. PLoS ONE 2013, 8, e64707. [Google Scholar] [CrossRef] [PubMed]
- Mattijssen, S.; Pruijn, G.J. Viperin, a key player in the antiviral response. Microbes Infect. 2012, 14, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Happel, C.; Ziegelbauer, J.M. Kaposi’s Sarcoma-Associated Herpesvirus MicroRNAs Target GADD45B To Protect Infected Cells from Cell Cycle Arrest and Apoptosis. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Shamulailatpam, P.; Raja, A.N.; Gottwein, E. Kaposi’s sarcoma-associated herpesvirus encodes a mimic of cellular miR-23. J. Virol. 2013, 87, 11821–11830. [Google Scholar] [CrossRef] [PubMed]
- Suffert, G.; Malterer, G.; Hausser, J.; Viiliainen, J.; Fender, A.; Contrant, M.; Ivacevic, T.; Benes, V.; Gros, F.; Voinnet, O.; et al. Kaposi’s sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog. 2011, 7, e1002405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Guo, X.K.; Gao, L.; Huang, C.; Li, N.; Jia, X.; Liu, W.; Feng, W.H. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 2014, 450–451, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Kim, H.A.; Suh, C.H.; Byun, H.O.; Jung, J.Y.; Sohn, S. The relevance of miRNA-21 in HSV-induced inflammation in a mouse model. Int. J. Mol. Sci. 2015, 16, 7413–7427. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; He, S.; Wang, J. MicroRNA-373 facilitates HSV-1 replication through suppression of type I IFN response by targeting IRF1. Biomed. Pharmacother. 2018, 97, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shi, X.; Zhang, X.; Wang, A.; Wang, L.; Yang, Y.; Deng, R.; Zhang, G.P. MicroRNA 373 Facilitates the Replication of Porcine Reproductive and Respiratory Syndrome Virus by Its Negative Regulation of Type I Interferon Induction. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, H.; Mitchelson, K.; Rao, H.; Luo, M.; Xie, L.; Sun, Y.; Zhang, L.; Lu, Y.; Liu, R.; et al. MicroRNAs-372/373 promote the expression of hepatitis B virus through the targeting of nuclear factor I/B. Hepatology 2011, 54, 808–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Di Bisceglie, A.M.; Ray, R.B. Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs the JAK/STAT signaling pathway. J. Virol. 2015, 89, 3356–3365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, J.; Tang, J.; Zhou, L.; Zhou, M. MicroRNA-649 promotes HSV-1 replication by directly targeting MALT1. J. Med. Virol. 2017, 89, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Q.; Li, Y.X.; Zhang, Y.; Li, X.; Tang, H. miR-101 regulates HSV-1 replication by targeting ATP5B. Antiviral Res. 2011, 89, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Diao, C.; Yang, X.; Yang, Z.; Liu, M.; Li, X.; Tang, H. ICP4-induced miR-101 attenuates HSV-1 replication. Sci. Rep. 2016, 6, 23205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulik, S.; Xu, J.; Reddy, P.B.; Rajasagi, N.K.; Gimenez, F.; Sharma, S.; Lu, P.Y.; Rouse, B.T. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am. J. Pathol. 2012, 181, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Pollara, G.; Henderson, S.; Gratrix, F.; Fabani, M.; Milne, R.S.; Gotch, F.; Boshoff, C. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010, 12, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20, 1500–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhela, S.; Mulik, S.; Gimenez, F.; Reddy, P.B.; Richardson, R.L.; Varanasi, S.K.; Jaggi, U.; Xu, J.; Lu, P.Y.; Rouse, B.T. Role of miR-155 in the pathogenesis of herpetic stromal keratitis. Am. J. Pathol. 2015, 185, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.; Caligiuri, K.A.; Gale, K.K.; Niu, Y.; Phillipson, C.S.; Booth, T.F.; Booth, S.A. Induction of Multiple miR-200/182 Members in the Brains of Mice Are Associated with Acute Herpes Simplex Virus 1 Encephalitis. PLoS ONE 2017, 12, e0169081. [Google Scholar] [CrossRef]
