
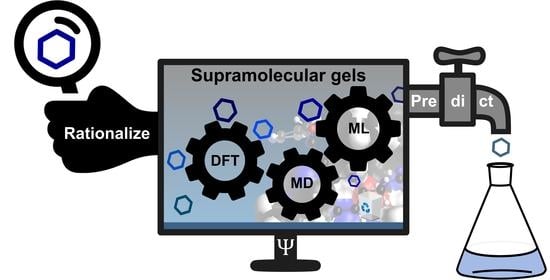
Computational Tools to Rationalize and Predict the Self-Assembly Behavior of Supramolecular Gels



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
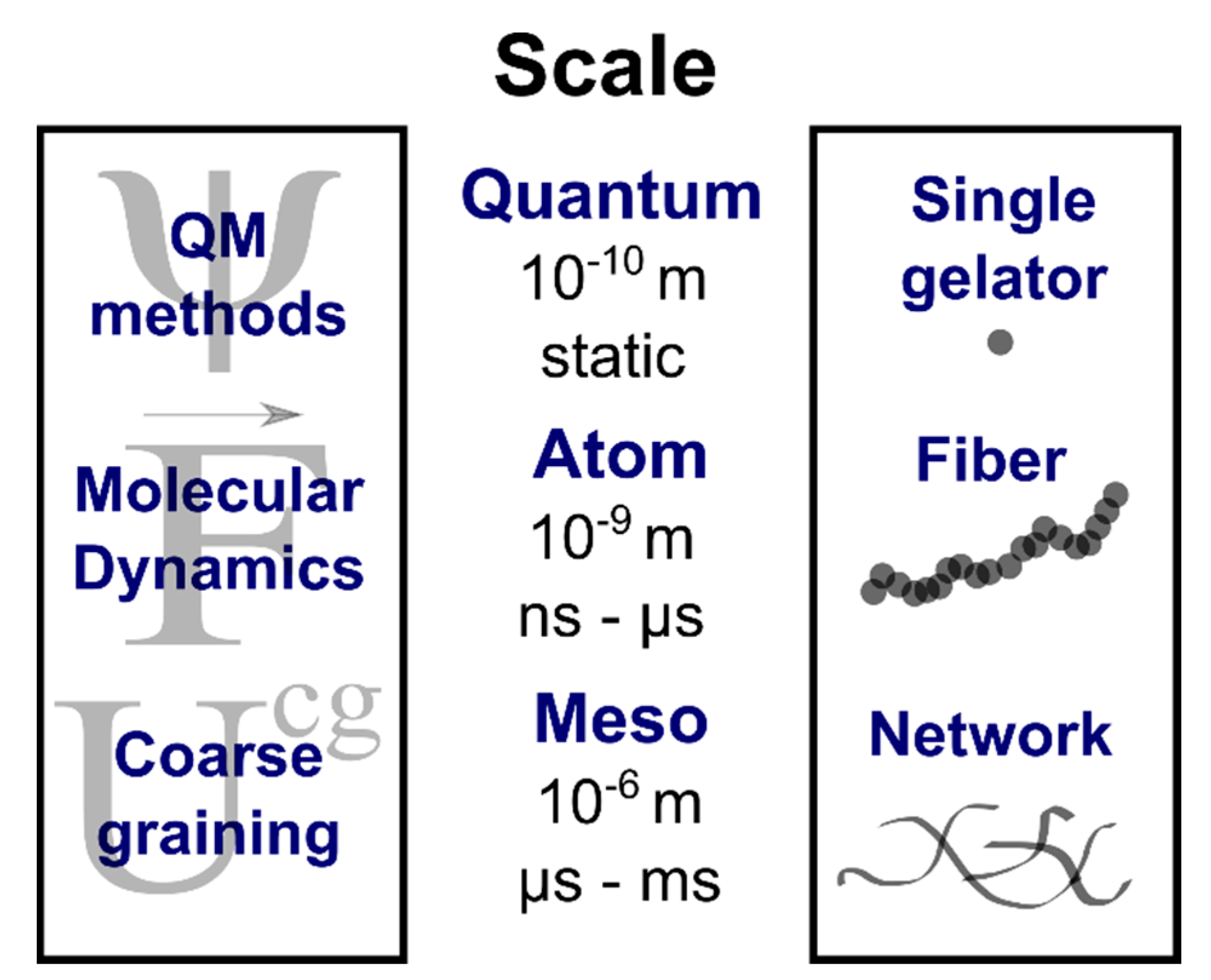
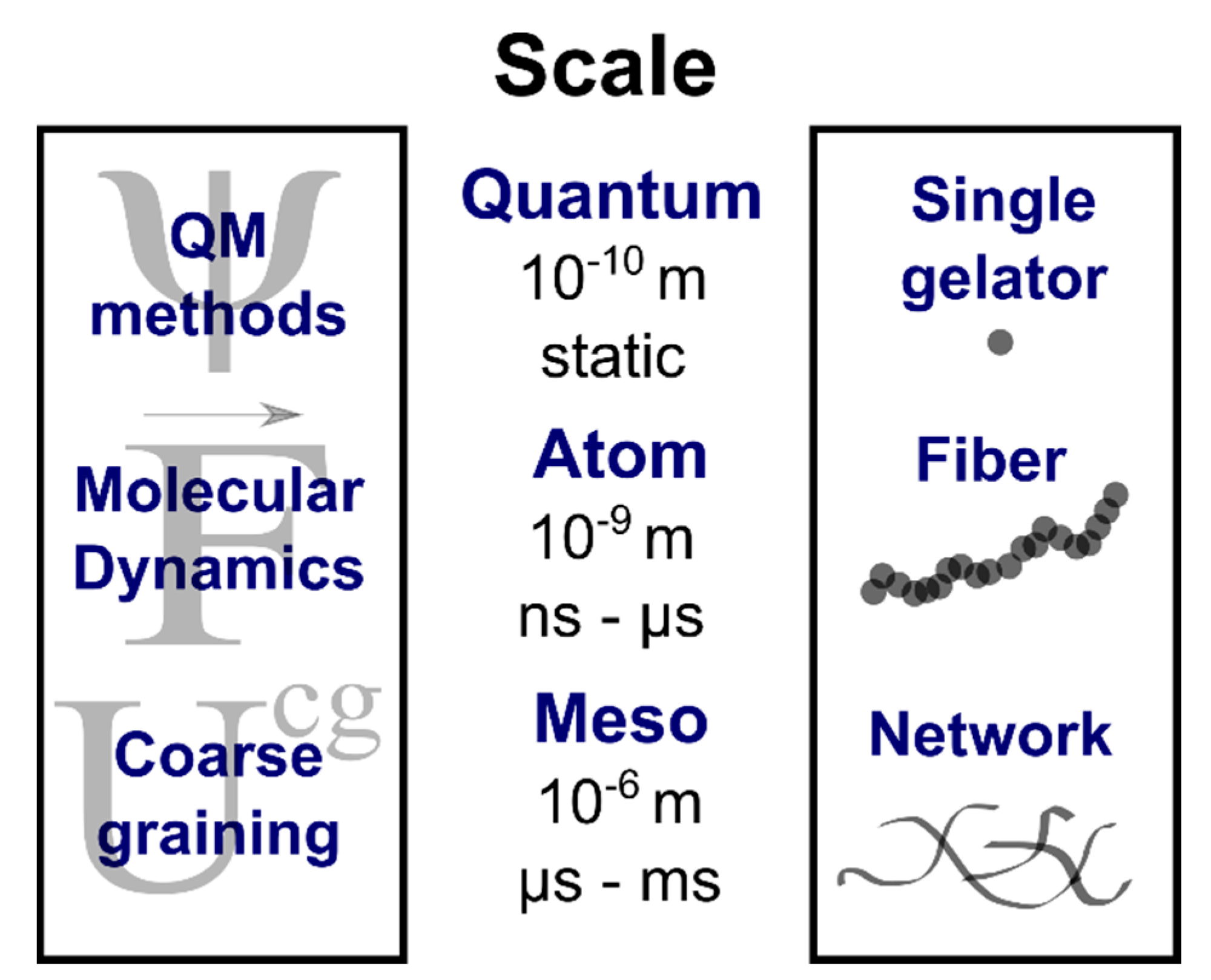
2. Rationalizing Supramolecular Gelation
2.1. Static Quantum Mechanical Calculations
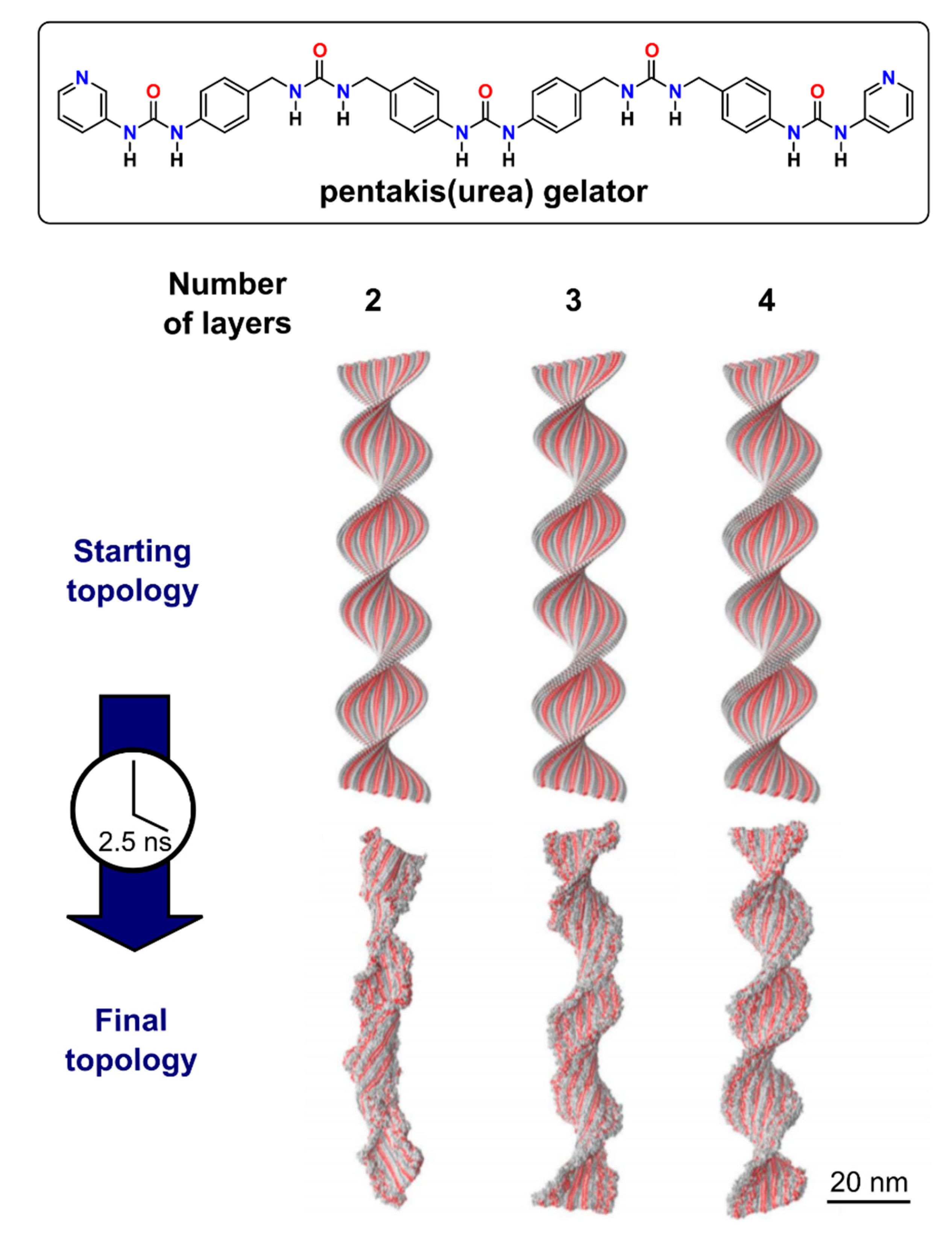
2.2. All-Atom Molecular Mechanics and Dynamic Simulations
2.3. United-Atom and Coarse-Grained Simulations
2.4. Other Methods
3. Predicting Supramolecular Gelation
3.1. Predicition through the Crystal Structure
3.2. Solvent Parameters
3.3. Molecular Dynamics and Machine Learning
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Draper, E.R.; Adams, D.J. Low-Molecular-Weight Gels: The State of the Art. Chem 2017, 3, 390–410. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.G. The Past, Present, and Future of Molecular Gels. What Is the Status of the Field, and Where Is It Going? J. Am. Chem. Soc. 2014, 136, 7519–7530. [Google Scholar] [CrossRef]
- Du, X.W.; Zhou, J.; Shi, J.F.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Chivers, P.R.A.; Smith, D.K. Shaping and structuring supramolecular gels. Nat. Rev. Mater. 2019, 4, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Yan, X.; Han, C.; Huang, F. Characterization of Supramolecular Gels. Chem. Soc. Rev. 2013, 42, 6697–6722. [Google Scholar] [CrossRef]
- Dawn, A.; Kumari, H. Low Molecular Weight Supramolecular Gels Under Shear: Rheology as the Tool for Elucidating Structure–Function Correlation. Chem. A Eur. J. 2017, 24, 762–776. [Google Scholar] [CrossRef]
- Escuder, B.; Llusar, M.; Miravet, J.F. Insight on the NMR Study of Supramolecular Gels and Its Application to Monitor Molecular Recognition on Self-Assembled Fibers. J. Org. Chem. 2006, 71, 7747–7752. [Google Scholar] [CrossRef] [PubMed]
- Kubota, R.; Nakamura, K.; Torigoe, S.; Hamachi, I. The Power of Confocal Laser Scanning Microscopy in Supramolecular Chemistry: In situ Real-time Imaging of Stimuli-Responsive Multicomponent Supramolecular Hydrogels. ChemistryOpen 2020, 9, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef]
- Al-Hamdani, Y.S.; Tkatchenko, A. Understanding Non-Covalent Interactions in Larger Molecular Complexes from First Principles. J. Chem. Phys. 2019, 150, 010901. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Lee, S.J.; Kang, S.; Lee, J.Y. Computational Approaches in Molecular Recognition, Self-assembly, Electron Transport, and Surface Chemistry. Supramol. Chem. 2007, 19, 229–241. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Y.; Qin, M.; Cao, Y.; Wang, W. Mechanics of single peptide hydrogelator fibrils. Nanoscale 2015, 7, 5638–5642. [Google Scholar] [CrossRef] [PubMed]
- Van Lommel, R.; Rutgeerts, L.A.J.; De Borggraeve, W.M.; De Proft, F.; Alonso, M. Rationalising Supramolecular Hydrogelation of Bis-Urea-Based Gelators through a Multiscale Approach. ChemPlusChem 2020, 85, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. A Review of Multiscale Computational Methods in Polymeric Materials. Polymers 2017, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, D.; Pavan, G.M. Molecular modelling of supramolecular polymers. Adv. Phys. X 2018, 3, 1436408. [Google Scholar] [CrossRef] [Green Version]
- Jensen, F. Introduction to Computational Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Calbo, J.; Sancho-García, J.C.; Ortí, E.; Aragó, J. Quantum-Chemical Insights into the Self-Assembly of Carbon-Based Supramolecular Complexes. Molecules 2018, 23, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Schwabe, T.; Mück-Lichtenfeld, C. Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of (bio)organic molecules. Org. Biomol. Chem. 2007, 5, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Cho, W.J.; Youn, I.S.; Lee, G.; Singh, N.J.; Kim, K.S. Density Functional Theory Based Study of Molecular Interactions, Recognition, Engineering, and Quantum Transport in π Molecular Systems. Acc. Chem. Res. 2014, 47, 3321–3330. [Google Scholar] [CrossRef]
