Copaifera langsdorffii Novel Putative Long Non-Coding RNAs: Interspecies Conservation Analysis in Adaptive Response to Different Biomes

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
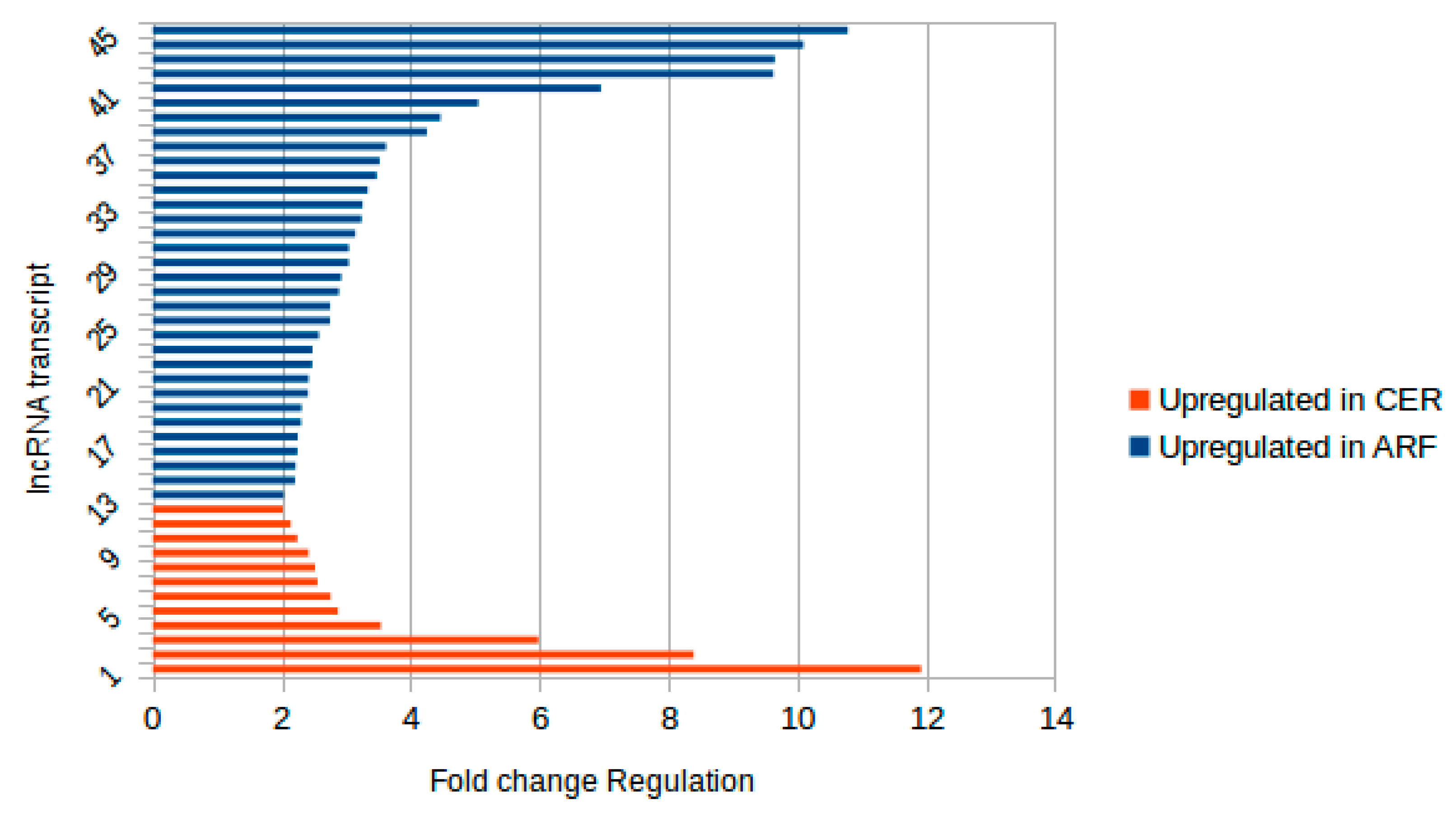
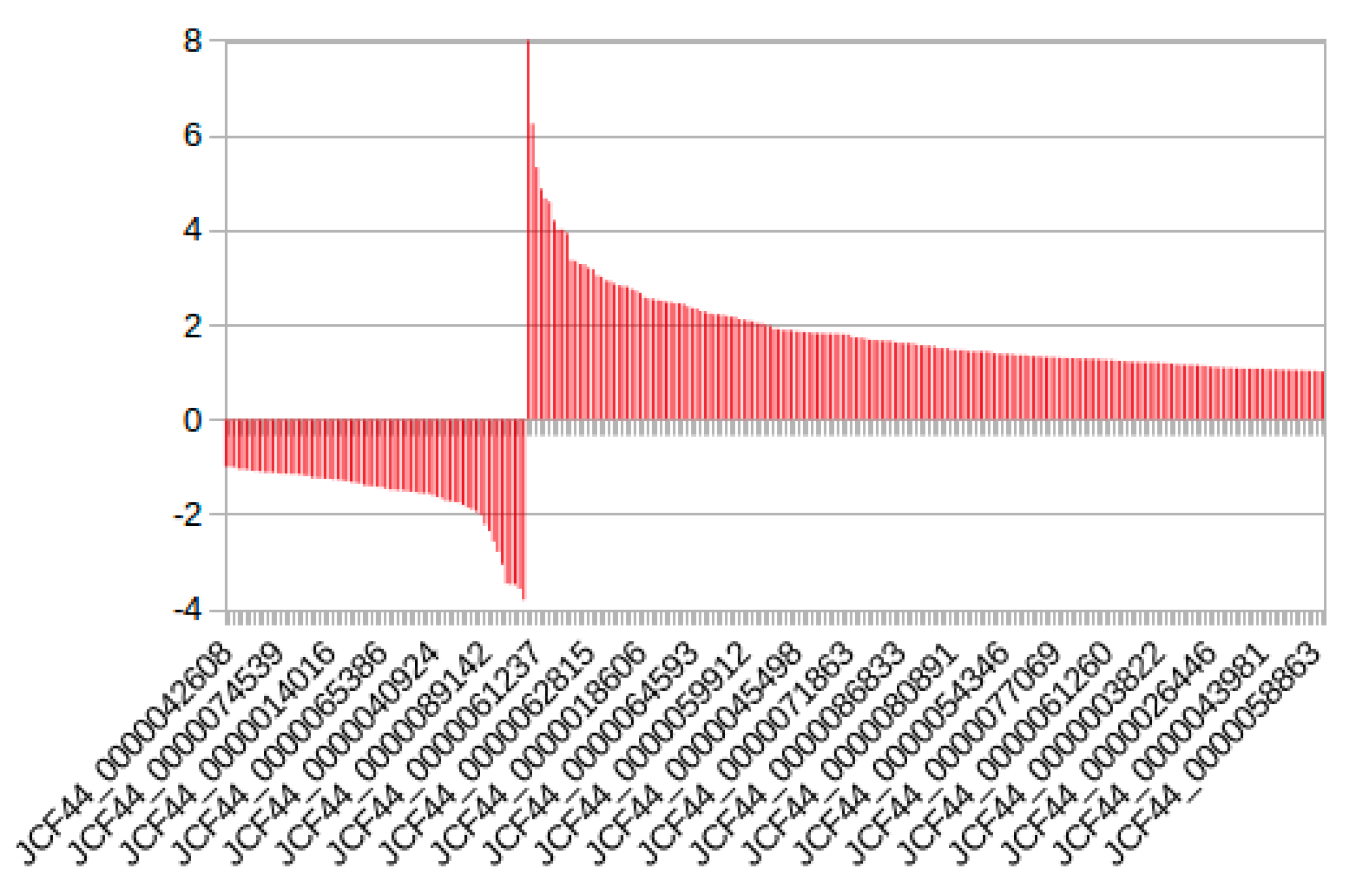
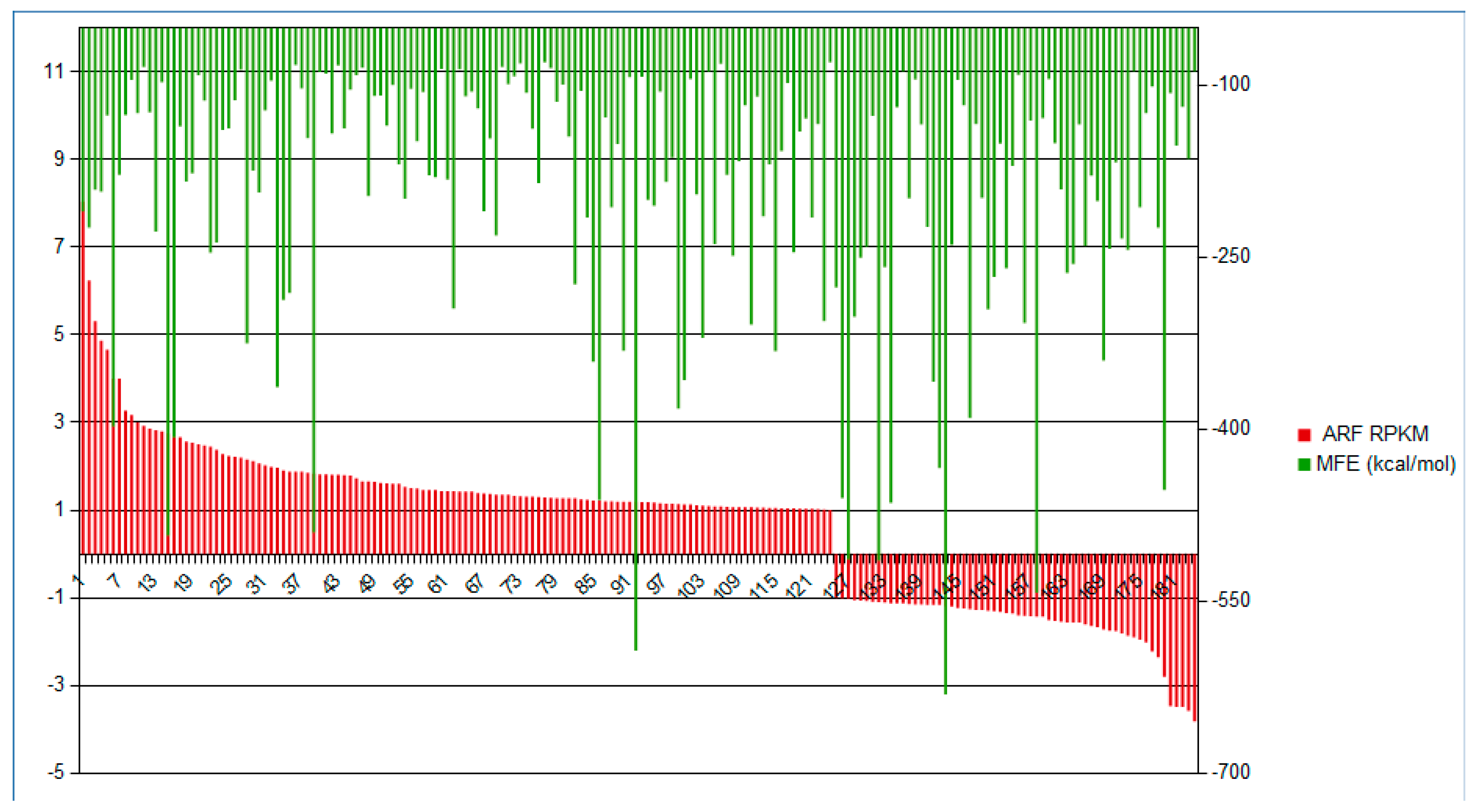
2.1. Identification of Novel and Differentially Expressed lncRNA
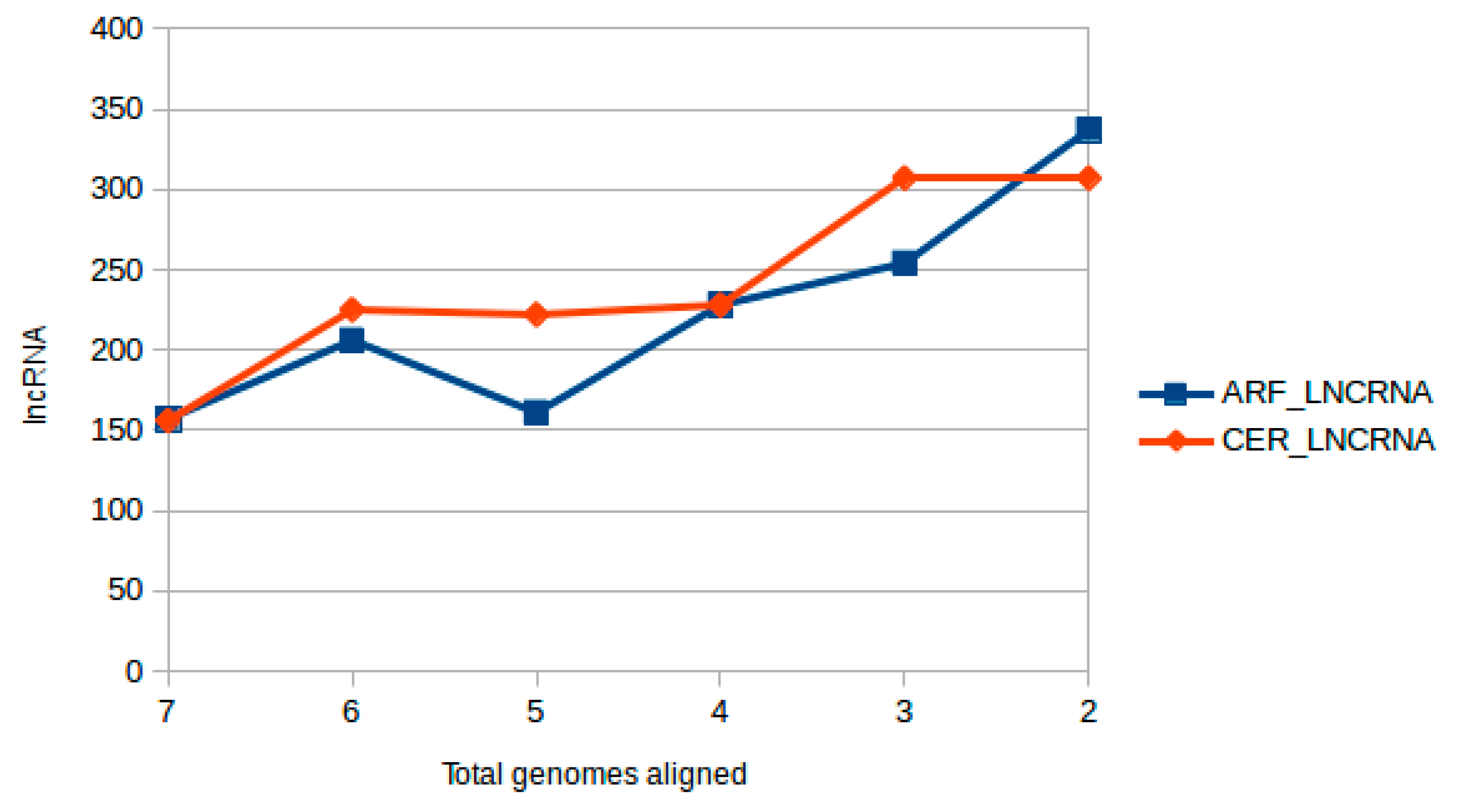
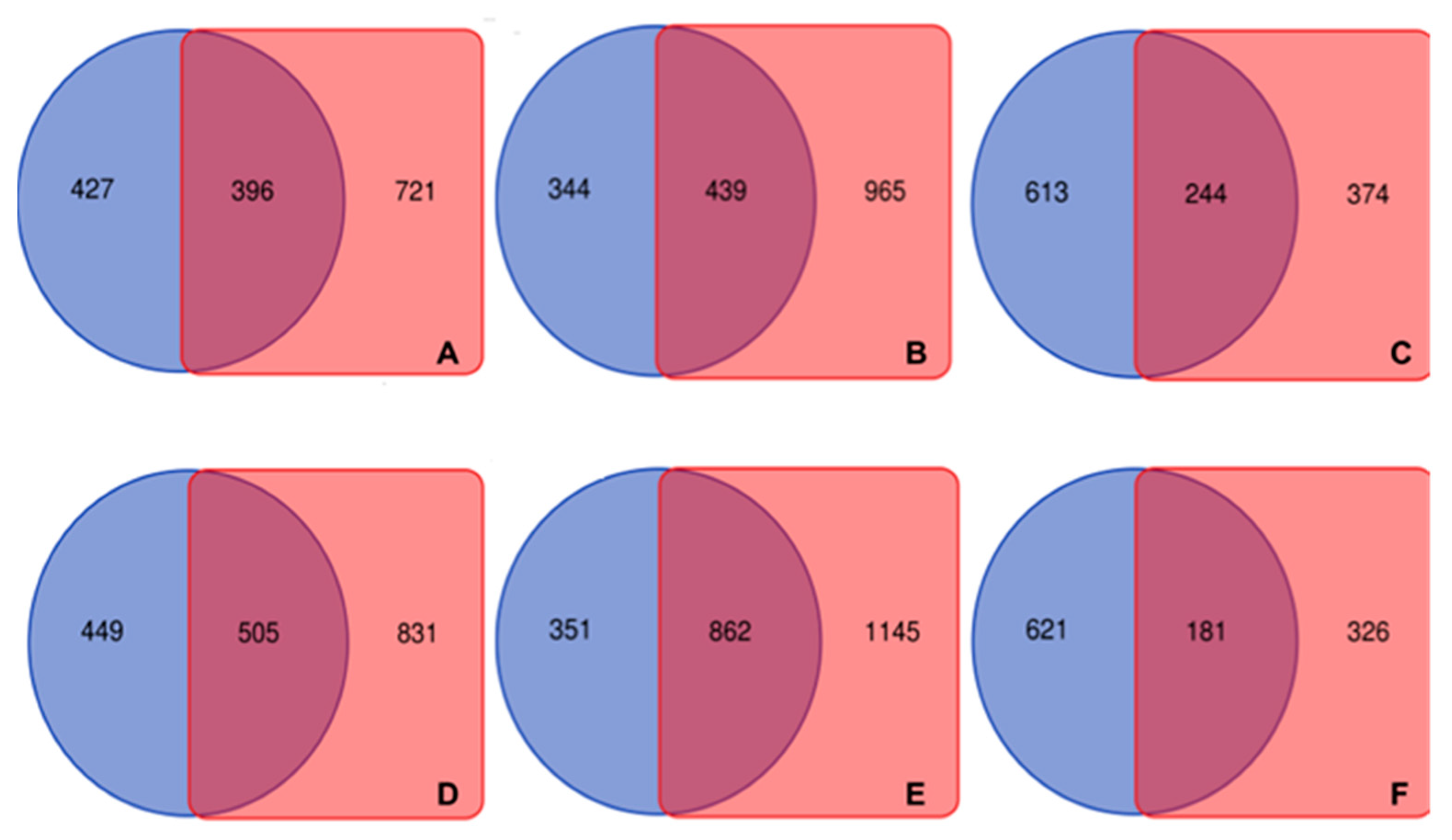
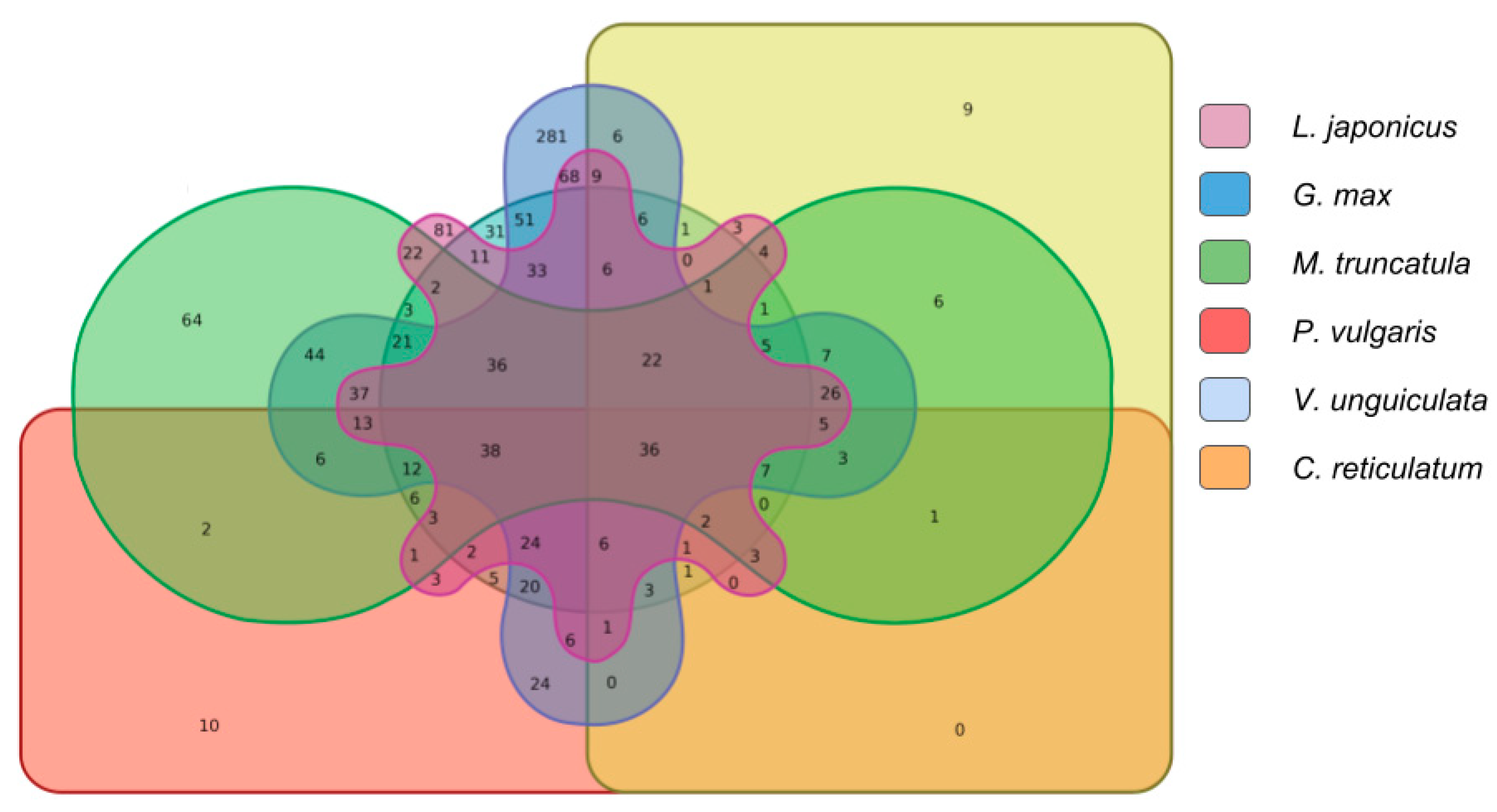
2.2. Interspecies lncRNA Conservation Analysis
2.2.1. Positional Conservation and Genome Alignment Analysis
2.2.2. Identification of Putative C. langsdorffii lncRNAs in EST Sequences of Other Fabaceae Species
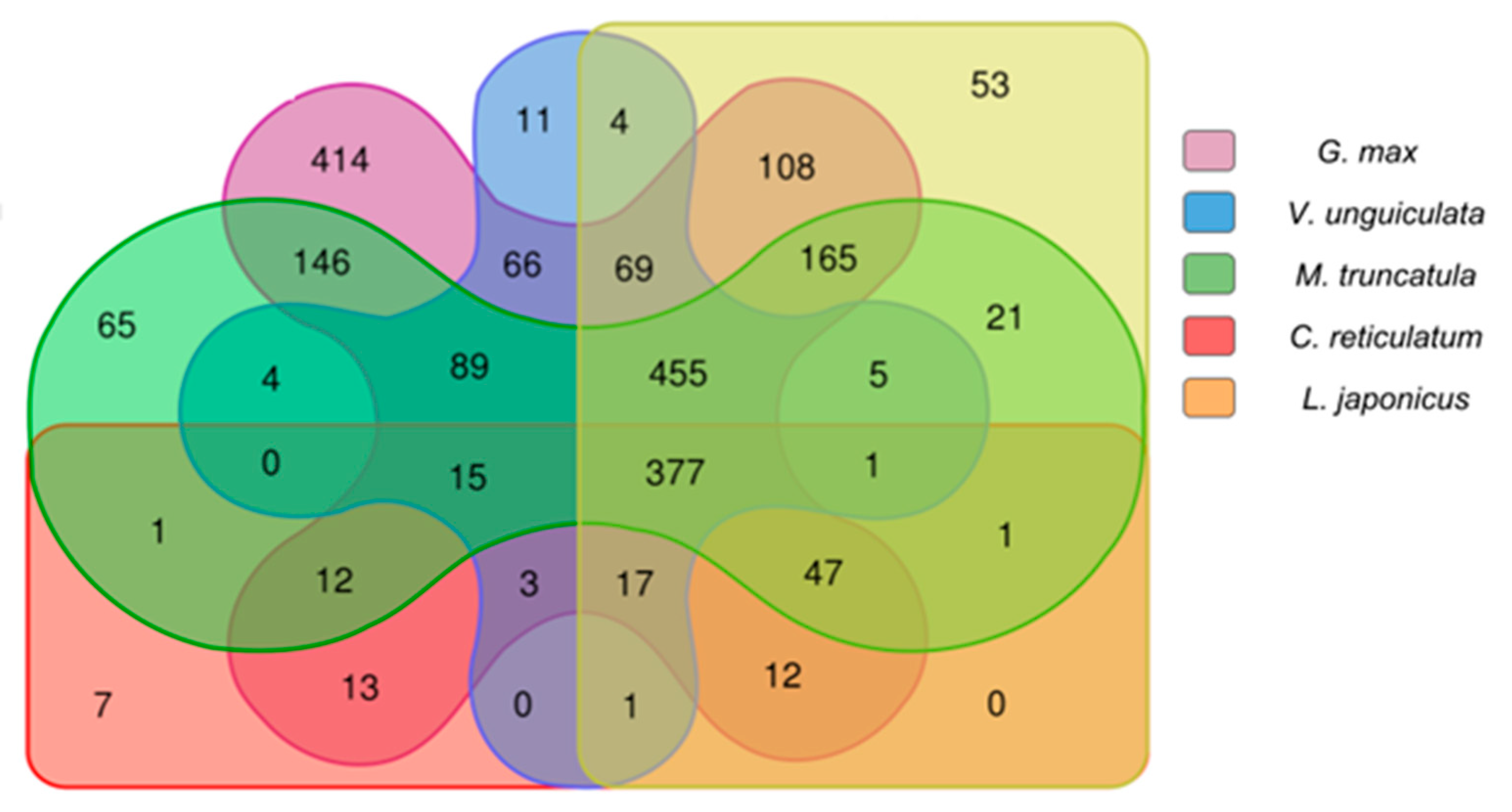
2.2.3. Comparison Analysis of Conserved Putative lncRNAs
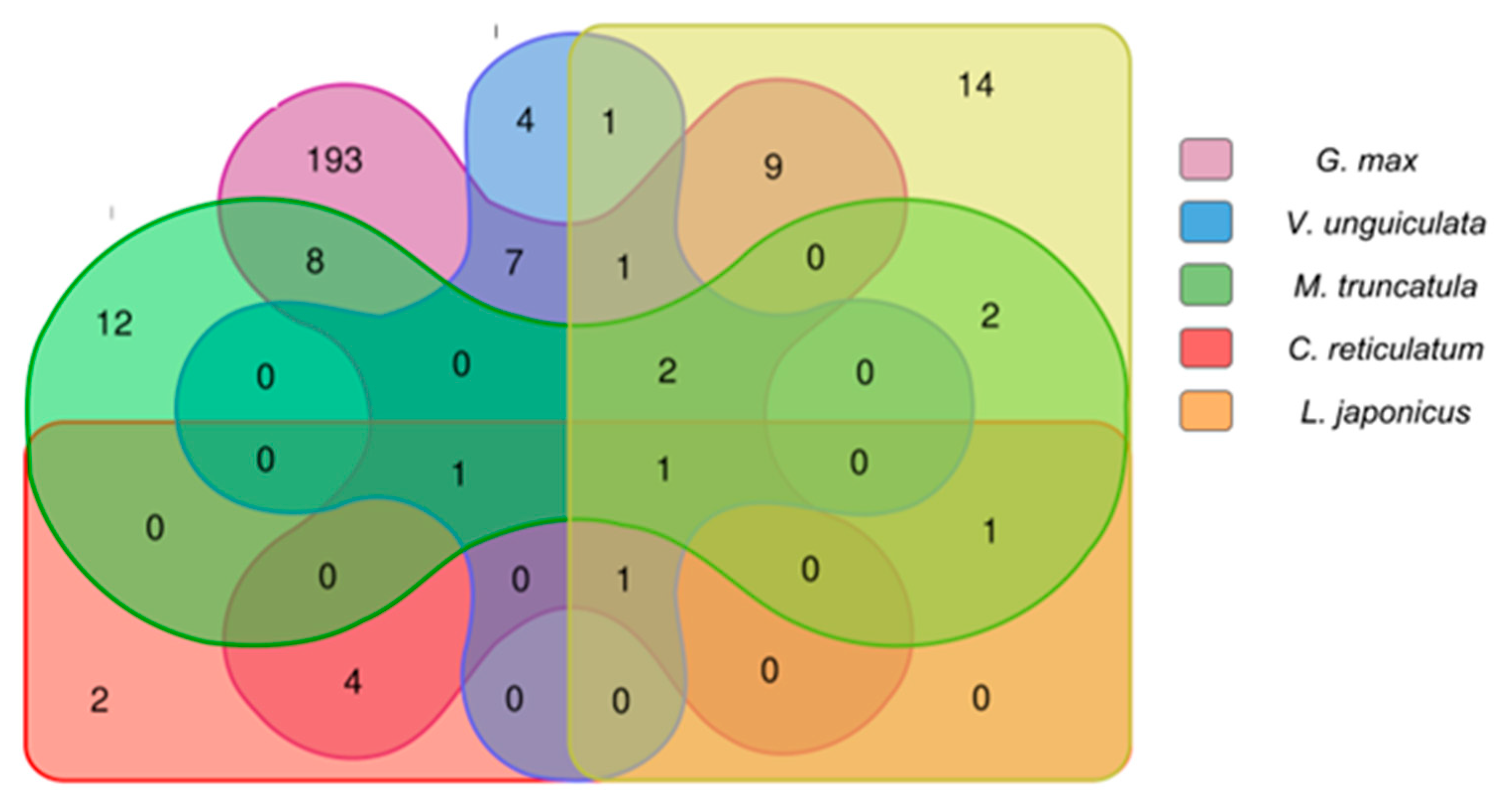
2.2.4. Expression Analysis of the Conserved Putative lncRNAs among Fabaceae
2.2.5. Identification of Known lncRNA
2.3. Stem-Loop Secondary Structure of Regulated Putative lncRNA
2.4. RT-qPCR Analysis of Copaíba lncRNA Expression
2.5. Computational Identification of miRNA and lncRNA Interactions
3. Discussion
4. Materials and Methods
4.1. Plant Material Collection
4.2. RNA Extraction to Sequencing
4.3. De Novo Transcriptome Assembly and Gene Expression Profiling
4.4. lncRNA Identification
4.5. Interspecies lncRNAs Conservation Analysis
4.6. Interspecies lncRNA Genome Conservation Analysis
4.7. Interspecies Conservation of Expressed lncRNA Analysis
4.8. Second Structure Modeling
4.9. RT-qPCR Analysis of Copaíba lncRNA Expression
4.10. Computational Prediction of miRNA and lncRNA Interaction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Junior, V.V.; Pinto, A.C. O GÊNERO Copaifera L. Quim. Nova 2002, 25, 273–286. [Google Scholar] [CrossRef]
- Breitbach, U.B.; Niehues, M.; Lopes, N.P.; Faria, J.E.; Brandão, M.G. Amazonian Brazilian medicinal plants described by CFP von Martius in the 19th century. J. Ethnopharmacol. 2013, 147, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Junior, V.V.; Rosas, E.; Carvalho, M.V.D.; Henriques, M.D.G.M.D.O.; Pinto, A.C. Chemical composition and anti-inflammatory activity of copaiba oils from Copaifera cearensis Huber ex Ducke, Copaifera reticulata Ducke and Copaifera multijuga Hayne—A comparative study. J. Ethnopharmacol. 2007, 112, 248–254. [Google Scholar]
- Basile, A.; Sertié, J.; Freitas, P.; Zanini, A. Anti-inflammatory activity of oleoresin from Brazilian Copaifera. J. Ethnopharmacol. 1988, 22, 101–109. [Google Scholar] [CrossRef]
- Falcão, H.D.S.; Lima, I.O.; Santos, V.L.D.; Dantas, H.D.F.; Diniz, M.D.F.; Barbosa-Filho, J.M.; Batista, L.M. Review of the plants with anti-inflammatory activity studied in Brazil. Rev. Bras. Farmacogn. 2005, 15, 381–391. [Google Scholar] [CrossRef]
- Souza, A.B.; Martins, C.H.; Souza, M.G.; Furtado, N.A.; Heleno, V.C.; de Sousa, J.P.; Rocha, E.M.; Bastos, J.K.; Cunha, W.R.; Veneziani, R.C.; et al. Antimicrobial activity of terpenoids from Copaifera langsdorffii Desf. against cariogenic bacteria. Phytother. Res. 2011, 25, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Paiva, L.; Rao, V.; Gramosa, N.; Silveira, E. Gastroprotective effect of Copaifera langsdorffii oleo-resin on experimental gastric ulcer models in rats. J. Ethnopharmacol. 1998, 62, 73–78. [Google Scholar] [CrossRef]
