

Clarithromycin and Pantoprazole Gastro-Retentive Floating Bilayer Tablet for the Treatment of Helicobacter Pylori: Formulation and Characterization

 ,
,  , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
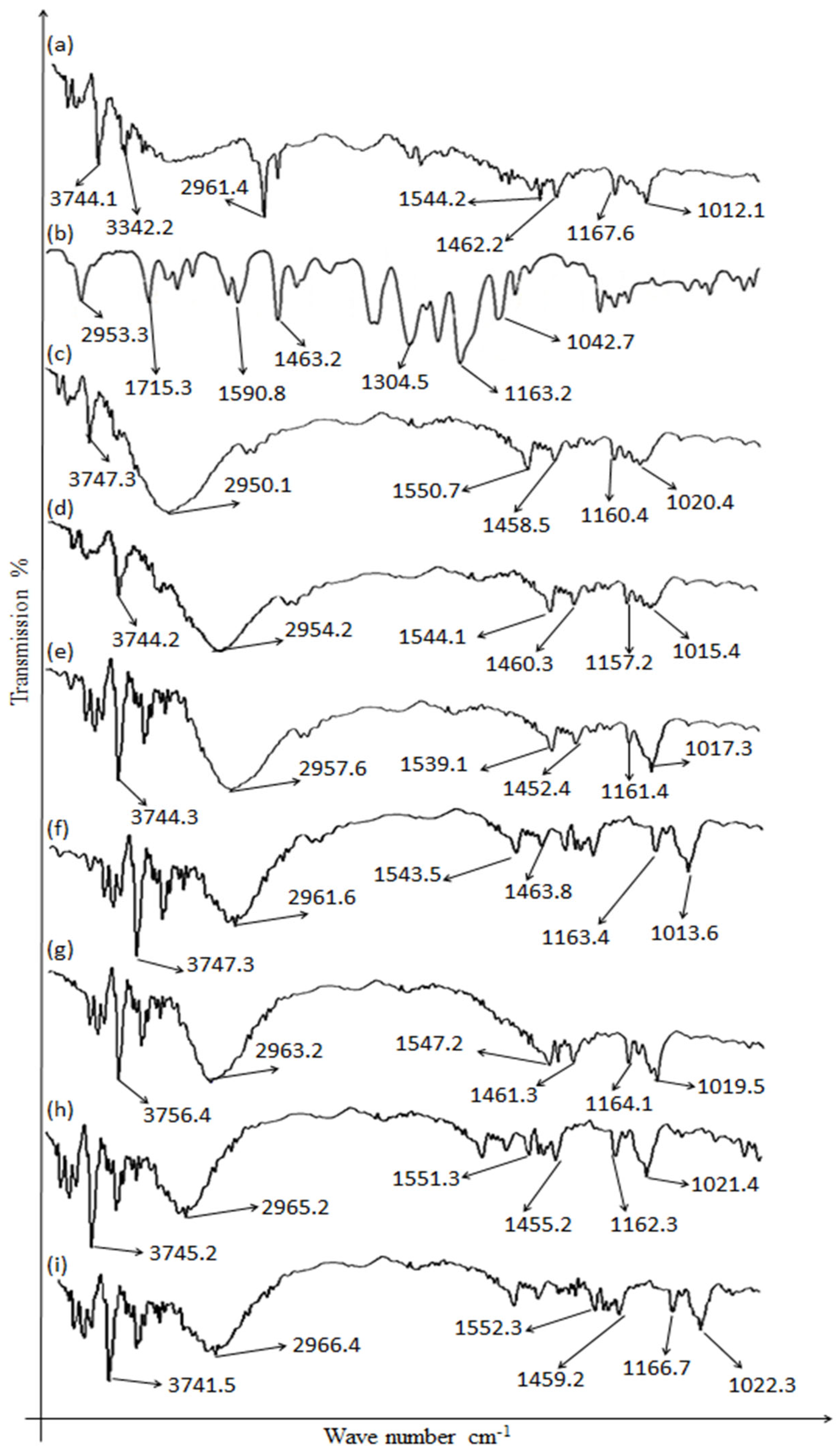
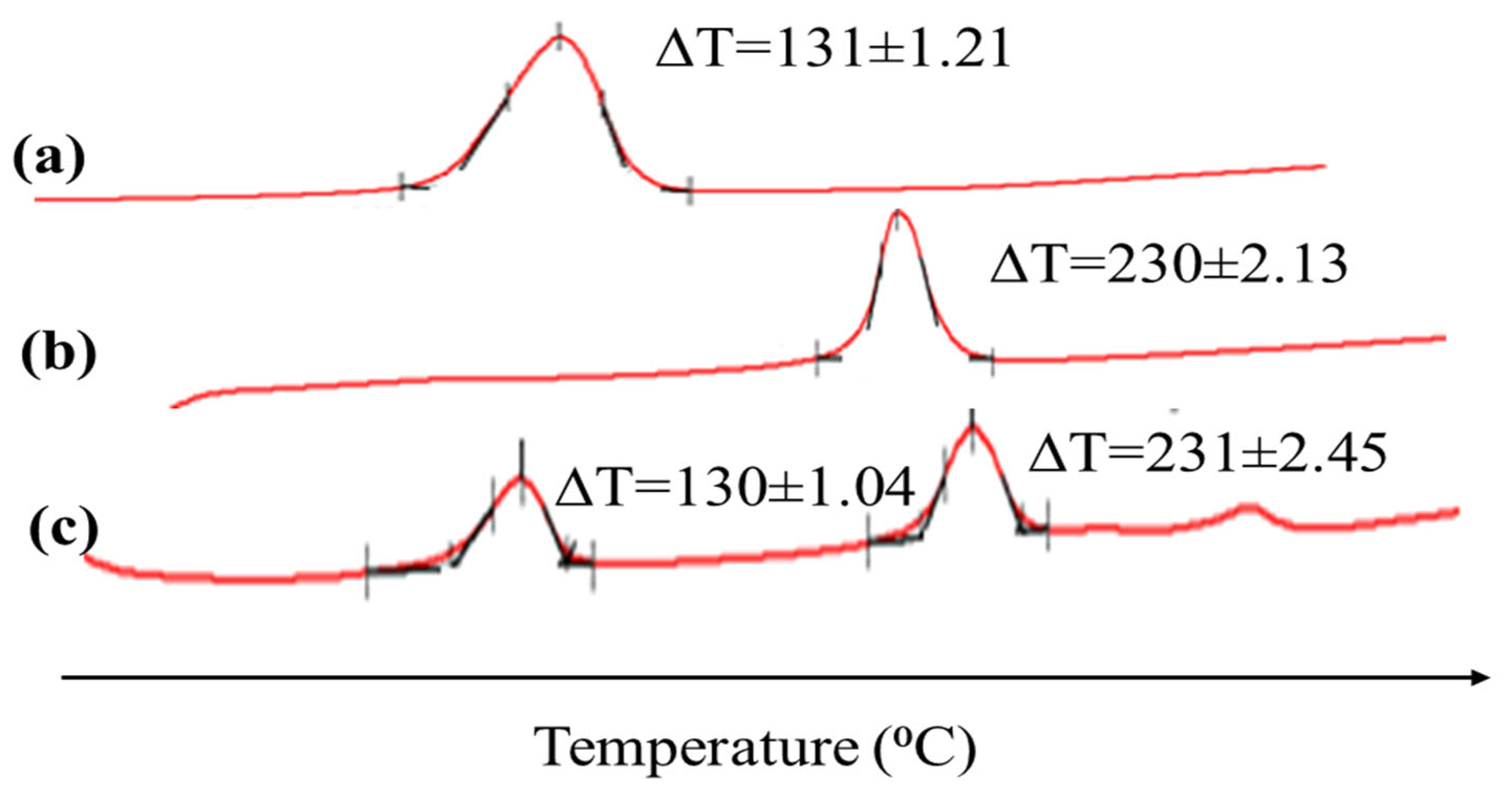
2.1. Drugs’ and Excipients’ Compatibility
2.2. Flow Behavior
2.3. Post-Compression Studies
2.3.1. Hardness
2.3.2. Uniformity of Weight
2.4. Uniformity of Drug Content
2.4.1. Dimensions
2.4.2. Friability
2.4.3. Tablet Floating Capacity
2.5. Swelling and Erosion Studies
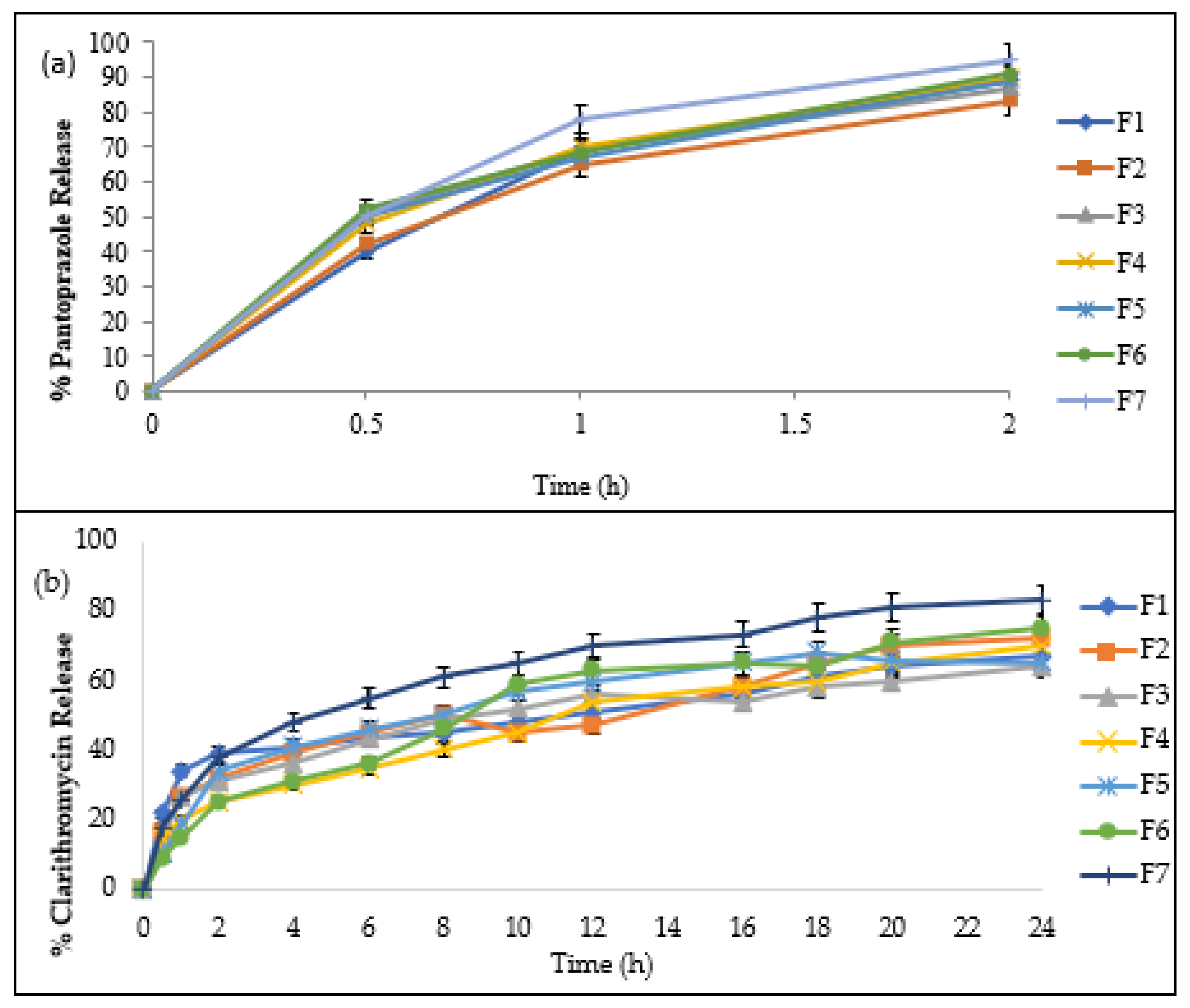
2.6. In Vitro Release of Drug
2.7. Release Kinetics of Drug
2.8. Stability Studies
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Formulation of Gastro Floating Bilayer Matrix Tablets
4.2.1. Preparation of Immediate-Release Layer of Pantoprazole Sodium
4.2.2. Sustained-Release Layer of Clarithromycin
4.3. Preparation of Bilayer Tablets
4.4. Pre-Compression Studies
4.4.1. Angle of Repose
4.4.2. Bulk Density
4.4.3. Tapped Density
4.4.4. Carr’s Compressibility Index
4.4.5. Hausner’s Ratio
4.5. Post-Compression Study
4.5.1. Hardness Test
4.5.2. Weight Variation
4.5.3. Tablets’ Dimensions
4.5.4. Friability
4.5.5. Floating Lag Time and Floating Time
4.6. Characterization of Drug and Excipients
4.6.1. Attenuated Total Reflectance/Fourier Transform Infrared Spectroscopy
4.6.2. Differential Scanning Calorimetry Analysis
4.7. Drug Content Uniformity
4.7.1. Drug Content of Clarithromycin
Procedure for Evaluating Drug Content
4.7.2. Drug Content of Pantoprazole
4.8. Swelling and Erosion Studies
4.9. SEM Analysis
4.10. In Vitro Release of Drug
4.11. Release Kinetics
4.11.1. Zero-Order Kinetics
4.11.2. First-Order Kinetics
4.11.3. Hixon–Crowell Model
4.11.4. Higuchi Model
4.11.5. Korsmeyer–Peppas model
4.12. Stability Studies
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patil, T.; Pawar, A.; Korake, S.; Patil, R.; Pawar, A.; Kamble, R. Green synthesis of polyacrylamide grafted Neem Gum for gastro retentive floating drug delivery of Ciprofloxacin Hydrochloride: In vitro and in vivo evaluation. J. Drug Deliv. Sci. Technol. 2022, 72, 103417. [Google Scholar] [CrossRef]