- Kristyán, S.; Pulay, P. Can (semi)local density functional theory account for the London dispersion forces? Chem. Phys. Lett. 1994, 229, 175–180. [Google Scholar] [CrossRef]
- Hobza, P.; Sponer, J.; Reschel, T. Density functional theory and molecular clusters. J. Comput. Chem. 1995, 16, 1315–1325. [Google Scholar] [CrossRef]
- DiLabio, G.A.; Otero-De-La-Roza, A. Noncovalent Interactions in Density Functional Theory. Rev. Comput. Chem. 2016, 1–97. [Google Scholar] [CrossRef] [Green Version]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Vydrov, O.A.; Van Voorhis, T. Nonlocal van der Waals density functional: The simpler the better. J. Chem. Phys. 2010, 133, 244103. [Google Scholar] [CrossRef] [Green Version]
- Calbo, J.; Ortí, E.; Sancho-Garcia, J.C.; Aragó, J. Accurate Treatment of Large Supramolecular Complexes by Double-Hybrid Density Functionals Coupled with Nonlocal van der Waals Corrections. J. Chem. Theory Comput. 2015, 11, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setnička, V.; Nový, J.; Böhm, S.; Sreenivasachary, N.; Urbanová, M.; Volka, K. Molecular Structure of Guanine-Quartet Supramolecular Assemblies in a Gel-State Based on a DFT Calculation of Infrared and Vibrational Circular Dichroism Spectra. Langmuir 2008, 24, 7520–7527. [Google Scholar] [CrossRef]
- Hoogsteen, K. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Crystallogr. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Li, J.; Fan, K.; Niu, L.; Li, Y.; Song, J. Effects of Salt on the Gelation Mechanism of a d-Sorbitol-Based Hydrogelator. J. Phys. Chem. B 2013, 117, 5989–5995. [Google Scholar] [CrossRef]
- Schön, E.-M.; Marqués-López, E.; Herrera, R.P.; Alemán, C.; Díaz, D.D. Exploiting Molecular Self-Assembly: From Urea-Based Organocatalysts to Multifunctional Supramolecular Gels. Chem. A Eur. J. 2014, 20, 10720–10731. [Google Scholar] [CrossRef] [Green Version]
- Draper, E.R.; Schweins, R.; Akhtar, R.; Groves, P.; Chechik, V.; Zwijnenburg, M.A.; Adams, D.J. Reversible Photoreduction as a Trigger for Photoresponsive Gels. Chem. Mater. 2016, 28, 6336–6341. [Google Scholar] [CrossRef] [Green Version]
- Vega-Granados, K.; Ramírez-Rodríguez, G.B.; Contreras-Montoya, R.; Ramírez, F.J.; Palomo, L.; Parra, A.; Delgado-López, J.M.; Lopez-Lopez, M.T.; De Cienfuegos, L.Á. Atmospheric water triggers supramolecular gel formation of novel low molecular weight maslinic and oleanolic triterpenic derivatives. Mater. Chem. Front. 2019, 3, 2637–2646. [Google Scholar] [CrossRef]
- Xu, F.; Pfeifer, L.; Crespi, S.; Leung, F.K.-C.; Stuart, M.C.A.; Wezenberg, S.J.; Feringa, B.L. From Photoinduced Supramolecular Polymerization to Responsive Organogels. J. Am. Chem. Soc. 2021, 143, 5990–5997. [Google Scholar] [CrossRef]
- Draper, E.R.; Greeves, B.J.; Barrow, M.; Schweins, R.; Zwijnenburg, M.A.; Adams, D.J. pH-Directed Aggregation to Control Photoconductivity in Self-Assembled Perylene Bisimides. Chem 2017, 2, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Gainar, A.; Lai, T.L.; Oliveras-González, C.; Pop, F.; Raynal, M.; Isare, B.; Bouteiller, L.; Linares, M.; Canevet, D.; Avarvari, N.; et al. Tuning the Organogelating and Spectroscopic Properties of a C 3-Symmetric Pyrene-Based Gelator through Charge Transfer. Chem. A Eur. J. 2021, 27, 2410–2420. [Google Scholar] [CrossRef]