- Silva, J.; Borges, V.R.D.A.; Pereira, L.D.C.B.; Ferrari, R.; de Mattos, R.M.; Barros, E.G.D.O.; Palmero, C.Y.; Fernandes, P.D.; de Carvalho, P.R.; Pereira de Sousa, V.; et al. The oil-resin of the tropical rainforest tree Copaifera langsdorffii reduces cell viability, changes cell morphology and induces cell death in human endometriotic stromal cultures. J. Pharm. Pharmacol. 2015, 67, 1744–1755. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.D.S. Atividades Cicatrizante e Antimicrobiana do Óleo-Resina de Copaiba (Copaifera langsdorffii) em Úlceras Cutâneas; Universidade de São Paulo: São Paulo, Brazil, 2011. [Google Scholar]
- Gushiken, L.F.S.; Hussni, C.A.; Bastos, J.K.; Rozza, A.L.; Beserra, F.P.; Vieira, A.J.; Padovani, C.R.; Lemos, M.; Polizello Junior, M.; Silva, J.J.M.D.; et al. Skin wound healing potential and mechanisms of the hydroalcoholic extract of leaves and oleoresin of Copaifera langsdorffii Desf. Kuntze in rats. Evid.-Based Complement. Altern. Med. 2017, 2017, 6589270. [Google Scholar] [CrossRef] [PubMed]
- Millas, A.; Siveira, J.V.; Barbosa, R.; Maria, B.; Koh, I.H.; Bittencourt, E. Implantes de scafolds de PLGA incorporados com oleo de resina do genero copaifera ssp. In Proceedings of the 14° Congresso da Sociedade Latino Americana de Biomateriais, Orgaos artificiais e Engenharia de Tecidos (SLABO), Maresisas, São Paulo, Brazil, 20–24 August 2017. [Google Scholar]
- Abed, R.A.; Cavasin, G.M.; Silva, H.H.G.D.; Geris, R.; Silva, I.G.D. Alterações morfohistológicas em larvas de Aedes aegypti (Linnaeus, 1762) (Diptera, Culicidae) causadas pela atividade larvicida do óleo-resina da planta medicinal Copaifera reticulata Ducke (Leguminosae). Rev. Patol. Trop. 2007, 36, 75–86. [Google Scholar] [CrossRef]
- Geris, R.; Silva, I.G.D.; Silva, H.H.G.D.; Barison, A.; Rodrigues-Filho, E.; Ferreira, A.G. Diterpenos de Copaifera reticulata Ducke com atividade larvicida contra Aedes aegypti (L.)(Diptera, Culicidae). Rev. Inst. Med. Trop. São Paulo 2008, 50, 26–28. [Google Scholar] [CrossRef]
- Santos, A.O.D.; Ueda-Nakamura, T.; Dias Filho, B.P.; Veiga Junior, V.F.; Pinto, A.C.; Nakamura, C.V. Antimicrobial activity of Brazilian copaiba oils obtained from different species of the Copaifera genus. Mem. Inst. Oswaldo Cruz 2008, 103, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Morelli, C.L.; Mahrous, M.; Belgacem, M.N.; Branciforti, M.C.; Bretas, R.E.S.; Bras, J. Natural copaiba oil as antibacterial agent for bio-based active packaging. Ind. Crops Prod. 2015, 70, 134–141. [Google Scholar] [CrossRef]
- Guimarães, A.; Cunha, E.A.; Matias, F.O.; Garcia, P.G.; Danopoulos, P.; Swikidisa, R.; Pinheiro, V.A.; Nogueira, R. Antimicrobial Activity of Copaiba (Copaifera officinalis) and Pracaxi (Pentaclethra macroloba) Oils against Staphylococcus Aureus: Importance in Compounding for Wound Care. Int. J. Pharm. Compd. 2016, 20, 58–62. [Google Scholar] [PubMed]
- Santos, A.O.; Ueda-Nakamura, T.; Dias Filho, B.P.; Junior, V.F.V.; Pinto, A.C.; Nakamura, C.V. Effect of Brazilian copaiba oils on Leishmania amazonensis. J. Ethnopharmacol. 2008, 120, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Calderon, L.D.A.; Silva-Jardim, I.; Zuliani, J.P.; Ciancaglini, P.; Silva, L.H.P.D.; Stábeli, R.G. Amazonian biodiversity: A view of drug development for leishmaniasis and malaria. J. Braz. Chem. Soc. 2009, 20, 1011–1023. [Google Scholar] [CrossRef]