- Karole, A.; Parvez, S.; Thakur, R.S.; Mudavath, S.L. Effervescent based nano-gas carrier enhanced the bioavailability of poorly aqueous soluble drug: A comprehensive mechanistic understanding. J. Drug Deliv. Sci. Technol. 2022, 69, 103167. [Google Scholar] [CrossRef]
- Tripathi, J.; Thapa, P.; Maharjan, R.; Jeong, S.H. Current State and Future Perspectives on Gastroretentive Drug Delivery Systems. Pharmaceutics 2019, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Vrettos, N.-N.; Roberts, C.J.; Zhu, Z. Gastroretentive Technologies in Tandem with Controlled-Release Strategies: A Potent Answer to Oral Drug Bioavailability and Patient Compliance Implications. Pharmaceutics 2021, 13, 1591. [Google Scholar] [CrossRef] [PubMed]
- Kotreka, U.K.; Adeyeye, M.C. Gastroretentive floating drug-delivery systems: A critical review. Crit. Rev. Ther. Drug Carr. Syst. 2011, 28, 47–99. [Google Scholar] [CrossRef]
- Pawar, V.K.; Kansal, S.; Garg, G.; Awasthi, R.; Singodia, D.; Kulkarni, G.T. Gastroretentive dosage forms: A review with special emphasis on floating drug delivery systems. Drug Deliv. 2011, 18, 97–110. [Google Scholar] [CrossRef]
- Jin, G.; Ngo, H.V.; Wang, J.; Cui, J.-H.; Cao, Q.-R.; Park, C.; Jung, M.; Lee, B.-J. Design and evaluation of in vivo bioavailability in beagle dogs of bilayer tablet consisting of immediate release nanosuspension and sustained release layers of rebamipide. Int. J. Pharm. 2022, 619, 121718. [Google Scholar] [CrossRef]
- Maddiboyina, B.; Hanumanaik, M.; Nakkala, R.K.; Jhawat, V.; Rawat, P.; Alam, A.; Foudah, A.I.; Alrobaian, M.M.; Shukla, R.; Singh, S.; et al. Formulation and evaluation of gastro-retentive floating bilayer tablet for the treatment of hypertension. Heliyon 2020, 6, e05459. [Google Scholar] [CrossRef]
- Efentakis, M.; Naseef, H.; Vlachou, M. Two- and three-layer tablet drug delivery systems for oral sustained release of soluble and poorly soluble drugs. Drug Dev. Ind. Pharm. 2010, 36, 903–916. [Google Scholar] [CrossRef]