- Lutz, G.; Jurak, I.; Kim, E.T.; Kim, J.Y.; Hackenberg, M.; Leader, A.; Stoller, M.L.; Fekete, D.M.; Weitzman, M.D.; Coen, D.M.; et al. Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors. Viruses 2017, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Noncoding RNA 2017, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.P.; Li, Y.J. MicroRNA-146a and human disease. Scand. J. Immunol. 2010, 71, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Bhela, S.; Mulik, S.; Reddy, P.B.; Richardson, R.L.; Gimenez, F.; Rajasagi, N.K.; Veiga-Parga, T.; Osmand, A.P.; Rouse, B.T. Critical role of microRNA-155 in herpes simplex encephalitis. J. Immunol. 2014, 192, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, H.; Yao, Y.; Smith, L.P.; Kgosana, L.; Green, J.; Petherbridge, L.; Baigent, S.J.; Nair, V. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek’s disease lymphomas. PLoS Pathog. 2011, 7, e1001305. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalsky, R.L.; Samols, M.A.; Plaisance, K.B.; Boss, I.W.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 2007, 81, 12836–12845. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; De Strooper, B. microRNA-132: A key noncoding RNA operating in the cellular phase of Alzheimer’s disease. FASEB J. 2017, 31, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Merika, M.; Williams, A.J.; Chen, G.; Collins, T.; Thanos, D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol. Cell 1998, 1, 277–287. [Google Scholar] [CrossRef]
- Dai, H.; Hou, K.; Cai, Z.; Zhou, Q.; Zhu, S. Low-level miR-646 in colorectal cancer inhibits cell proliferation and migration by targeting NOB1 expression. Oncol. Lett. 2017, 14, 6708–6714. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, M.; Feng, Y.; Xu, Y.F.; Huang, Y.F.; Che, J.P.; Wang, G.C.; Yao, X.D.; Zheng, J.H. Downregulated miR-646 in clear cell renal carcinoma correlated with tumour metastasis by targeting the nin one binding protein (NOB1). Br. J. Cancer 2014, 111, 1188–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Tang, W.M.; Zhang, H.; Li, Y.Q.; Peng, Y.; Wang, J.; Liu, G.N.; Huang, X.T.; Zhao, J.J.; Li, G.; et al. miR-646 inhibited cell proliferation and EMT-induced metastasis by targeting FOXK1 in gastric cancer. Br. J. Cancer 2017, 117, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Cunningham, D.M.; Smit, M.W.; Park, Y.; Fritz, D.; Wilusz, J.; Katze, M.G. Selective translation of eukaryotic mRNAs: Functional molecular analysis of GRSF-1, a positive regulator of influenza virus protein synthesis. J. Virol. 2002, 76, 10417–10426. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.W.; Wilusz, J.; Katze, M.G. Regulation of eukaryotic protein synthesis: Selective influenza viral mRNA translation is mediated by the cellular RNA-binding protein GRSF-1. Proc. Natl. Acad. Sci. USA 1999, 96, 6694–6699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, M.C.; Lopez, S.R.; Caputi, M. Members of the heterogeneous nuclear ribonucleoprotein H family activate splicing of an HIV-1 splicing substrate by promoting formation of ATP-dependent spliceosomal complexes. J. Biol. Chem. 2007, 282, 13617–13626. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Weber, F.; Croce, C.; Liu, C.G.; Liao, X.; Pellett, P.E. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J. Virol. 2008, 82, 9065–9074. [Google Scholar] [CrossRef] [PubMed]