- Soares, D.C.; Portella, N.A.; Ramos, M.F.D.S.; Siani, A.C.; Saraiva, E.M. Trans-β-caryophyllene: An effective antileishmanial compound found in commercial copaiba oil (Copaifera spp.). Evid.-Based Complement. Altern. Med. 2013, 2013, 761323. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.S.; Souza, A.B.; Tekwani, B.L.; Ambrósio, S.R.; Veneziani, R.C. Synthesis and biological evaluation of polyalthic acid derivatives for the treatment of neglected diseases. Bioorg. Med. Chem. Lett. 2015, 25, 5529–5531. [Google Scholar] [CrossRef] [PubMed]
- de Moraes, A.R.D.P.; Tavares, G.D.; Rocha, F.J.S.; de Paula, E.; Giorgio, S. Effects of nanoemulsions prepared with essential oils of copaiba-and andiroba against Leishmania infantum and Leishmania amazonensis infections. Exp. Parasitol. 2018, 187, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Sâmia, R.; de Oliveira, R.; Moscardini, V.; Carvalho, G. Effects of aqueous extracts of Copaifera langsdorffii (Fabaceae) on the growth and reproduction of Spodoptera frugiperda (JE Smith) (Lepidoptera: Noctuidae). Neotrop. Entomol. 2016, 45, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Alencar, J.D.C. Estudos silviculturais de uma população natural de Copaifera multijuga Hayne-Leguminosae, na Amazônia Central. 2-Produção de óleo-resina. Acta Amazon. 1982, 12, 75–89. [Google Scholar] [CrossRef]
- Carvalho, M.C.; Gomide, L.R.; Santos, R.M.D.; Scolforo, J.R.S.; Carvalho, L.M.T.D.; Mello, J.M.D. Modeling ecological niche of tree species in Brazilian tropical area. Cerne 2017, 23, 229–240. [Google Scholar] [CrossRef]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shao, C.; Jin, Y.; Wang, H.; Meng, Y. Long non-coding RNAs: A novel endogenous source for the generation of Dicer-like 1-dependent small RNAs in Arabidopsis thaliana. RNA Biol. 2014, 11, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Ariel, F.; Romero-Barrios, N.; Jégu, T.; Benhamed, M.; Crespi, M. Battles and hijacks: Noncoding transcription in plants. Trends Plant Sci. 2015, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Kashi, K.; Henderson, L.; Bonetti, A.; Carninci, P. Discovery and functional analysis of lncRNAs: Methodologies to investigate an uncharacterized transcriptome. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, G.; Goyal, N.; Sharma, S.; Upadhyay, S.K.; Singh, K. Present scenario of long non-coding RNAs in plants. Non-Coding RNA 2017, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Mohammadin, S.; Edger, P.P.; Pires, J.C.; Schranz, M.E. Positionally-conserved but sequence-diverged: Identification of long non-coding RNAs in the Brassicaceae and Cleomaceae. BMC Plant Biol. 2015, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Shafiq, S.; Li, J.; Sun, Q. Functions of plants long non-coding RNAs. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Kotake, Y.; Ohhata, T. Long non-coding RNAs involved in cancer development and cell fate determination. Curr. Drug Targets 2012, 13, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Bardou, F.; Ariel, F.; Simpson, C.G.; Romero-Barrios, N.; Laporte, P.; Balzergue, S.; Brown, J.W.; Crespi, M. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev. Cell 2014, 30, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Bazin, J.; Bailey-Serres, J. Emerging roles of long non-coding RNA in root developmental plasticity and regulation of phosphate homeostasis. Front. Plant Sci. 2015, 6, 400. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, L.; Li, D.; Zhu, L.; Hu, S. Long non-coding RNAs and their biological roles in plants. Genom. Proteom. Bioinform. 2015, 13, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Amor, B.B.; Wirth, S.; Merchan, F.; Laporte, P.; d’Aubenton Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.; Lucas, A.; Deragon, J.M.; et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2008, 19, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNAs in Populus under nitrogen deficiency. Mol. Genet. Genom. 2016, 291, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Gai, Y.-P.; Yuan, S.-S.; Zhao, Y.-N.; Zhao, H.-N.; Zhang, H.-L.; Ji, X.-L. A Novel LncRNA, MuLnc1, Associated with Environmental Stress in Mulberry (Morus multicaulis). Front. Plant Sci. 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- He, Y. Chromatin regulation of flowering. Trends Plant Sci. 2012, 17, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Rowley, M.J.; Böhmdorfer, G.; Wierzbicki, A.T. A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol. Cell 2013, 49, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Vance, K.W.; Ponting, C.P. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014, 30, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of chromatin structure by long noncoding RNA. Trends Cell Biol. 2015, 25, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Ariel, F.; Jegu, T.; Latrasse, D.; Romero-Barrios, N.; Christ, A.; Benhamed, M.; Crespi, M. Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin-driven chromatin loop. Mol. Cell 2014, 55, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-D.; Sung, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Sung, S. Vernalization-triggered intragenic chromatin loop formation by long noncoding RNAs. Dev. Cell 2017, 40, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Lau, O.S.; Deng, X.W. Light-regulated transcriptional networks in higher plants. Nat. Rev. Genet. 2007, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Quan, M.; Du, Q.; Zhang, D. The interactions between the long non-coding RNA NERDL and its target gene affect wood formation in Populus tomentosa. Front. Plant Sci. 2017, 8, 1035. [Google Scholar] [CrossRef] [PubMed]
- Jabnoune, M.; Secco, D.; Lecampion, C.; Robaglia, C.; Shu, Q.; Poirier, Y. A rice cis-natural antisense RNA acts as a translational enhancer for its cognate mRNA and contributes to phosphate homeostasis and plant fitness. Plant Cell 2013, tpc-113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Han, Z.; Guo, Q.; Liu, Y.; Zheng, Y.; Wu, F.; Jin, W. Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 2014, 9, e98958. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Liang, Z.; Ge, M.; Qi, W.; Zhang, T.; Lin, F.; Peng, Z.; Zhao, H. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (Zea mays L.). BMC Genom. 2016, 17, 350. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Arikit, S.; Zhai, J.; Meyers, B.C. Biogenesis and function of rice small RNAs from non-coding RNA precursors. Curr. Opin. Plant Biol. 2013, 16, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Wang, Z.-M.; Wang, M.; Wang, X.-J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, W.; Yang, Y.; Li, X.; Chen, T.; Liu, T.; Ma, N.; Yang, X.; Liu, R.; Zhang, B. Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci. Rep. 2015, 5, 16946. [Google Scholar] [CrossRef] [PubMed]
- Szcześniak, M.W.; Rosikiewicz, W.; Makałowska, I. CANTATAdb: A collection of plant long non-coding RNAs. Plant Cell Physiol. 2015, 57, e8. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601. [Google Scholar] [CrossRef] [PubMed]
- Hezroni, H.; Perry, R.B.-T.; Meir, Z.; Housman, G.; Lubelsky, Y.; Ulitsky, I. A subset of conserved mammalian long non-coding RNAs are fossils of ancestral protein-coding genes. Genome Biol. 2017, 18, 162. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protocols 2012, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An improved genome release (version Mt4. 0) for the model legume Medicago truncatula. BMC Genom. 2014, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.; Capella-Gutiérrez, S.; Rendón-Anaya, M.; Hernández-Oñate, M.; Minoche, A.E.; Erb, I.; Câmara, F.; Prieto-Barja, P.; Corvelo, A.; Sanseverino, W.; et al. Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Amatriain, M.; Mirebrahim, H.; Xu, P.; Wanamaker, S.I.; Luo, M.; Alhakami, H.; Alpert, M.; Atokple, I.; Batieno, B.J.; Boukar, O.; et al. Genome resources for climate-resilient cowpea, an essential crop for food security. Plant J. 2017, 89, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Nawaz, K.; Parween, S.; Roy, R.; Sahu, K.; Kumar Pole, A.; Khandal, H.; Srivastava, R.; Kumar Parida, S.; Chattopadhyay, D. Draft genome sequence of Cicer reticulatum L., the wild progenitor of chickpea provides a resource for agronomic trait improvement. DNA Res. 2016, 24, 1–10. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Sizing up long non-coding RNAs: Do lncRNAs have secondary and tertiary structure? BioArchitecture 2012, 2, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014, 30, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.; Mattick, J.S. Structure and Function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Steffens, S.; Thiel, H.-J.; Behrens, S.-E. The RNA-Dependent RNA polymerases of different members of the family flaviviridae exhibit similar properties in vitro. J. Gen. Virol. 1999, 80, 2583–2590. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Qu, K.; Ouyang, Z.; Kertesz, M.; Li, J.; Tibshirani, R.; Makino, D.; Nutter, R.; Segal, E.; Chang, H. Genome-wide measurement of RNA folding energies. Mol. Cell 2012, 48, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rice, K.; Wang, Y.; Chen, W.; Zhong, Y.; Nakayama, Y.; Zhou, Y.; Klibanski, A. Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: Isoform structure, expression, and functions. Endocrinology 2010, 151, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.; Hennelly, S.P.; Sanbonmatsu, K. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 2012, 40, 5034–5051. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.; Hofacker, I. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Ma, Y.-K.; Chen, T.; Wang, M.; Wang, X.-J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; Van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Karakülah, G.; Kurtoğlu, K.Y.; Unver, T. PeTMbase: A database of plant endogenous target mimics (eTMs). PLoS ONE 2016, 11, e0167698. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.W.; Deng, S.; Xu, H.; Mao, H.Z.; Liu, J.; Niu, Q.W.; Wang, H.; Chua, N.H. A noncoding RNA transcribed from the AGAMOUS (AG) second intron binds to CURLY LEAF and represses AG expression in leaves. New Phytol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nithin, C.; Thomas, A.; Basak, J.; Bahadur, R.P. Genome-wide identification of miRNAs and lncRNAs in Cajanus cajan. BMC Genom. 2017, 18, 878. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. Lost in translation? microRNAs at the rough ER. Trends Plant Sci. 2017, 22, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Meng, X.; Li, X.; Illing, N.; Ingle, R.A.; Wang, J.; Chen, M. PceRBase: A database of plant competing endogenous RNA. Nucleic Acids Re. 2017, 45, D1009–D1014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.V.; Vo, K.T.X.; Hong, W.-J.; Jung, K.-H.; Jeon, J.-S. Defense response to pathogens through epigenetic regulation in rice. J. Plant Biol. 2018, 61, 1–10. [Google Scholar] [CrossRef]
- Wang, L.; Xia, X.; Jiang, H.; Lu, Z.; Cui, J.; Cao, F.; Jin, B. Genome-wide identification and characterization of novel NAs in Ginkgo biloba. Trees 2018, 32, 1429–1442. [Google Scholar] [CrossRef]
- Aitken, S.N.; Yeaman, S.; Holliday, J.A.; Wang, T.; Curtis-McLane, S. Adaptation, migration or extirpation: Climate change outcomes for tree populations. Evol. Appl. 2008, 1, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Song, Z.; Zhu, C.; Tao, C.; Kang, L.; Liu, W.; He, F.; Yan, J.; Sang, T. Systematic comparison of lncRNAs with protein coding MRNAs in population expression and their response to environmental change. BMC Plant Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Trindade, I.; Capitão, C.; Dalmay, T.; Fevereiro, M.P.; Dos Santos, D.M. miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 2010, 231, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, Q.; Wang, Q.; Wang, X.; Liu, J.; Li, M.; Huang, L.; Kang, Z. Target of tae-miR408, a chemocyanin-like protein gene (TaCLP1), plays positive roles in wheat response to high-salinity, heavy cupric stress and stripe rust. Plant Mol. Biol. 2013, 83, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Hajyzadeh, M.; Turktas, M.; Khawar, K.M.; Unver, T. miR408 overexpression causes increased drought tolerance in chickpea. Gene 2015, 555, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Hong, P.; Wu, J.Y.; Chen, X.B.; Ye, X.G.; Pan, Y.Y.; Wang, J.; Zhang, X.S. The tae-miR408-mediated control of TaTOC1 gene transcription is required for the regulation of heading time in wheat (Triticum aestivum L.). Plant Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Yu, Y.; Feng, Y.-Z.; Zhou, Y.-F.; Zhang, F.; Yang, Y.-W.; Lei, M.-Q.; Zhang, Y.-C.; Chen, Y.-Q. miRNA MiR408 regulates grain yield and photosynthesis via a phytocyanin protein. Plant Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, L.; Wang, Y.; Li, H.; Li, S.; Zhao, H.; Zhang, H. Constitutive expression of miR408 improves biomass and seed yield in Arabidopsis. Front. Plant Sci. 2018, 8, 2114. [Google Scholar] [CrossRef] [PubMed]