- Parsa, H.; Zangivand, A.A.; Hajimaghsoudi, L. The effect of pentoxifylline on chronic venous ulcers. Wounds Compend. Clin. Res. Pract. 2012, 24, 190–194. [Google Scholar]
- Cui, G.; Yuan, A.; Li, Z. Occurrences and phenotypes of RIPK3-positive gastric cells in Helicobacter pylori infected gastritis and atrophic lesions. Dig. Liver Dis. 2022, 54, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Valladales-Restrepo, L.F.; Correa-Sánchez, Y.; Aristizábal-Carmona, B.S.; Machado-Alba, J.E. Treatment regimens used in the management of Helicobacter pylori in Colombia. Braz. J. Infect. Dis. 2022, 26, 102331. [Google Scholar] [CrossRef] [PubMed]
- Hussein, R.A.; Al-Ouqaili, M.T.S.; Majeed, Y.H. Detection of clarithromycin resistance and 23SrRNA point mutations in clinical isolates of Helicobacter pylori isolates: Phenotypic and molecular methods. Saudi J. Biol. Sci. 2021, 29, 513–520. [Google Scholar] [CrossRef]
- Rodvold, K.A. Clinical Pharmacokinetics of Clarithromycin. Clin. Pharmacokinet. 1999, 37, 385–398. [Google Scholar] [CrossRef]
- Jain, N.; Jain, P.; Parmar, T.; Parkhe, G.; Jain, S.K. Preparation and Evaluation of Bi-Layer Tablets of Pantorazole and Clarithromycin. J. Pharm. Educ. Res. 2016, 5, 75–85. [Google Scholar]
- Peppas, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Solunke, R.S.; Khade, G.A.; Krishnmurthy; Deshmukh, M.T.; Shete, R.V.; Kore, K.J. Development and Evaluation of Pantoprazole Sodium Floating Gel. Res. J. Pharm. Technol. 2020, 13, 682. [Google Scholar] [CrossRef]
- Guarnizo-Herrero, V.; Torrado-Salmerón, C.; Pabón, N.S.T.; Durán, G.T.; Morales, J.; Torrado-Santiago, S. Study of Different Chitosan/Sodium Carboxymethyl Cellulose Proportions in the Development of Polyelectrolyte Complexes for the Sustained Release of Clarithromycin from Matrix Tablets. Polymers 2021, 13, 2813. [Google Scholar] [CrossRef]
- Prakash, G.; Chandra, S.A.; Sandhya, P.; Bidur, C.; Samir, D. Pharmacopoeial comparison of in-process and finished product quality control test for pharmaceutical tablets. GSC Biol. Pharm. Sci. 2020, 11, 155–165. [Google Scholar] [CrossRef]
- British Pharmacopoeia. Her Majesty’s Stationery Office; Atlantic House: London, UK, 2009; Volume 1, p. 2011416. [Google Scholar]