- Santhakumar, D.; Forster, T.; Laqtom, N.N.; Fragkoudis, R.; Dickinson, P.; Abreu-Goodger, C.; Manakov, S.A.; Choudhury, N.R.; Griffiths, S.J.; Vermeulen, A.; et al. Combined agonist-antagonist genome-wide functional screening identifies broadly active antiviral microRNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 13830–13835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Suemasa, F.; Sagara, H.; Nakamura, S.; Ino, Y.; Kobayashi, K.; Hiramatsu, H.; Haraguchi, T.; Kurokawa, K.; Todo, T.; et al. miR-199a Inhibits Secondary Envelopment of Herpes Simplex Virus-1 Through the Downregulation of Cdc42-specific GTPase Activating Protein Localized in Golgi Apparatus. Sci Rep 2017, 7, 6650. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Gao, Y.; Zhou, Q.; Zhang, Q.; Peng, Y.; Tian, K.; Wang, J.; Zheng, X. Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene 2014, 536, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Li, H.; Wu, X.; Covarrubias, M.; Scherer, L.; Meinking, K.; Luk, B.; Chomchan, P.; Alluin, J.; Gombart, A.F.; et al. Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res. 2012, 40, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Kozak, R.A.; Majer, A.; Biondi, M.J.; Medina, S.J.; Goneau, L.W.; Sajesh, B.V.; Slota, J.A.; Zubach, V.; Severini, A.; Safronetz, D.; et al. MicroRNA and mRNA Dysregulation in Astrocytes Infected with Zika Virus. Viruses 2017, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Carnero, E.; Sutherland, J.D.; Fortes, P. Adenovirus and miRNAs. Biochim. Biophys. Acta 2011, 1809, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Hackenberg, M.; Kim, J.Y.; Pesola, J.M.; Everett, R.D.; Preston, C.M.; Wilson, A.C.; Coen, D.M. Expression of Herpes Simplex Virus 1 MicroRNAs in Cell Culture Models of Quiescent and Latent Infection. J. Virol. 2014, 88, 2337–2339. [Google Scholar] [CrossRef] [PubMed]
- Davari, M.; Soheili, Z.S.; Samiei, S.; Sharifi, Z.; Pirmardan, E.R. Overexpression of miR-183/-96/-182 triggers neuronal cell fate in Human Retinal Pigment Epithelial (hRPE) cells in culture. Biochem. Biophys. Res. Commun. 2017, 483, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.H.; Chen, W.X.; Li, S.; He, X.Y.; Zhang, Z.M.; Li, M.; Cao, R.S.; Hao, B.; Zhang, H.J.; Qiu, H.Q.; et al. MicroRNA-183 promotes proliferation and invasion in oesophageal squamous cell carcinoma by targeting programmed cell death 4. Br. J. Cancer 2014, 111, 2003–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Witmer, P.D.; Lumayag, S.; Kovacs, B.; Valle, D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J. Biol. Chem. 2007, 282, 25053–25066. [Google Scholar] [CrossRef] [PubMed]
- Dambal, S.; Shah, M.; Mihelich, B.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Hara, T.; Choi, Y.; Subramanian, M.; Francis, P.; Bilke, S.; Walker, R.L.; Pineda, M.; Zhu, Y.; Yang, Y.; et al. A p21-ZEB1 complex inhibits epithelial-mesenchymal transition through the microRNA 183-96-182 cluster. Mol. Cell. Biol. 2014, 34, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 Proteome during HSV-1 Infection. PLoS Pathog. 2015, 11, e1005059. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Seo, D.; Kim, D.; Hong, Y.; Chang, H.; Baek, D.; Kim, V.N.; Lee, S.; Ahn, K. Temporal Landscape of MicroRNA-Mediated Host-Virus Crosstalk during Productive Human Cytomegalovirus Infection. Cell Host Microbe 2015, 17, 838–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, T.J.; Arnold, J.D.; Spector, D.H.; Yeo, G.W. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J. Virol. 2012, 86, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Everett, R.D. Analysis of the functional interchange between the IE1 and pp71 proteins of human cytomegalovirus and ICP0 of herpes simplex virus 1. J. Virol. 2015, 89, 3062–3075. [Google Scholar] [CrossRef] [PubMed]