- Draper, E.R.; Wilbraham, L.; Adams, D.J.; Wallace, M.; Schweins, R.; Zwijnenburg, M.A. Insight into the self-assembly of water-soluble perylene bisimide derivatives through a combined computational and experimental approach. Nanoscale 2019, 11, 15917–15928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piana, F.; Case, D.H.; Ramalhete, S.M.; Pileio, G.; Facciotti, M.; Day, G.M.; Khimyak, Y.Z.; Angulo, J.; Brown, R.C.D.; Gale, P.A. Substituent interference on supramolecular assembly in urea gelators: Synthesis, structure prediction and NMR. Soft Matter 2016, 12, 4034–4043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Dubey, M.; Kumar, A.; Pandey, D.S. A Saponification-Triggered Gelation of Ester-Based Zn(ii) Complex through Conformational Transformations. Chem. Commun. 2014, 50, 10086–10089. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.C.; Li, W.; Yin, X.L.; Xie, J.M. A comprehensive theoretical study of the hydrogen bonding interactions and microscopic solvation structures of a pyridyl-urea-based hydrogelator in aqueous solution. Comput. Theor. Chem. 2013, 1006, 76–84. [Google Scholar] [CrossRef]
- Meng, S.C.; Tang, Y.Q.; Yin, Y.; Yin, X.L.; Xie, J.M. A theoretical study of molecular conformations and gelation ability of N,N′-dipyridyl urea compounds in ethanol solution: DFT calculations and MD simulations. RSC Adv. 2013, 3, 18115. [Google Scholar] [CrossRef]
- Wezenberg, S.J.; Croisetu, C.M.; Stuart, M.C.A.; Feringa, B.L. Reversible gel–sol photoswitching with an overcrowded alkene-based bis-urea supergelator. Chem. Sci. 2016, 7, 4341–4346. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Bjornsson, R.; Damodaran, K.K. Role of N–Oxide Moieties in Tuning Supramolecular Gel-State Properties. Gels 2020, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Hashemnejad, S.M.; Huda, M.M.; Rai, N.; Kundu, S. Molecular Insights into Gelation of Di-Fmoc-l-Lysine in Organic Solvent–Water Mixtures. ACS Omega 2017, 2, 1864–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foloppe, N.; MacKerell, A.D., Jr. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- van Esch, J.H.; Schoonbeek, F.; de Loos, M.; Kooijman, H.; Spek, A.L.; Kellogg, R.M.; Feringa, B.L. Cyclic Bis-Urea Compounds as Gelators for Organic Solvents. Chem. A Eur. J. 1999, 5, 937–950. [Google Scholar] [CrossRef]
- Mallia, V.A.; George, M.; Blair, D.L.; Weiss, R.G. Robust Organogels from Nitrogen-Containing Derivatives of (R)-12-Hydroxystearic Acid as Gelators: Comparisons with Gels from Stearic Acid Derivatives†. Langmuir 2009, 25, 8615–8625. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederix, P.W.J.M.; Patmanidis, I.; Marrink, S.J. Molecular simulations of self-assembling bio-inspired supramolecular systems and their connection to experiments. Chem. Soc. Rev. 2018, 47, 3470–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre-Requena, J.V.; Saldías, C.; Inostroza-Rivera, R.; Díaz, D.D. Understanding Hydrogelation Processes through Molecular Dynamics. J. Mater. Chem. B 2019, 7, 1652–1673. [Google Scholar] [CrossRef]
- Knapp, B.; Frantal, S.; Cibena, M.; Schreiner, W.; Bauer, P. Is an Intuitive Convergence Definition of Molecular Dynamics Simulations Solely Based on the Root Mean Square Deviation Possible? J. Comput. Biol. 2011, 18, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Williams, R.J.; Tang, C.; Coppo, P.; Collins, R.F.; Turner, M.L.; Saiani, A.; Ulijn, R.V. Fmoc-Diphenylalanine Self Assembles to a Hydrogel via a Novel Architecture Based on π–π Interlocked β-Sheets. Adv. Mater. 2008, 20, 37–41. [Google Scholar] [CrossRef]
- Drechsler, S.; Balog, S.; Kilbinger, A.F.M.; Casalini, T. The influence of substituents on gelation and stacking order of oligoaramid—Based supramolecular networks. Soft Matter 2019, 15, 7250–7261. [Google Scholar] [CrossRef]