- Patel, S.S.; Patel, N.M. Flowability Testing of Directly Compressible Excipients According to British Pharmacopoeia. J. Pharm. Res. 2009, 8, 66. [Google Scholar] [CrossRef]
- Kumar, V.S.; Rijo, J.; Sabitha, M. Guargum and Eudragit® coated curcumin liquid solid tablets for colon specific drug delivery. Int. J. Biol. Macromol. 2018, 110, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Dev, D.; Prasad, D.N.; Hans, M. Sustained release drug delivery system with the role of natural polymers: A review. J. Drug Deliv. Ther. 2019, 9, 913–923. [Google Scholar]
- Naseem, F.; Shah, S.U.; Rashid, S.A.; Farid, A.; Almehmadi, M.; Alghamdi, S. Metronidazole Based Floating Bioadhesive Drug Delivery System for Potential Eradication of H. pylori: Preparation and In Vitro Characterization. Polymers 2022, 14, 519. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Jeong, S.H. Effects of Formulation and Process Variables on Gastroretentive Floating Tablets with A High-Dose Soluble Drug and Experimental Design Approach. Pharmaceutics 2018, 10, 161. [Google Scholar] [CrossRef]
- Sharma, V.; Devi, J. Design of Cellulose Derivative and Alginate Based Smart Polymers to Develop Stomach Specific Floating Drug Delivery System. Curr. Smart Mater. 2021, 5, 129–149. [Google Scholar] [CrossRef]
- Mittal, S.; Pawar, S. Design and evaluation of buccal mucoadhesive tablets of pantoprazole sodium. Eur. J. Pharm. Med. Res. 2018, 5, 514–522. [Google Scholar]
- Rajeswari, S.; Kudamala, S.; Murthy, K.V.R. Development, Formulation and Evaluation of A Bilayer Gastric Retentive Floating Tablets of Ranitidine Hcl and Clarithromycin using Natural Polymers. Int. J. Pharm. Pharm. Sci. 2017, 9, 164–177. [Google Scholar] [CrossRef][Green Version]
- Alam, M.; Shah, K.U.; Khan, K.A.; Nawaz, A.; Bibi, H.; Razaque, G.; Niazi, Z.R.; Alfatama, M. Formulation and in vitro Evaluation of Effervescent Bilayer Floating Controlled Release Tablets of Clarithromycin and Famotidine. Res. J. Pharm. Technol. 2021, 14, 4391–4398. [Google Scholar] [CrossRef]