- Stief, A.; Altmann, S.; Hoffmann, K.; Pant, B.D.; Scheible, W.-R.; Bäurle, I. Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 2014, tpc-114. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Niu, Q.-W.; Ng, K.-H.; Chua, N.-H. The role of miR156/SPL s modules in Arabidopsis lateral root development. Plant J. 2015, 83, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xu, Y.; Shi, M.; Lai, Y.; Wu, X.; Wang, H.; Zhu, Z.; Poethig, R.S.; Wu, G. Repression of miR156 by miR159 regulates the timing of the juvenile-to-adult transition in Arabidopsis. Plant Cell 2017, tpc-00975. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Han, J.; Zhou, G.; Xu, Y.; Ding, Y.; Shi, M.; Guo, C.; Wu, G. Silencing of miR156 confers enhanced resistance to brown planthopper in rice. Planta 2018, 248, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xie, K.; Xiong, L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 2014, 65, 2119–2135. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Xie, K.; Hou, X. MIR164 Gene That Controls Plant Root System Development and Fertility and Use Thereof. U.S. Patent No. 8,802,929, 12 August 2014. [Google Scholar]
- Jun, W.; Junhong, Z.; Menghui, H.; Minhui, Z.; Zaikang, T. Expression analysis of miR164 and its target gene NAC1 in response to low nitrate availability in Betula luminifera. Hereditas 2015, 38, 155–162. [Google Scholar]
- Li, J.; Lai, T.; Song, H.; Xu, X. miR164 is involved in delaying senescence of strawberry (Fragaria ananassa) fruit by negatively regulating NAC transcription factor genes under low temperature. Russ. J. Plant Physiol. 2017, 64, 251–259. [Google Scholar] [CrossRef]
- Lu, X.; Dun, H.; Lian, C.; Zhang, X.; Yin, W.; Xia, X. The role of peu-miR164 and its target PeNAC genes in response to abiotic stress in Populus euphratica. Plant Physiol. Biochem. 2017, 115, 418–438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Xia, H.; Cao, T.; Yang, Y.; Zhao, S.; Hou, L.; Zhang, Y.; Li, C.; Zhang, X.; Wang, X. Small RNA and degradome deep sequencing reveals peanut microRNA roles in response to pathogen infection. Plant Mol. Biol. Rep. 2015, 33, 1013–1029. [Google Scholar] [CrossRef]
- Hanemian, M.; Barlet, X.; Sorin, C.; Yadeta, K.A.; Keller, H.; Favery, B.; Simon, R.; Thomma, B.P.; Hartmann, C.; Crespi, M.; et al. Arabidopsis CLAVATA1 and CLAVATA2 receptors contribute to Ralstonia solanacearum pathogenicity through a miR169-dependent pathway. New Phytol. 2016, 211, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Serivichyaswat, P.T.; Susila, H.; Ahn, J.H. Elongated hypocotyl 5-homolog (HYH) negatively regulates expression of the ambient temperature-responsive microRNA gene MIR169. Front. Plant Sci. 2017, 8, 2087. [Google Scholar] [CrossRef] [PubMed]
- Moradi, K.; Khalili, F. Assessment of pattern expression of miR172 and miR169 in response to drought stress in Echinacea purpurea L. Biocatal. Agric. Biotechnol. 2018, in press. [Google Scholar] [CrossRef]