- Parisi, E.; Garcia, A.M.; Marson, D.; Posocco, P.; Marchesan, S. Supramolecular Tripeptide Hydrogel Assembly with 5-Fluorouracil. Gels 2019, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Eckes, K.M.; Mu, X.; Ruehle, M.A.; Ren, P.; Suggs, L.J. β Sheets Not Required: Combined Experimental and Computational Studies of Self-Assembly and Gelation of the Ester-Containing Analogue of an Fmoc-Dipeptide Hydrogelator. Langmuir 2014, 30, 5287–5296. [Google Scholar] [CrossRef]
- Mu, X.; Eckes, K.M.; Nguyen, M.M.; Suggs, L.J.; Ren, P. Experimental and Computational Studies Reveal an Alternative Supramolecular Structure for Fmoc-Dipeptide Self-Sssembly. Biomacromolecules 2012, 13, 3562–3571. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.M.; Eckes, K.M.; Suggs, L.J. Charge and sequence effects on the self-assembly and subsequent hydrogelation of Fmoc-depsipeptides. Soft Matter 2014, 10, 2693–2702. [Google Scholar] [CrossRef] [Green Version]
- Lee, O.-S.; Stupp, S.I.; Schatz, G.C. Atomistic Molecular Dynamics Simulations of Peptide Amphiphile Self-Assembly into Cylindrical Nanofibers. J. Am. Chem. Soc. 2011, 133, 3677–3683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Z.; Zhao, S.; Hong, K.H.; Zhang, M.-Y.; Song, L.; Yu, F.; Luo, G.; He, Y.-P. Pyromellitic-Based Low Molecular Weight Gelators and Computational Studies of Intermolecular Interactions: A Potential Additive for Lubricant. Langmuir 2021, 37, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Wychowaniec, J.K.; Patel, R.; Leach, J.; Mathomes, R.; Chhabria, V.; Patil-Sen, Y.; Hidalgo-Bastida, A.; Forbes, R.T.; Hayes, J.M.; ElSawy, M.A. Aromatic Stacking Facilitated Self-Assembly of Ultrashort Ionic Complementary Peptide Sequence: β-Sheet Nanofibers with Remarkable Gelation and Interfacial Properties. Biomacromolecules 2020, 21, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.D.; Simmons, H.T.D.; Horner, K.E.; Liu, K.; Thompson, R.L.; Steed, J.W. Braiding, branching and chiral amplification of nanofibres in supramolecular gels. Nat. Chem. 2019, 11, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.D.; Kennedy, S.R.; Walker, M.; Yufit, D.S.; Steed, J.W. Scrolling of Supramolecular Lamellae in the Hierarchical Self-Assembly of Fibrous Gels. Chem 2017, 3, 603–628. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.B.; Albertazzi, L.; Voets, I.K.; Leenders, C.M.; Palmans, A.R.; Pavan, G.M.; Meijer, E.W. Consequences of chirality on the dynamics of a water-soluble supramolecular polymer. Nat. Commun. 2015, 6, 6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knani, D.; Alperstein, D. Simulation of DBS, DBS-COOH, and DBS-CONHNH2 as Hydrogelators. J. Phys. Chem. A 2017, 121, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Angelerou, M.G.F.; Frederix, P.W.J.M.; Wallace, M.; Yang, B.; Rodger, A.; Adams, D.J.; Marlow, M.; Zelzer, M. Supramolecular Nucleoside-Based Gel: Molecular Dynamics Simulation and Characterization of Its Nanoarchitecture and Self-Assembly Mechanism. Langmuir 2018, 34, 6912–6921. [Google Scholar] [CrossRef] [PubMed]
- Draper, E.R.; Dietrich, B.; McAulay, K.; Brasnett, C.; Abdizadeh, H.; Patmanidis, I.; Marrink, S.J.; Su, H.; Cui, H.; Schweins, R.; et al. Using Small-Angle Scattering and Contrast Matching to Understand Molecular Packing in Low Molecular Weight Gels. Matter 2020, 2, 764–778. [Google Scholar] [CrossRef] [Green Version]
- Dunfield, L.G.; Burgess, A.W.; Scheraga, H.A. Energy parameters in polypeptides. 8. Empirical potential energy algorithm for the conformational analysis of large molecules. J. Phys. Chem. 1978, 82, 2609–2616. [Google Scholar] [CrossRef]