- Müller, D.; Fimbinger, E.; Brand, C. Algorithm for the determination of the angle of repose in bulk material analysis. Powder Technol. 2021, 383, 598–605. [Google Scholar] [CrossRef]
- Kamal, M.M.; Salawi, A.; Lam, M.; Nokhodchi, A.; Abu-Fayyad, A.; El Sayed, K.A.; Nazzal, S. Development and characterization of curcumin-loaded solid self-emulsifying drug delivery system (SEDDS) by spray drying using Soluplus® as solid carrier. Powder Technol. 2020, 369, 137–145. [Google Scholar] [CrossRef]
- Chaudhari, K.D.; Nimbalwar, M.G.; Singhal, N.S.; Panchale, W.A.; Manwar, J.V.; Bakal, R.L. Comprehensive review on characterizations and application of gastro-retentive floating drug delivery system. GSC Adv. Res. Rev. 2021, 7, 35–44. [Google Scholar] [CrossRef]
- Özalp, Y.; Onayo, M.M.; Nailla, J. Evaluation of lactose-based direct tabletting agents’ compressibility behaviour using a compaction simulator. Turk. J. Pharm. Sci. 2020, 17, 367. [Google Scholar] [CrossRef]
- Venkatesh, D.N.; Meyyanathan, S.; Shanmugam, R.; Zielinska, A.; Campos, J.; Ferreira, J.; Souto, E. Development, in vitro release and in vivo bioavailability of sustained release nateglinide tablets. J. Drug Deliv. Sci. Technol. 2019, 55, 101355. [Google Scholar] [CrossRef]
- Thakur, S.; Ramya, K.; Shah, D.K.; Raj, K. Floating Drug Delivery System. J. Drug Deliv. Ther. 2021, 11, 125–130. [Google Scholar] [CrossRef]
- Rao, G.K.; Mandapalli, P.K.; Manthri, R.; Reddy, V.P. Development and in vivo evaluation of gastroretentive delivery systems for cefuroxime axetil. Saudi Pharm. J. 2013, 21, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Bukhari, N.I.; Karim, S.; Hafiz, M.A.; Hayat, U. Floating tablets of minocycline hydrochloride: Formulation, in-vitro evaluation and optimization. Future J. Pharm. Sci. 2017, 3, 131–139. [Google Scholar] [CrossRef]
- Yin, L.; Qin, C.; Chen, K.; Zhu, C.; Cao, H.; Zhou, J.; He, W.; Zhang, Q. Gastro-floating tablets of cephalexin: Preparation and in vitro/in vivo evaluation. Int. J. Pharm. 2013, 452, 241–248. [Google Scholar] [CrossRef]
- Sharaf, M.; Arif, M.; Khan, S.; Abdalla, M.; Shabana, S.; Chi, Z.; Liu, C. Co-delivery of hesperidin and clarithromycin in a nanostructured lipid carrier for the eradication of Helicobacter pylori In vitro. Bioorganic Chem. 2021, 112, 104896. [Google Scholar] [CrossRef]
