1. Introduction
MicroRNAs (miRNAs) are a well characterized family of short non-coding RNAs [
1,
2]. These regulatory molecules are cleaved from premature transcripts by Dicer family proteins to form ~22 nucleotide (nt) molecules. They function to negatively regulate gene expression of messenger RNAs (mRNAs) in a sequence dependent manner [
1,
3], to inhibit translational initiation [
4] via disruption of cap–tail interactions [
5,
6], and to promote exonuclease-mediated mRNA degradation [
7,
8]. It is now appreciated that nearly all genes and cellular programs important for the development and progression of complex diseases are regulated by miRNAs [
9].
Traditionally, each miRNA precursor was believed to give rise to a single mature miRNA sequence, but recent work [
10,
11,
12,
13,
14] has revealed a more complex system at play. The use of short RNA sequencing has revealed that a miRNA precursor constitutively produces a cloud of mature miRNAs from a given precursor arm, instead of only one. These isomiRs differ from the archetypal sequence (the one annotated in public reference databases such as miRBase [
2]) on their 5′- and/or -3′ end. Our previous analysis of 452 deep sequencing datasets from the GEUVADIS project [
15] showed that isomiR profiles are able to classify individual samples based on characteristics, such as: sex, race, and population [
11]. More recently, we showed that isomiRs can distinguish amongst tissue type and disease subtypes and that isoforms of the same miRNA can target largely non-overlapping groups of mRNAs [
10,
16].
While the functional importance of isomiRs has not been fully understood, the shift on their 5′-end alters the miRNA seed sequence, resulting in changes in target profiles. Evidence for the functional consequences of this shift is accumulating. In [
10], we showed that transfection of miR-183-5p isomiRs −2|−2 and +2|+2 into MDA-MB-231 cells shows a less than 15% overlap in targeted mRNAs, as compared to the archetypal miR-183-5p 0|0 isomiR. A more recent study [