- Oussaief, L.; Fendri, A.; Chane-Woon-Ming, B.; Poirey, R.; Delecluse, H.J.; Joab, I.; Pfeffer, S. Modulation of MicroRNA Cluster miR-183-96-182 Expression by Epstein-Barr Virus Latent Membrane Protein 1. J. Virol. 2015, 89, 12178–12188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, A.; Gaya, A.; Martinez, A.; Urbano-Ispizua, A.; Pons, A.; Balague, O.; Gel, B.; Abrisqueta, P.; Lopez-Guillermo, A.; Artells, R.; et al. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood 2008, 111, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.L.; Wang, Z.; Yu, X.; Mertz, J.E. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein-Barr virus latent-lytic switch in a cell-type-specific manner. J. Virol. 2010, 84, 6139–6152. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.H.; Kraus, R.J.; Dickerson, S.J.; Lim, H.J.; Jones, R.J.; Yu, X.; Mertz, J.E.; Kenney, S.C. ZEB1 and c-Jun levels contribute to the establishment of highly lytic Epstein-Barr virus infection in gastric AGS cells. J. Virol. 2007, 81, 10113–10122. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Z.; Mertz, J.E. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 2007, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Skalsky, R.L.; Kennedy, E.M.; Furuse, Y.; Whisnant, A.W.; Flores, O.; Schultz, K.L.; Putnam, N.; Barrows, N.J.; Sherry, B.; et al. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J. Virol. 2014, 88, 8065–8076. [Google Scholar] [CrossRef] [PubMed]
- Aguado, L.C.; Schmid, S.; Sachs, D.; Shim, J.V.; Lim, J.K.; tenOever, B.R. MicroRNA Function Is Limited to Cytokine Control in the Acute Response to Virus Infection. Cell Host Microbe 2015, 18, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Pourchet, A.; Modrek, A.S.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens 2017, 6, 24. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| miRNA | Up-/Downregulated | Target | Possible Roles | Model | References | Cellular Process | |
|---|---|---|---|---|---|---|---|
| miR-23a | Down- then Upregulated | IRF1 | Inhibition of innate immune response and cell survival | HeLa | [66] | APOPTOSIS | |
| miR-649 | Downregulated | MALT1 | Inhibition of innate and adaptive immune response | HeLa | [78] | ||
| miR-101 | Upregulated | ATP5B | Blocking DNA packaging and capsid maturation | HeLa | [79] | INHIBITION OF VIRAL REPLICATION | |
| Upregulated | GRSF1 | Inhibition of viral protein synthesis | HeLa | [80] | |||
| miR-132 | Upregulated | Ras-GAP | Immuno-inflammatory response leading to neovascularization and stromal keratitis lesions | Murine corneas | [81] | ANTIVIRAL IMMUNITY | |
| Upregulated | p300 | Inhibition of innate immune response | Monocytes (THP-1 cell line) | [82] | |||
| miR-146a | Upregulated | Complement factor H | Evasion of HSV-1 from the innate immune response | Human neuronal-glial cells | [83] | ||
| miR-373 | Upregulated | IRF1 | Inhibition of innate immune response | HeLa and patients with herpetic gingivostomatitis | [74] | ||
| miR-155 | Deficiency | SOCS1 | Regulation of T cell differentiation | In vivo mouse model of herpes simplex encephalitis | [84] | ||
| Upregulated | Unknown | - | In vivo acute viral encephalitis model—mouse brain | [85] | TARGET NOT KNOWN | ||
| miR-183/96/182 | Upregulated | Unknown | - | Primary fibroblasts and neurons | [86] | ||
| miR-15b, miR-26a, miR-141, miR-183/96/182, miR-200a, b, c, miR-429 1 | Upregulated | Unknown | - | In vivo acute viral encephalitis model—mouse brain | [85] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cokarić Brdovčak, M.; Zubković, A.; Jurak, I. Herpes Simplex Virus 1 Deregulation of Host MicroRNAs. Non-Coding RNA 2018, 4, 36. https://doi.org/10.3390/ncrna4040036
Cokarić Brdovčak M, Zubković A, Jurak I. Herpes Simplex Virus 1 Deregulation of Host MicroRNAs. Non-Coding RNA. 2018; 4(4):36. https://doi.org/10.3390/ncrna4040036
Chicago/Turabian StyleCokarić Brdovčak, Maja, Andreja Zubković, and Igor Jurak. 2018. "Herpes Simplex Virus 1 Deregulation of Host MicroRNAs" Non-Coding RNA 4, no. 4: 36. https://doi.org/10.3390/ncrna4040036
APA StyleCokarić Brdovčak, M., Zubković, A., & Jurak, I. (2018). Herpes Simplex Virus 1 Deregulation of Host MicroRNAs. Non-Coding RNA, 4(4), 36. https://doi.org/10.3390/ncrna4040036