- Japelaghi, R.H.; Haddad, R.; Garoosi, G.A. Rapid and efficient isolation of high quality nucleic acids from plant tissues rich in polyphenols and polysaccharides. Mol. Biotechnol. 2011, 49, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Paytuvi Gallart, A.; Hermoso Pulido, A.; Anzar Martinez de Lagrán, I.; Sanseverino, W.; Aiese Cigliano, R. GREENC: A Wiki-based database of plant lncRNAs. Nucleic Acids Res. 2015, 44, D1161–D1166. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, M.; Kanamori, H.; Komatsu, S.; Namiki, N.; Mukai, Y.; Kurita, K.; Kamatsuki, K.; Ikawa, H.; Yano, R.; Ishimoto, M.; et al. The Glycine max cv. Enrei genome for improvement of Japanese soybean cultivars. Int. J. Genom. 2015, 2015, 358127. [Google Scholar]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharwal, M.; Wilkerson, M.D.; Lawrence, C.J.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucleic Acids Res. 2007, 36, D959–D965. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2011, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Transcripts | Longest Contig per Cluster | CPC lncRNA Prediction | PLEK lncRNA Prediction | Overlapped lncRNA Predicted | One-to-One Correspondence (RPKM > 1) |
|---|---|---|---|---|---|---|
| ARF | 138,175 | 94,815 | 67,251 | 86,608 | 64,801 | 8020 |
| CER | 199,556 | 140,011 | 102,804 | 129,710 | 99,570 | 8020 |
| LncRNA ID | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| Candidate 1 | AATGCAATACAGCAACCTCTAAACC | GGAGGCACCTGGTGTATTGG |
| Candidate 2 | TCATATCAATGCGGCACTCAA | TGTCTTCAGCTGCCCTTTCTG |
| Candidate 3 | AGCAATTGCGGTTGGTATCC | TGGTACCTTTTCATGTTGCTTTCA |
| Candidate 4 | TCAGGCAGCAGAGGAAGAATC | CACCCAGTTCATGCAACCAA |
| Candidate 5 | CGCCAAATGTCCGCAGAT | GGACTTGCCCGCTATGCA |
| Candidate 6 | AGCAATTGCGGTTGGTATCC | TGGTACCTTTTCATGTTGCTTTCA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danilevicz, M.F.; Moharana, K.C.; Venancio, T.M.; Franco, L.O.; Cardoso, S.R.S.; Cardoso, M.; Thiebaut, F.; Hemerly, A.S.; Prosdocimi, F.; Ferreira, P.C.G. Copaifera langsdorffii Novel Putative Long Non-Coding RNAs: Interspecies Conservation Analysis in Adaptive Response to Different Biomes. Non-Coding RNA 2018, 4, 27. https://doi.org/10.3390/ncrna4040027
Danilevicz MF, Moharana KC, Venancio TM, Franco LO, Cardoso SRS, Cardoso M, Thiebaut F, Hemerly AS, Prosdocimi F, Ferreira PCG. Copaifera langsdorffii Novel Putative Long Non-Coding RNAs: Interspecies Conservation Analysis in Adaptive Response to Different Biomes. Non-Coding RNA. 2018; 4(4):27. https://doi.org/10.3390/ncrna4040027
Chicago/Turabian StyleDanilevicz, Monica F., Kanhu C. Moharana, Thiago M. Venancio, Luciana O. Franco, Sérgio R. S. Cardoso, Mônica Cardoso, Flávia Thiebaut, Adriana S. Hemerly, Francisco Prosdocimi, and Paulo C. G. Ferreira. 2018. "Copaifera langsdorffii Novel Putative Long Non-Coding RNAs: Interspecies Conservation Analysis in Adaptive Response to Different Biomes" Non-Coding RNA 4, no. 4: 27. https://doi.org/10.3390/ncrna4040027
APA StyleDanilevicz, M. F., Moharana, K. C., Venancio, T. M., Franco, L. O., Cardoso, S. R. S., Cardoso, M., Thiebaut, F., Hemerly, A. S., Prosdocimi, F., & Ferreira, P. C. G. (2018). Copaifera langsdorffii Novel Putative Long Non-Coding RNAs: Interspecies Conservation Analysis in Adaptive Response to Different Biomes. Non-Coding RNA, 4(4), 27. https://doi.org/10.3390/ncrna4040027