13] demonstrated that the different seed sequences of miR-140-3p 5′-isomiRs functionally target distinct downstream genes that the archetypal sequence lacks the ability to regulate. In the context of type 2 diabetes, isomiRs of the miR-375 locus target an overlapping, but distinct complement of beta cell genes, as compared to the archetypal sequence [
17]. Finally, a third group also recently demonstrated that miR-142-3p 5′-isomiRs have a diverging, yet overlapping effect on actin dynamics in immortalized human microvascular endothelial cells [
12]. These studies suggest that isomiRs are not only an actual feature of a miRNA locus, but that their presence can considerably increase the repertoire of genes regulated by a specific locus. By extension, isomiRs are expected to play important roles in disease etiology and, thus, their functions warrant detailed studies. To this end, accurate detection of isomiRs and accurate quantification of their abundance is vital.
Among the many current technologies [
18], RNA sequencing is the only technique that affords such detection and quantification in an unbiased manner. However, owing to its high cost and the complexity of the required computational analysis, this approach is not widely accessible. Moreover, it is often necessary to characterize only one, or a small group of isomiRs, rather than the full complement of isomiRs within the sample. Multiple detection methods are available to efficiently and accurately quantify miRNAs [
18], but these approaches have not been tested for their ability to quantify specific isomiRs. Therefore, there is currently an unmet need to determine how effective these approaches are in distinguishing varying isomiRs.