- Yang, L.; Tan, C.-H.; Hsieh, M.-J.; Wang, J.; Duan, Y.; Cieplak, P.; Caldwell, J.; Kollman, P.A.; Luo, R. New-Generation Amber United-Atom Force Field. J. Phys. Chem. B 2006, 110, 13166–13176. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Potoff, J.J.; Siepmann, J.I. Monte Carlo Calculations for Alcohols and Their Mixtures with Alkanes. Transferable Potentials for Phase Equilibria. 5. United-Atom Description of Primary, Secondary, and Tertiary Alcohols. J. Phys. Chem. B 2001, 105, 3093–3104. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United-Atom Description ofn-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Stubbs, J.M.; Potoff, J.J.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 6. United-Atom Description for Ethers, Glycols, Ketones, and Aldehydes. J. Phys. Chem. B 2004, 108, 17596–17605. [Google Scholar] [CrossRef]
- Tjörnhammar, R.; Edholm, O. Reparameterized United Atom Model for Molecular Dynamics Simulations of Gel and Fluid Phosphatidylcholine Bilayers. J. Chem. Theory Comput. 2014, 10, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Stober, S.T.; Abrams, C.F. Effects of Optical Purity and Finite System Size on Self-Assembly of 12-Hydroxystearic Acid in Hexane: Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 9223–9233. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.M.; Rai, N. Probing Early-Stage Aggregation of Low Molecular Weight Gelator in an Organic Solvent. J. Phys. Chem. B 2020, 124, 2277–2288. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Badaczewska-Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- Noid, W.G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139, 090901. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The Martini Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, D.H.; Singh, G.; Bennett, W.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Lee, O.-S.; Cho, V.; Schatz, G.C. Modeling the Self-Assembly of Peptide Amphiphiles into Fibers Using Coarse-Grained Molecular Dynamics. Nano Lett. 2012, 12, 4907–4913. [Google Scholar] [CrossRef]
- Basavalingappa, V.; Guterman, T.; Tang, Y.; Nir, S.; Lei, J.; Chakraborty, P.; Schnaider, L.; Reches, M.; Wei, G.; Gazit, E. Expanding the Functional Scope of the Fmoc-Diphenylalanine Hydrogelator by Introducing a Rigidifying and Chemically Active Urea Backbone Modification. Adv. Sci. 2019, 6, 1900218. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Hall, C.K. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 16180–16185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, I.W.; Markegard, C.B.; Chu, B.K.; Nguyen, H.D. The Role of Electrostatics and Temperature on Morphological Transitions of Hydrogel Nanostructures Self-Assembled by Peptide Amphiphiles Via Molecular Dynamics Simulations. Adv. Healthc. Mater. 2013, 2, 1388–1400. [Google Scholar] [CrossRef]
- Xiong, Q.; Jiang, Y.; Cai, X.; Yang, F.; Li, Z.; Han, W. Conformation Dependence of Diphenylalanine Self-Assembly Structures and Dynamics: Insights from Hybrid-Resolution Simulations. ACS Nano 2019, 13, 4455–4468. [Google Scholar] [CrossRef]
- Badaczewska-Dawid, A.E.; Kolinski, A.; Kmiecik, S. Computational reconstruction of atomistic protein structures from coarse-grained models. Comput. Struct. Biotechnol. J. 2020, 18, 162–176. [Google Scholar] [CrossRef]