- Latha, S.; Selvamani, P.; Suganya, G.; Raj, D.B.T.G.; Pal, T.K. Preparation and In-vitro Evaluation of Pantoprazole Sodium Magnetic Microspheres by Emulsion Solvent Evaporation Method. BioNanoScience 2021, 11, 643–647. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
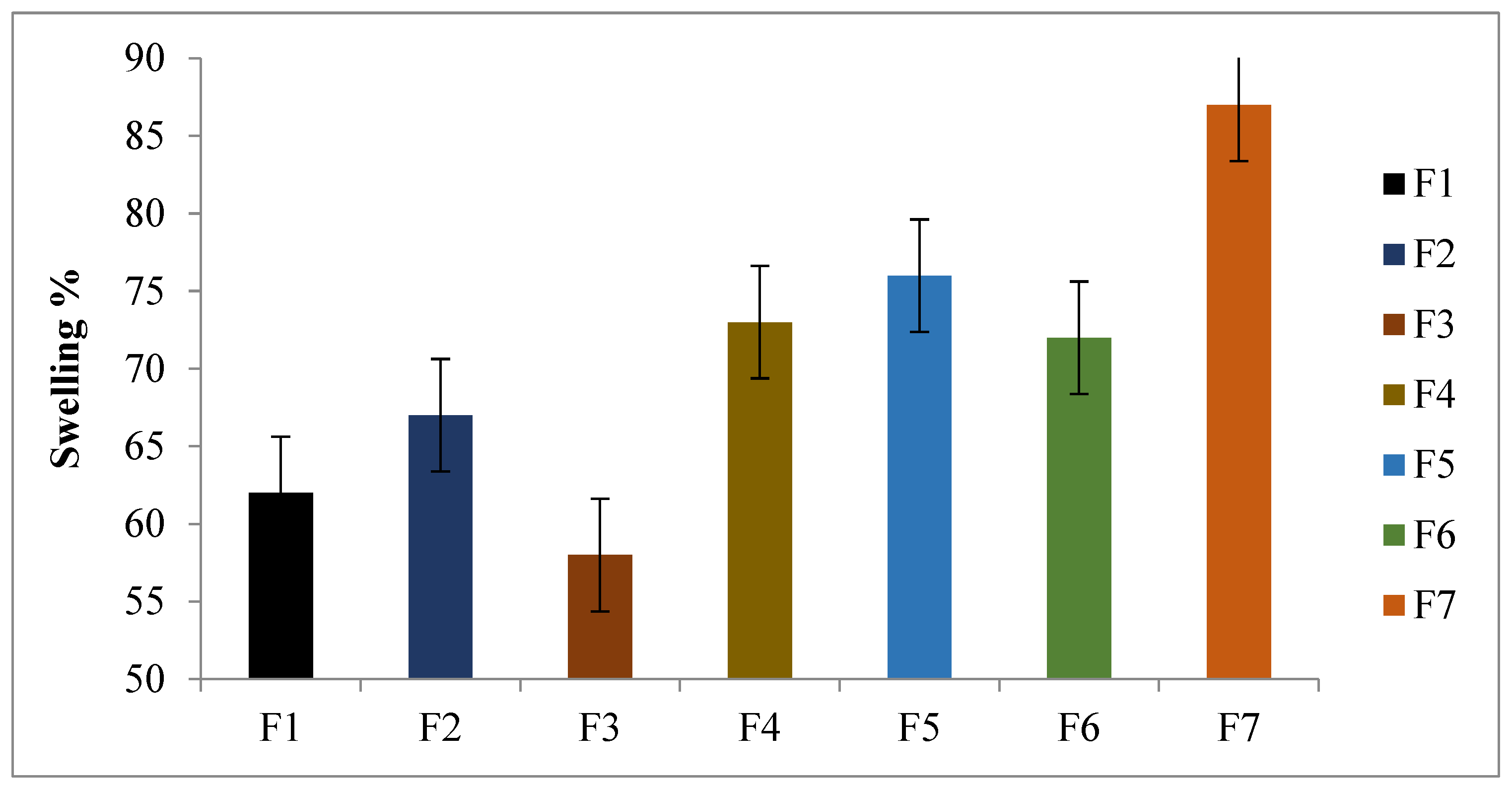{kind=link}
{kind=link}
{kind=link}
| F. Layer-IR | Bulk Density (mg/cm3) | Tapped Density (mg/cm3) | Compressibility Index (%) | Hausner’s Ratio | Angle of Repose (θ) |
|---|---|---|---|---|---|
| F1 IR | 0.862 ± 0.021 | 0.940 ± 0.036 | 8.298 ± 0.0122 | 1.09 ± 0.031 | 29.47 |
| F2 IR | 0.868 ± 0.007 | 0.937 ± 0.028 | 7.364 ± 0.0115 | 1.08 ± 0.034 | 29.62 |
| F3 IR | 0.871 ± 0.030 | 0.936 ± 0.023 | 6.944 ± 0.025 | 1.075 ± 0.078 | 29.70 |
| F1 FL | 0.797 ± 0.023 | 0.921 ± 0.005 | 13.46 ± 0.018 | 1.156 ± 0.012 | 29.71 |
| F2 FL | 0.783 ± 0.082 | 0.919 ± 0.014 | 14.80 ± 0.0115 | 1.174 ± 0.043 | 29.42 |
| F3 FL | 0.819 ± 0.029 | 0.899 ± 0.070 | 8.899 ± 0.0173 | 1.098 ± 0.056 | 30.14 |
| F4 FL | 0.833 ± 0.018 | 0.961 ± 0.037 | 13.32 ± 0.0015 | 1.154 ± 0.041 | 30.53 |
| F5 FL | 0.784 ± 0.031 | 0.865 ± 0.009 | 9.364 ± 0.052 | 1.103 ± 0.032 | 30.02 |
| F6 FL | 0.850 ± 0.023 | 0.941 ± 0.011 | 9.671 ± 0.001 | 1.107 ± 0.019 | 30.31 |
| F7 FL | 0.847 ± 0.016 | 0.925 ± 0.081 | 8.432 ± 0.026 | 1.092 ± 0.063 | 30.13 |
| F. Code | Thickness (mm) | Hardness (N/cm2) | Weight Uniformity (mg) | Friability (%) | Drug Content (%) | |
|---|---|---|---|---|---|---|
| Clarithromycin | Pantoprazole | |||||
| F1 | 2.75 ± 0.1 | 68 ± 1.2 | 599.1 ± 2.4 | 0.73 ± 0.02 | 98.2 ± 1.3 | 100.5 ± 1.5 |
| F2 | 2.68 ± 0.3 | 69 ± 0.4 | 601.7 ± 3.1 | 0.79 ± 0.05 | 99.1 ± 1.1 | 97.9 ± 1.4 |
| F3 | 2.65 ± 0.1 | 68 ± 1.1 | 598.3 ± 3.2 | 0.64 ± 0.04 | 98.7 ± 1.4 | 99.3 ± 1.7 |
| F4 | 2.73 ± 0.2 | 73 ± 0.9 | 604.1 ± 1.6 | 0.58 ± 0.09 | 100.1 ± 1.6 | 98.1 ± 1.3 |
| F5 | 2.69 ± 0.3 | 75 ± 1.3 | 602.8 ± 1.9 | 0.79 ± 0.05 | 97.9 ± 1.8 | 99.8 ± 1.2 |
| F6 | 2.70 ± 0.4 | 71 ± 1.2 | 600.5 ± 1.1 | 0.72 ± 0.04 | 99.6 ± 1.5 | 98.5 ± 1.6 |