Here, we set out to determine the discriminatory ability of two of the more commonly used miRNA detection methods: the Exiqon LNA miRCuRY quantitative polymerase chain reaction (qPCR) and the ThermoFisher Taqman microRNA qPCR. Neither Exiqon nor ThermoFisher claim that these commercial qPCR methods are capable of discriminating individual isomiRs. However, we set out to extend on demonstrated and advertised success in discriminating closely related variants of hsa-let-7 [
18]. We wanted to determine whether such discriminatory capacities extend to isomiRs with single nucleotide endpoint differences, by extending previous attempts to address this question [
19]. Using four isomiRs from the miR-21-5p locus, we investigated the capacity of these assays to detect these miRNA isoforms, using synthetic RNAs and total RNA extracted from model cell lines.
3. Discussion
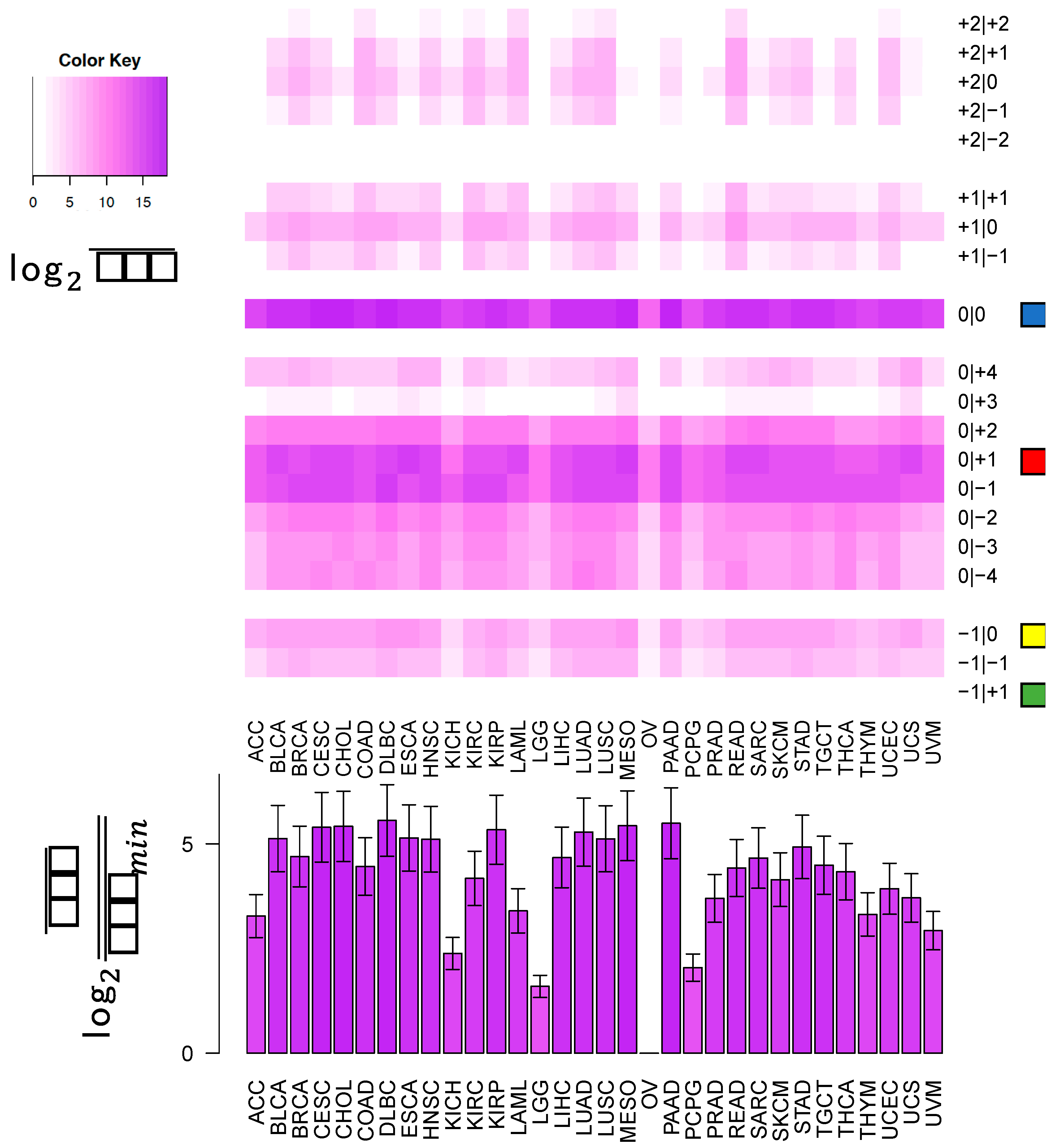
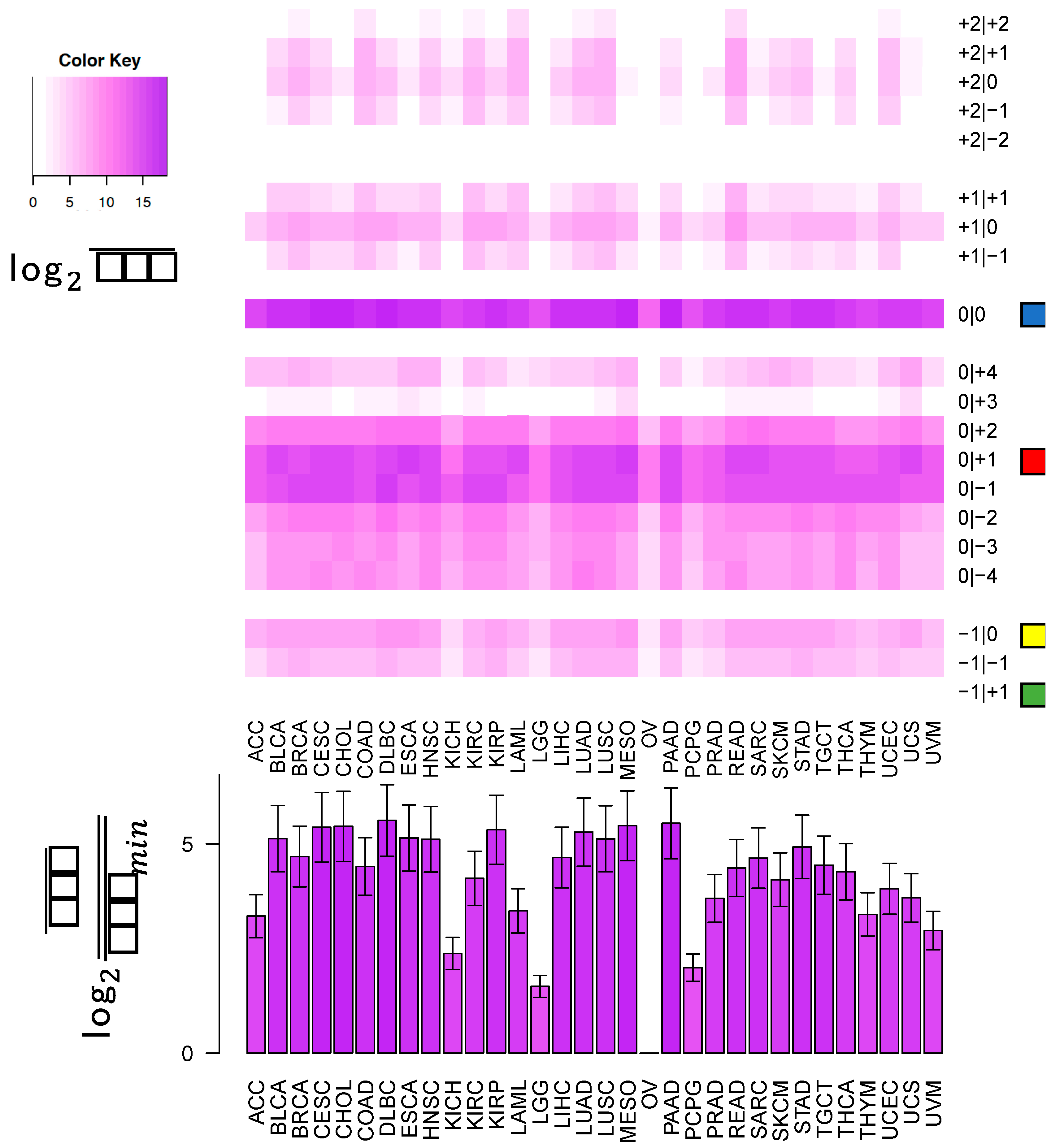
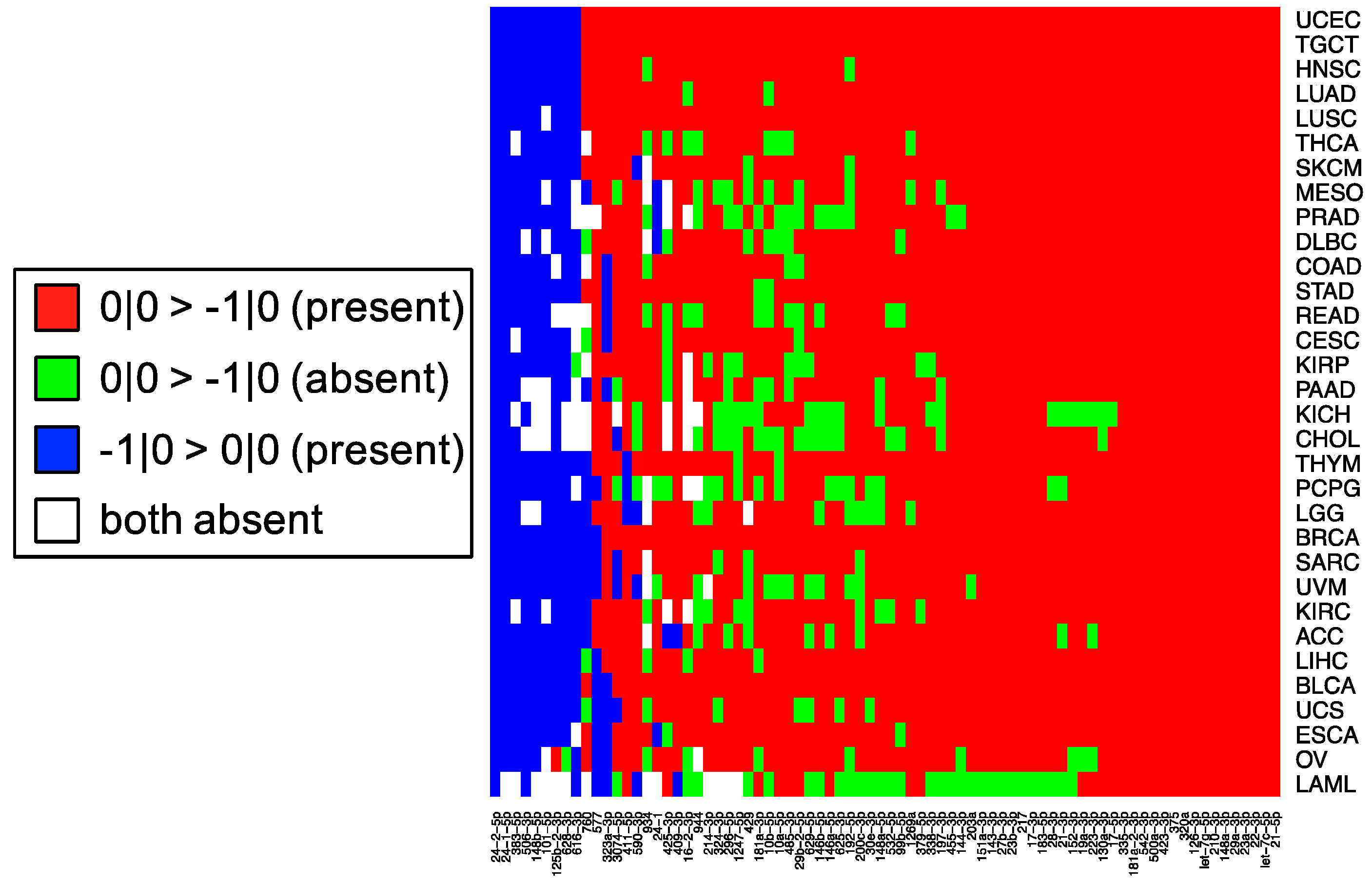
In this study, we sought to determine whether two commonly-used qPCR assays can discriminate amongst isoforms of the same miRNA. The two approaches tested here: LNA and Taqman qPCR: are widely used. Being able to efficiently and accurately quantify isomiR abundance is fast becoming important as researchers begin to delve into studies of the isomiRs’ functional and diagnostic capacities. To assess this discriminatory capacity, we characterized the ability of these methods to quantify differences in both 3′ and 5′ hsa-miR-21-5p isomiRs, when these isomiRs are in isolation and when these isomiRs are in combination in both synthetic and real cell contexts. miR-21 was chosen for this study, because it is ubiquitous and shows high expression across a wide range of tissue types (
Figure 1). miR-21-5p is also one of the most widely characterized miRNAs with reported functions in diverse contexts [
19]. The specific isomiRs of miR-21-5p were chosen for their high sequence similarity, because they represent the range of abundances (
Figure 1) over which miR-21-5p isomiRs are found, from most abundant to completely absent.