- Otero-De-La-Roza, A.; Johnson, E.R.; Contreras-Garcia, J. Revealing non-covalent interactions in solids: NCI plots revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165–12172. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Boto, R.Á.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.-P.; Maday, Y.; Contreras-García, J. NCIPLOT4: Fast, Robust, and Quantitative Analysis of Noncovalent Interactions. J. Chem. Theory Comput. 2020, 16, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.C.; Contreras-García, J.; Hénon, E. Accurately Extracting the Signature of Intermolecular Interactions Present in the NCI plot of the Reduced Density Gradient versus Electron Density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Laplaza, R.; Peccati, F.; Boto, R.A.; Quan, C.; Carbone, A.; Piquemal, J.P.; Maday, Y.; Contreras-García, J. NCIPLOT and the Analysis of Noncovalent Interactions using the Reduced Density Gradient. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1497. [Google Scholar] [CrossRef]
- Li, Z.H.; Yang, H.L.; Adam, K.M.; Yao, H.; Wei, T.B.; Zhang, Y.M.; Lin, Q. Theoretical and Experimental Insights into the Self-Assembly and Ion Response Mechanisms of Tripodal Quinolinamido-Based Supramolecular Organogels. ChemPlusChem 2021, 86, 146–154. [Google Scholar] [CrossRef]
- Bettens, T.; Lacanau, V.; Van Lommel, R.; De Maeseneer, T.; Vandeplassche, W.; Bertouille, J.; Brancart, J.; Barlow, T.M.A.; Woller, T.; Van den Brande, N.; et al. Towards the Understanding of Halogenation in Peptide Hydrogels: A Quantum Chemical Approach. Mater. Adv. 2021. [Google Scholar] [CrossRef]
- Contreras-García, J.; Boto, R.A.; Izquierdo-Ruiz, F.; Reva, I.; Woller, T.; Alonso, M. A Benchmark for the Non-Covalent Interaction (NCI) index or… is it Really all in the Geometry? Theor. Chem. Acc. 2016, 135, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Pedre, B.; Van Bergen, L.A.H.; Palló, A.; Rosado, L.A.; Dufe, V.T.; Van Molle, I.; Wahni, K.; Erdogan, H.; Alonso, M.; De Proft, F.; et al. The active site architecture in peroxiredoxins: A case study on Mycobacterium tuberculosis AhpE. Chem. Commun. 2016, 52, 10293–10296. [Google Scholar] [CrossRef]
- Van Bergen, L.A.H.; Alonso, M.; Palló, A.; Nilsson, L.; De Proft, F.; Messens, J. Revisiting sulfur H-bonds in proteins: The example of peroxiredoxin AhpE. Sci. Rep. 2016, 6, 30369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.K.; Steed, J.W. Supramolecular gel phase crystallization: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.L.; Pearson, E.; Yufit, D.S.; Steed, J.W.; Edkins, K. Supramolecular Gelation as the First Stage in Ostwald’s Rule. Cryst. Growth Des. 2018, 18, 7690–7700. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Lebedytė, I.; Yufit, D.S.; Damodaran, K.K.; Steed, J.W. Selective Gelation of N-(4-pyridyl)nicotinamide by Copper(ii) Salts. CrystEngComm 2015, 17, 8130–8138. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Llansola, F.; Hermida-Merino, D.; Nieto-Ortega, B.; Ramírez, F.J.; Navarrete, J.T.L.; Casado, J.; Hamley, I.W.; Escuder, B.; Hayes, W.; Miravet, J.F. Self-Assembly Studies of a Chiral Bisurea-Based Superhydrogelator. Chem. A Eur. J. 2012, 18, 14725–14731. [Google Scholar] [CrossRef]
- Woodley, S.M.; Catlow, R. Crystal Structure Prediction from First Principles. Nat. Mater. 2008, 7, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Nyman, J.; Reutzel-Edens, S.M. Crystal structure prediction is changing from basic science to applied technology. Faraday Discuss. 2018, 211, 459–476. [Google Scholar] [CrossRef]
- Ryan, K.; Lengyel, J.; Shatruk, M. Crystal Structure Prediction via Deep Learning. J. Am. Chem. Soc. 2018, 140, 10158–10168. [Google Scholar] [CrossRef]
- Maddox, J. Crystals from first principles. Nature 1988, 335, 201. [Google Scholar] [CrossRef]
- Price, S.L. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 2013, 43, 2098–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.M.; Day, G.M.; Paterson, M.J.; Byrne, P.; Clarke, N.; Steed, K. Structure Calculation of an Elastic Hydrogel from Sonication of Rigid Small Molecule Components. Angew. Chem. Int. Ed. 2008, 47, 1058–1062. [Google Scholar] [CrossRef]