| F7 | 2.68 ± 0.3 | 72 ± 1.2 | 602.3 ± 2.0 | 0.63 ± 0.07 | 100.7 ± 1.1 | 97.4 ± 1.8 |
| F. Codes | Floating Lag Time (min:s) | Total Floating Duration (h) |
|---|---|---|
| F1 | 0:24 ± 0.17 | 18 |
| F2 | 0:16 ± 0.13 | 17 |
| F3 | 0:43 ± 0.10 | 18 |
| F4 | 0:12 ± 0.06 | 18 |
| F5 | 0:39 ± 0.15 | 22 |
| F6 | 1:38 ± 0.14 | 23 |
| F7 | 1:01 ± 0.14 | 24 |
| F. Code | Zero-Order | 1st-Order | Higuchi | Hixon–Crowell | Korsmeyer–Peppas | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| K ± SD | R2 | K ± SD | R2 | K ± SD | R2 | K ± SD | R2 | K ± SD | R2 | n | |
| F1 | 1.13 ± 0.113 | 0.976 | 1.32 ± 0.132 | 0.534 | 2.29 ± 0.132 | 0.954 | 1.54 ± 0.134 | 0.992 | 0.143 ± 0.034 | 0.975 | 0.61 |
| F2 | 1.32 ± 0.119 | 0.987 | 1.13 ± 0.167 | 0.643 | 1.43 ± 0.243 | 0.954 | 1.34 ± 0.232 | 0.978 | 0.162 ± 0.332 | 0.981 | 0.61 |
| F3 | 1.14 ± 0.142 | 0.967 | 1.19 ± 0.132 | 0.623 | 1.54 ± 0.423 | 0.976 | 1.65 ± 0.154 | 0.989 | 0.045 ± 0.343 | 0.970 | 0.49 |
| F4 | 1.17 ± 0.114 | 0.976 | 1.32 ± 0.176 | 0.554 | 1.24 ± 1.254 | 0.945 | 1.23 ± 0.234 | 0.978 | 0.065 ± 0.123 | 0.984 | 0.515 |
| F5 | 1.28 ± 0.128 | 0.979 | 1.28 ± 0.121 | 0.525 | 2.32 ± 0.034 | 0.976 | 1.54 ± 0.245 | 0.986 | 0.056± 0.045 | 0.981 | 0.535 |
| F6 | 1.26 ± 0.126 | 0.987 | 1.19 ± 0.134 | 0.643 | 1.54 ± 0.132 | 0.923 | 1.25 ± 0.254 | 0.987 | 0.112 ± 0.234 | 0.980 | 0.49 |
| F7 | 1.18 ± 0.229 | 0.983 | 1.32 ± 0.122 | 0.648 | 1.13 ± 0.124 | 0.987 | 1.54 ± 0.165 | 0.990 | 0.054 ± 0.316 | 0.982 | 0.553 |
| Time (Month) | Evaluation Parameters | |||||
|---|---|---|---|---|---|---|
| Drug Content | FLT (s) | TFT (h) | % Drug Release | |||
| Cln | Ptz | Cln | Ptz | |||
| 0 | 100.7 ± 1.1 | 97.4 ± 1.8 | 60 | >24 | 99.4 | 97.3 |
| 1 | 99.5 ± 1.3 | 97.1 ± 1.4 | 58 | >24 | 99.3 | 96.5 |
| 2 | 99.3 ± 0.9 | 96.6 ± 1.1 | 55 | >24 | 98.7 | 95.2 |
| 3 | 99.1 ± 1.5 | 96.2 ± 2.1 | 59 | >24 | 99.1 | 96.4 |
| Ingredients | F1 IR (mg) | F2 IR (mg) | F3 IR (mg) |
|---|---|---|---|
| Pantoprazole sodium | 40 | 40 | 40 |
| Sodium starch glycolate (Primojel) | 05 | 10 | 15 |
| Avicel 102 | 44 | 39 | 34 |
| Magnesium stearate | 05 | 05 | 05 |
| Talcum powder | 06 | 06 | 06 |