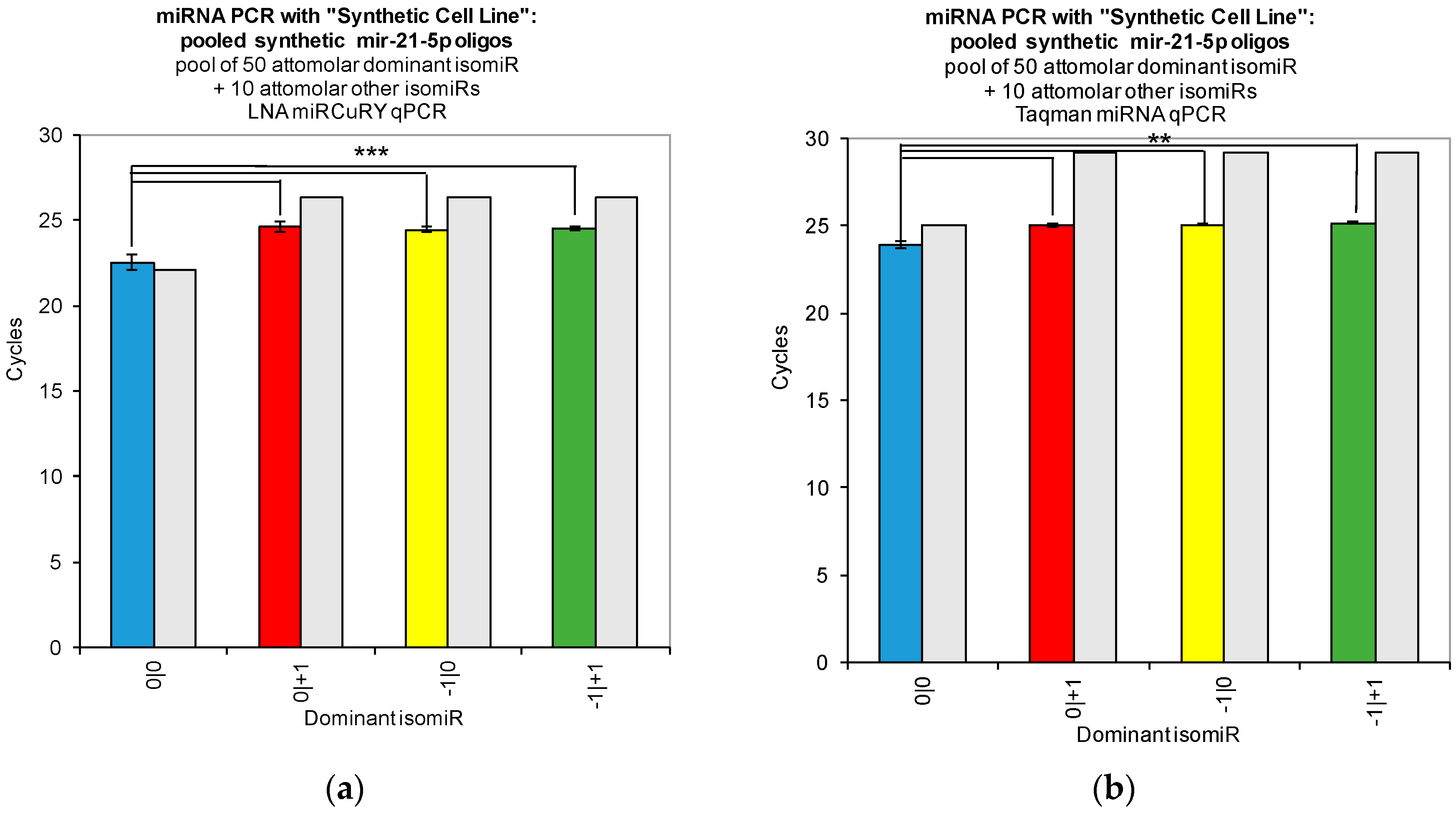
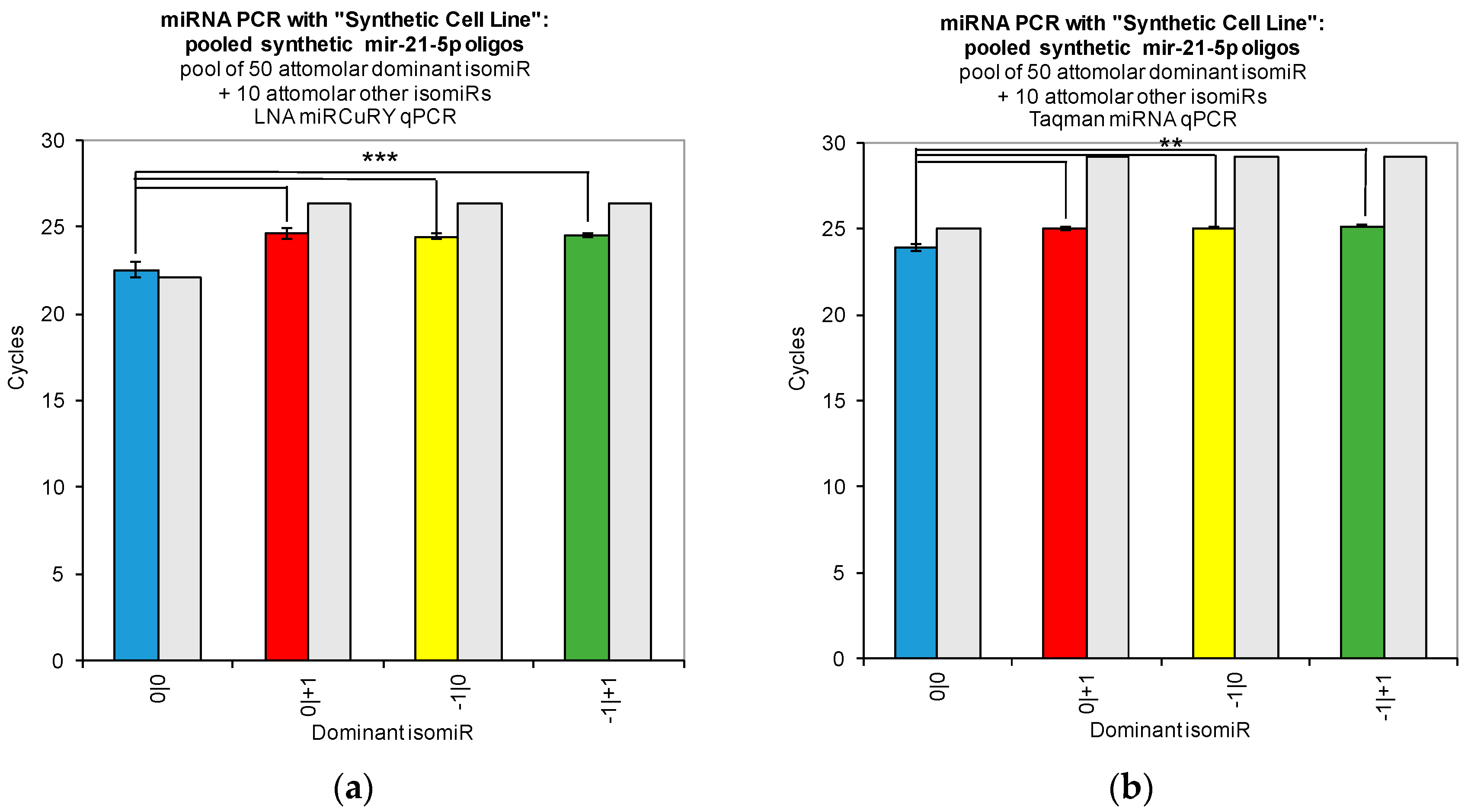
Our work reveals large variations in the assays’ ability to detect changes in the concentration of individual isomiRs (
Figure 2). Both tested qPCR assays showed greatest sensitivity for detection of the isomiR sequence for which they were originally designed (0|0 isomiR) (
Figure 2). In
Figure 2, we show that the two qPCR assays are able to detect concentration differences in the sequences for which they are designed, as evidenced by the fact that we were able to recover a standard curve for the hsa-miR-21-5p 0|0 synthetic RNA dilutions with each qPCR method. However, use of these probes to assay synthetic RNA representing any of the closely related isomiRs reveals that these structural variants do contribute to qPCR signals coming from these probes, as evidenced by the non-zero Ct signal observed for the alternate standard curves (
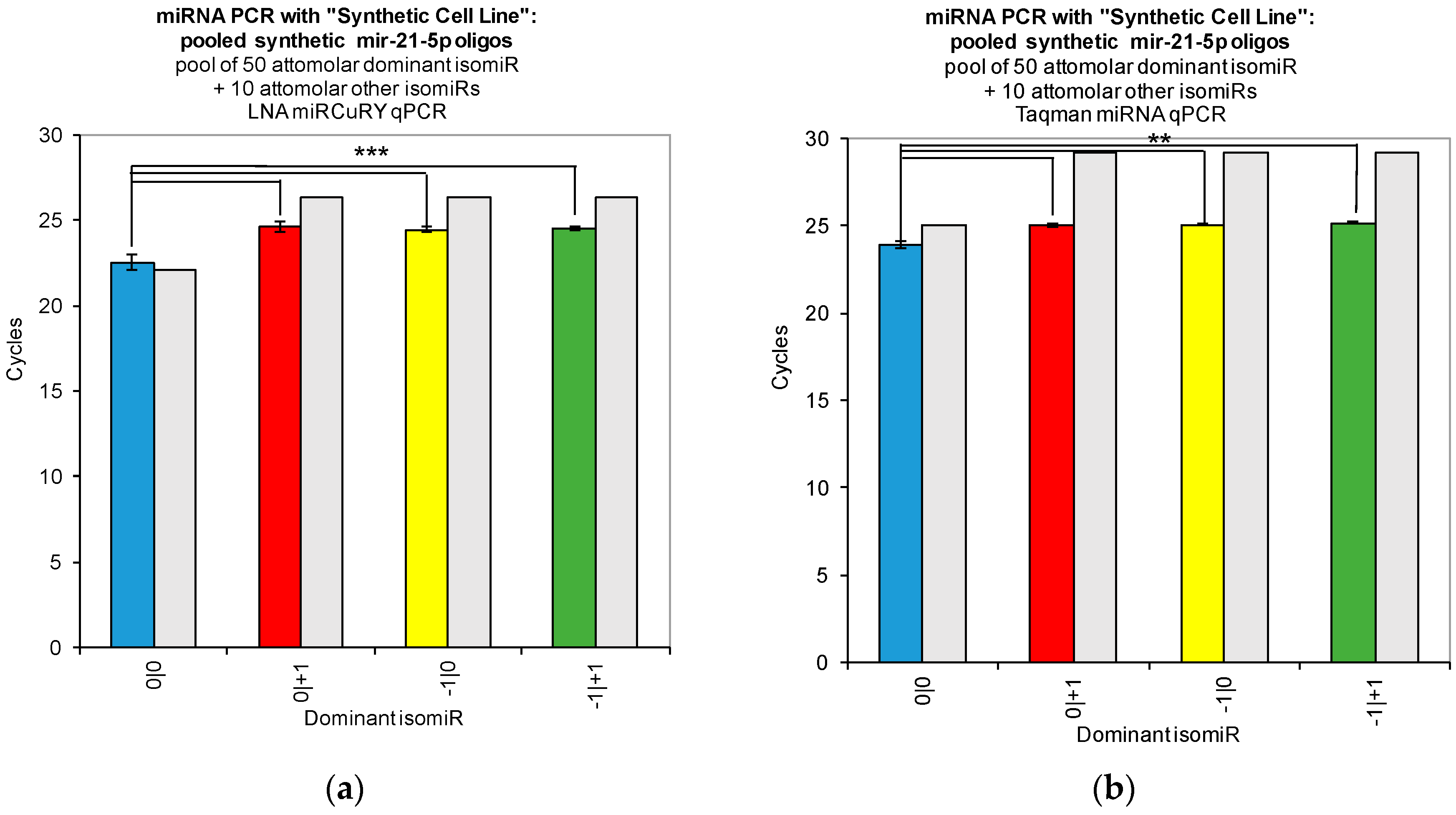
Figure 2). Moreover, when we combined multiple isomiRs into a “synthetic” cell line, we observed significant cross-reactivity between closely related isomiRs and the qPCR probes designed only for the canonical isomiR (0|0) (
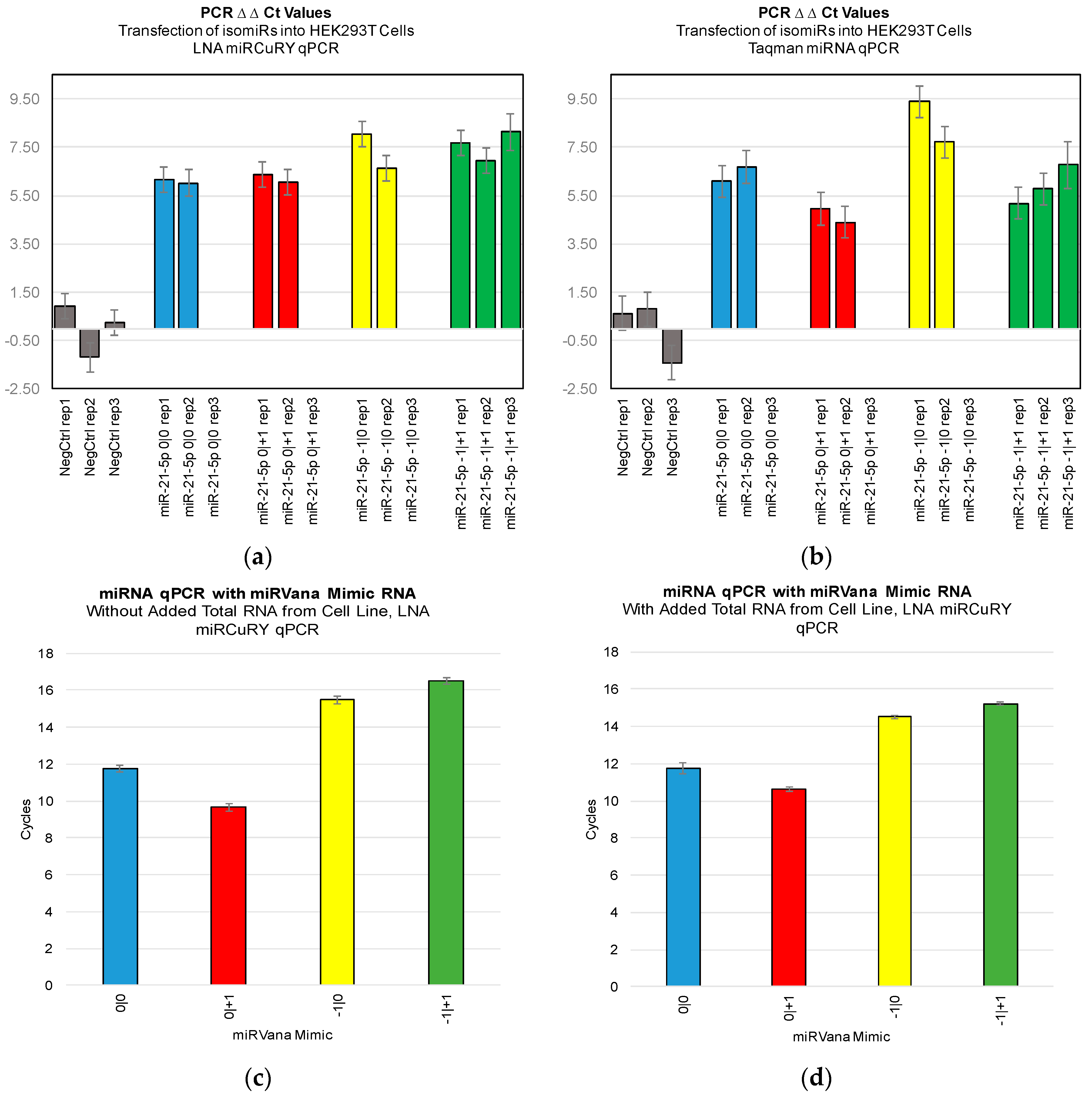
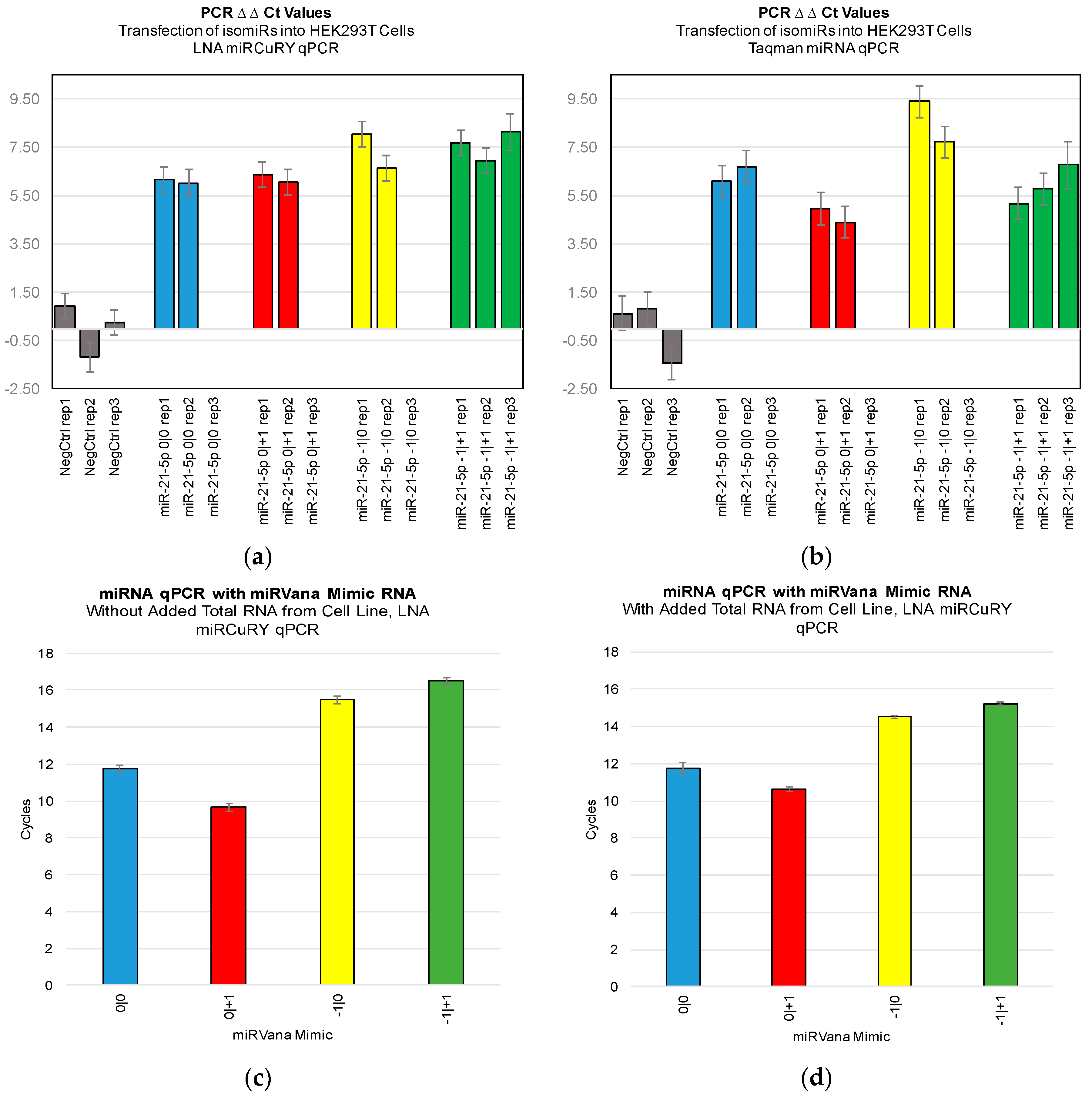