- Adams, D.J.; Morris, K.; Chen, L.; Serpell, L.C.; Bacsa, J.; Day, G.M. The delicate balance between gelation and crystallisation: Structural and computational investigations. Soft Matter 2010, 6, 4144–4156. [Google Scholar] [CrossRef]
- Khavasi, H.R.; Esmaeili, M. Is Gelation Behavior Predictable through a Crystal Engineering Approach? A Case Study in Four Similar Coordination Compounds. Langmuir 2019, 35, 4660–4671. [Google Scholar] [CrossRef]
- Adalder, T.K.; Dastidar, P. Crystal Engineering Approach toward Selective Formation of an Asymmetric Supramolecular Synthon in Primary Ammonium Monocarboxylate (PAM) Salts and Their Gelation Studies. Cryst. Growth Des. 2014, 14, 2254–2262. [Google Scholar] [CrossRef]
- Zurcher, D.M.; McNeil, A.J. Tools for Identifying Gelator Scaffolds and Solvents. J. Org. Chem. 2015, 80, 2473–2478. [Google Scholar] [CrossRef]
- Veits, G.K.; Carter, K.K.; Cox, S.J.; McNeil, A.J. Developing a Gel-Based Sensor Using Crystal Morphology Prediction. J. Am. Chem. Soc. 2016, 138, 12228–12233. [Google Scholar] [CrossRef]
- BIOVA Materials Studio. Dassault Systèmes; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Hisaki, I.; Kometani, E.; Tohnai, N.; Miyata, M. Gelation or crystallization? Subtle balance of structural factors for assembly of DBA derivatives with methyl esters. CrystEngComm 2015, 17, 8079–8084. [Google Scholar] [CrossRef]
- Houton, K.A.; Morris, K.L.; Chen, L.; Schmidtmann, M.; Jones, J.T.A.; Serpell, L.C.; Lloyd, G.O.; Adams, D.J. On Crystal versus Fiber Formation in Dipeptide Hydrogelator Systems. Langmuir 2012, 28, 9797–9806. [Google Scholar] [CrossRef] [PubMed]
- Barker, E.C.; Martin, A.D.; Garvey, C.J.; Goh, C.Y.; Jones, F.; Mocerino, M.; Skelton, B.W.; Ogden, M.I.; Becker, T. Thermal annealing behaviour and gel to crystal transition of a low molecular weight hydrogelator. Soft Matter 2017, 13, 1006–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, A.R.; Smith, D.K. Solvent Effects on Supramolecular Gel-Phase Materials: Two-Component Dendritic Gel. Langmuir 2004, 20, 10851–10857. [Google Scholar] [CrossRef]
- Partridge, K.S.; Smith, D.K.; Dykes, G.M.; McGrail, P.T. Supramolecular dendritic two-component gel. Chem. Commun. 2001, 4, 319–320. [Google Scholar] [CrossRef]
- Niu, L.; Song, J.; Li, J.; Tao, N.; Lu, M.; Fan, K. Solvent effects on the gelation performance of melamine and 2-ethylhexylphosphoric acid mono-2-ethylhexyl ester in water–organic mixtures. Soft Matter 2013, 9, 7780–7786. [Google Scholar] [CrossRef]
- Zhu, G.; Dordick, J.S. Solvent Effect on Organogel Formation by Low Molecular Weight Molecules. Chem. Mater. 2006, 18, 5988–5995. [Google Scholar] [CrossRef]
- Lin, P.; Zhang, N.-X.; Li, J.-J.; Zhang, J.; Liu, J.-H.; Zhang, B.; Song, J. To gel or not to gel: A prior prediction of gelation in solvent mixtures. Chin. Chem. Lett. 2017, 28, 771–776. [Google Scholar] [CrossRef]
- Lan, Y.; Corradini, M.G.; Weiss, R.G.; Raghavan, S.R.; Rogers, M.A. To gel or not to gel: Correlating molecular gelation with solvent parameters. Chem. Soc. Rev. 2015, 44, 6035–6058. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Ballabh, A. Design, synthesis, and application of a new series of organogelator using crystal engineering approach and solvent parameter study: A synergetic approach. J. Mol. Liq. 2021, 322, 114520. [Google Scholar] [CrossRef]
- Iqbal, S.; Khan, A.A. Supramolecular self-assembly and physical-gel formation in disc-like liquid crystals: A scalable predictive model for gelation and an application in photovoltaics. RSC Adv. 2019, 9, 6335–6345. [Google Scholar] [CrossRef] [Green Version]