| Total weight | 100 | 100 | 100 |
| Ingredients (mg) | F1 CR | F2 CR | F3 CR | F4 CR | F5 CR | F6 CR | F7 CR |
|---|---|---|---|---|---|---|---|
| Clarithromycin HCl | 250 | 250 | 250 | 250 | 250 | 250 | 250 |
| Chitosan | 150 | --- | --- | 75 | --- | 75 | 50 |
| HPMC K15M | --- | 150 | --- | 75 | 75 | --- | 50 |
| Sodium alginate | --- | --- | 150 | --- | 75 | 75 | 50 |
| NaHCO3 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| PVP K30 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Talcum powder | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Magnesium stearate | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Avicel 102 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| Weight/tablet | 500 | 500 | 500 | 500 | 500 | 500 | 500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, G.; Nawaz, A.; Latif, M.S.; Shah, K.U.; Ahmad, S.; Javed, F.; Alfatama, M.; Abd Ghafar, S.A.; Lim, V. Clarithromycin and Pantoprazole Gastro-Retentive Floating Bilayer Tablet for the Treatment of Helicobacter Pylori: Formulation and Characterization. Gels 2023, 9, 43. https://doi.org/10.3390/gels9010043
Ullah G, Nawaz A, Latif MS, Shah KU, Ahmad S, Javed F, Alfatama M, Abd Ghafar SA, Lim V. Clarithromycin and Pantoprazole Gastro-Retentive Floating Bilayer Tablet for the Treatment of Helicobacter Pylori: Formulation and Characterization. Gels. 2023; 9(1):43. https://doi.org/10.3390/gels9010043
Chicago/Turabian StyleUllah, Ghufran, Asif Nawaz, Muhammad Shahid Latif, Kifayat Ullah Shah, Saeed Ahmad, Fatima Javed, Mulham Alfatama, Siti Aisyah Abd Ghafar, and Vuanghao Lim. 2023. "Clarithromycin and Pantoprazole Gastro-Retentive Floating Bilayer Tablet for the Treatment of Helicobacter Pylori: Formulation and Characterization" Gels 9, no. 1: 43. https://doi.org/10.3390/gels9010043
APA StyleUllah, G., Nawaz, A., Latif, M. S., Shah, K. U., Ahmad, S., Javed, F., Alfatama, M., Abd Ghafar, S. A., & Lim, V. (2023). Clarithromycin and Pantoprazole Gastro-Retentive Floating Bilayer Tablet for the Treatment of Helicobacter Pylori: Formulation and Characterization. Gels, 9(1), 43. https://doi.org/10.3390/gels9010043