Figure 3). Furthermore, we were not able to discriminate cell line RNA in which we transfected 0|0 isomiR from those in which we transfected these same closely related isomiRs (
Figure 4a,b). Importantly, these differences can be attributed to detection of the transfected RNA, as qPCR of the transfected RNA alone: in the presence and absence of total RNA from cell lines: showed a high Ct signal for each of the four isomiRs (
Figure 4c,d).
Our findings suggest that traditional qPCR assays are not adequately specific to quantify distinct miRNA isomiRs. This should not be surprising. Both LNA and Taqman miRNA qPCR assays have been shown to be highly specific for miRNA isomiRs that differ on internal base pairs, such as the let-7 family. For example, recent work showed that these qPCR methods possess exceptional discriminatory capacity for internal sequence differences, giving rise to their ability to distinguish individual miRNAs in the whole cell milieu [
18]. We conjecture that this specificity arises from the fact that internal base differences are disruptive to probe-target interactions. However, when the differences lie solely at the ends of the target sequences, the full discriminatory power of these assays is diminished due to flexibility in the structures of target-probe pairs. Consequently, changes in the abundance of isomiR variants whose sequences differ only slightly from those of the archetype miRNA cannot be quantified using the current incarnation of these assays. Moreover, since the assays appear to be optimized for the 0|0 isomiR, any changes in the abundance of nearly-similar isomiRs will manifest themselves as “cross-talk” that will skew the quantification of the 0|0 isomiR. We believe these results hold for the qPCR probes designed for the 0|0 isomiR, and infer that qPCR probes designed for other isomiRs will exhibit the same tendency to cross-react, rendering testing of alternate probes superfluous.
This cross-reactivity is highlighted by the fact that the −1|+1 isomiR is not an endogenously expressed isomiR in any of the 32 TCGA tissues profiled (
Figure 1). Nonetheless, the qPCR probes tested are capable of detecting artificially elevated levels of this rare isomiR. In fact, a substantial background signal is observed in the 0|0 probe signal, both in synthetic and real cell contexts, before and after transfection of this isomiR (
Figure 2,
Figure 3 and
Figure 4). If one were to attempt to probe −1|+1 levels using a qPCR probe, in order to confirm its absence from a sample and thereby heighten suspicion of a specific disease (see [
16] for example), one would potentially encounter background signals that render the problem of confirming lack of a specific isomiR non-trivial. The reverse is true as well; in order to confirm the presence of a specific isomiR, one would need to be sure that any signal observed on qPCR captured the abundance of the isomiR itself, and not closely related sequences, which here are shown to produce non-trivial background noise.
These observations raise an important question for researchers: when preforming miRNA qPCR assays, what molecule is being profiled and what is that molecule’s true abundance? Our experiments indicate that individual LNA and Taqman probes will be influenced by “cross-talk” and in actuality, profile multiple isomiRs among those that are expressed from a distinct miRNA arm, rather than a single isomiR. As we have shown previously [
11] and in
Figure 5, the most abundant isomiR is not always the 0|0 isomiR, and in many cases, these differences will vary based upon the tissue type. Moreover, recent findings suggest that specific isomiRs will change in a manner that is correlated with an individual’s sex and race as well as the nature of the tissue at hand, the tissue’s state, and the disease subtype [
11,
16]. However, in light of our findings, we reason that these investigations would be exceptionally difficult to verify using these commercial qPCR methods.
IsomiRs have also recently been described in functional roles that are drastically different from those of the mature miRNA to which they are most closely related. We showed that isomiRs of miR-183-3p only share 15% of their differentially affected targets after transfection [
10]. Furthermore, other groups have elucidated functional roles for isomiRs of miR-140-3p [
13], miR-375 [
17] and miR-142-3p [
12] that differ from those roles established for the mature miRNA. Taken together, these observations have set the precedent that different isomiRs from the same locus will have functionally distinct roles. While much more work is needed to functionally evaluate the role of different isomiRs, being able to experimentally distinguish amongst them is of great importance. Our observations suggest that the two commercial qPCR technologies tested here have limited ability to distinguish amongst various isomiRs.
While our work reveals some of the limitations of commercial miRNA qPCR assays, one alternative approach to address this problem has been explored. This method, named “dumbbell PCR” (dbPCR) [
21], relies on the addition of a second stem-loop PCR step to the 5′-end of the isomiR, in order to improve the discriminatory capacity of the traditional Taqman qPCR method. While the authors do demonstrate the ability to detect both 5′ and 3′ isomiRs of miR-16, they do not compare their method to commercial qPCR assays. Such a method represents a promising tool for use in future experiments with isomiRs. However, despite the potential utility of dbPCR, it comes with the limitation of the added expense of custom-made qPCR probes and time to optimize the PCR reactions.
As the idea of functional roles of isomiRs gains traction, being able to accurately, specifically and inexpensively profile these molecules will become even more pressing. We recognize that sequencing approaches provide the technical specificity necessary to confidently capture sequences that differ in their 5′ or 3′ endpoints, but simultaneously understand that such approaches are not always feasible because of the high costs and relative difficulty in the analysis of the generated data. However, while we look forward to the next generation of short RNA qPCR methods, we believe that the safest current approach to the study of isomiRs is short RNA sequencing.
4. Materials and Methods
4.1. Synthetic RNAs
Four synthetic RNA oligonucleotides (Integrated DNA Technologies, Coralville, IA, USA), representing the four hsa-miR-21-5p isomiRs were designed (
Table 1). Each synthetic RNA oligonucleotide bore standard modifications as are seen in the cell: a 5′-phosphate group with a 3′-hydroxyl group. We created four “synthetic cell lines” from these isomiRs, by combining 50 microliters of a 10
–14 M starting concentration of the “dominant” isomiR together with 10 microliters of a 10
–14 M starting concentration of the other three isomiRs, in H
2O. We repeated this process an additional three times to create a synthetic cell line with each of the four isomiRs being “dominant” in turn.
4.2. qPCR Assays
We performed qPCR according to manufacturer specifications, using two popular qPCR methods: LNA miRCuRY (Exiqon, Woburn, MA, USA) and Taqman (ThermoFisher, Waltham, MA, USA). All assays were run on an Applied Biosystems StepOne qPCR machine. For both LNA and Taqman, we performed qPCR using the commercially available probe from each company that is marketed as detecting hsa-miR-21-5p. These probes are designed to detect the canonical hsa-miR-21-5p sequence, or the 0|0 isomiR.
For each qPCR assay, we performed a standard curve experiment of serial dilutions of each isomiR, from a starting concentration of 10–13 M (100 fm) RNA stepping by a dilution factor of 10 down to a concentration of 10–18 M (1 am). This range was selected to represent the physiological range of isomiR concentrations we have previously observed in sequencing data (not shown). For each synthetic oligo dilution, qPCR was run in triplicate from a single reverse transcriptase (RT) reaction.
LNA miRCuRY qPCR and Taqman miRNA qPCR were then repeated as above, using each of these synthetic cell lines as starting material and using the qPCR probe designed for the 0|0 isomiR in each case. Triplicate qPCR was run from a single RT starting sample.
4.3. Cell Culture and Transfection
To assess the discriminatory capacity of these assays in real cell contexts, we transfected mimics of each of the four isomiRs into HEK293T cells. Two of the mimics (hsa-miR-21-5p 0|0 and 0|+1) were purchased directly from ThermoFisher (ThermoFisher, Waltham, MA, USA), while the other two isomiRs (hsa-miR-21-5p −1|0 and −1|+1) were custom ordered. For comparison, we also transfected a short RNA negative control of a scrambled RNA sequence. Transfections were performed using Lipofectamine RNAi-Max. The cells were exposed to transfection reagents for 5 h prior to removing the transfection medium and replacing it with fresh Dulbecco Modified Eagle Medium (DMEM) supplemented with 10% FBS and penicillin/streptomycin. For each transfection, we used 1 microliter of 50 micromolar stock. After 48 h, we extracted RNA and performed LNA miRCuRY and Taqman miRNA assays as above. Transfection was performed in triplicate: each transfection yielded a single RT sample with which qPCR was run in triplicate. For normalization, we ran Taqman miRNA qPCR using a U6 probe (ThermoFisher, Waltham, MA, USA). We performed cell culture using standard techniques.
4.4. Short RNA-Seq Data Analysis
In order to discuss the implications of our qPCR findings, we downloaded and analyzed 10,274 short RNA-seq samples from TCGA, corresponding to samples that have not been marked for removal from one of the 32 TCGA projects due to incorrect filing (these samples are annotated as such in the file_annotations.txt file included with each download). We downloaded these data through the online data portal at:
http://gdc-portal.nci.nih.gov. We then computed isomiR expression using the distributed miRNA isoform data, as in [
11,
16], and further subjected the computed reads to Threshold-seq, in order to determine a dynamic threshold for inclusion of isomiRs in downstream analysis [
22]. Finally, we built expression matrices for isomiRs from each of the 32 TCGA projects, retaining only those isomiRs within a specific TCGA project that exceed Threshold-seq filtering level in at least one sample and retaining samples of any original type (specifically: tumor, normal, blood based, or metastasis). We subsequently computed the mean expression of isomiRs using these matrices, and plotted the results using the heatmap.2() function of the gplots package in R.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}