- Diehn, K.K.; Oh, H.; Hashemipour, R.; Weiss, R.G.; Raghavan, S.R. Insights into organogelation and its kinetics from Hansen solubility parameters. Toward a priori predictions of molecular gelation. Soft Matter 2014, 10, 2632. [Google Scholar] [CrossRef]
- Yan, N.; Xu, Z.; Diehn, K.K.; Raghavan, S.R.; Fang, Y.; Weiss, R.G. How Do Liquid Mixtures Solubilize Insoluble Gelators? Self-Assembly Properties of Pyrenyl-Linker-Glucono Gelators in Tetrahydrofuran–Water Mixtures. J. Am. Chem. Soc. 2013, 135, 8989–8999. [Google Scholar] [CrossRef]
- Delbecq, F.; Adenier, G.; Ogue, Y.; Kawai, T. Gelation properties of various long chain amidoamines: Prediction of solvent gelation via machine learning using Hansen solubility parameters. J. Mol. Liq. 2020, 303, 112587. [Google Scholar] [CrossRef]
- Hansen, C.M. 50 Years with Solubility Parameters—Past and Future. Prog. Org. Coat. 2004, 51, 77–84. [Google Scholar] [CrossRef]
- Raynal, M.; Bouteiller, L. Organogel formation rationalized by Hansen solubility parameters. Chem. Commun. 2011, 47, 8271–8273. [Google Scholar] [CrossRef]
- Bonnet, J.; Suissa, G.; Raynal, M.; Bouteiller, L. Organogel formation rationalized by Hansen solubility parameters: Dos and don’ts. Soft Matter 2014, 10, 3154–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, J.; Suissa, G.; Raynal, M.; Bouteiller, L. Organogel Formation Rationalized by Hansen Solubility Parameters: Influence of Gelator Structure. Soft Matter 2015, 11, 2308–2312. [Google Scholar] [CrossRef] [Green Version]
- Nunes, D.R.; Raynal, M.; Isare, B.; Albouy, P.-A.; Bouteiller, L. Organogel formation rationalized by Hansen solubility parameters: Improved methodology. Soft Matter 2018, 14, 4805–4809. [Google Scholar] [CrossRef] [PubMed]
- Nunes, D.R.; Reche-Tamayo, M.; Ressouche, E.; Raynal, M.; Isare, B.; Foury-Leylekian, P.; Albouy, P.-A.; Brocorens, P.; Lazzaroni, R.; Bouteiller, L. Organogel Formation Rationalized by Hansen Solubility Parameters: Shift of the Gelation Sphere with the Gelator Structure. Langmuir 2019, 35, 7970–7977. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Auzanneau, F.-I.; Corradini, M.G.; Grover, G.; Weiss, R.G.; Rogers, M.A. Molecular Nuances Governing the Self-Assembly of 1,3:2,4-Dibenzylidene-d-sorbitol. Langmuir 2017, 33, 10907–10916. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wu, S.; Rogers, M.A. Harnessing Hansen solubility parameters to predict organogel formation. J. Mater. Chem. 2012, 22, 12651–12658. [Google Scholar] [CrossRef]
- Lan, Y.Q.; Corradini, M.G.; Liu, X.; May, T.E.; Borondics, F.; Weiss, R.G.; Rogers, M.A. Comparing and Correlating Solubility Parameters Governing the Self-Assembly of Molecular Gels Using 1,3:2,4-Dibenzylidene Sorbitol as the Gelator. Langmuir 2014, 30, 14128–14142. [Google Scholar] [CrossRef]
- Lan, Y.; Corradini, M.G.; Rogers, M.A. Do Molecular Gelators Cluster in Hansen Space? Cryst. Growth Des. 2014, 14, 4811–4818. [Google Scholar] [CrossRef]
- Lan, Y.; Lv, M.; Guo, S.; Nasr, P.; Ladizhansky, V.; Vaz, R.F.; Corradini, M.G.; Hou, T.; Ghazani, S.M.; Marnangoni, A.; et al. Molecular motifs encoding self-assembly of peptide fibers into molecular gels. Soft Matter 2019, 15, 9205–9214. [Google Scholar] [CrossRef]
- Moreira, I.P.; Scott, G.G.; Ulijn, R.V.; Tuttle, T. Computational prediction of tripeptide-dipeptide co-assembly. Mol. Phys. 2018, 117, 1151–1163. [Google Scholar] [CrossRef]
- Frederix, P.; Ulijn, R.V.; Hunt, N.T.; Tuttle, T. Virtual Screening for Dipeptide Aggregation: Toward Predictive Tools for Peptide Self-Assembly. J. Phys. Chem. Lett. 2011, 2, 2380–2384. [Google Scholar] [CrossRef] [PubMed]
- Frederix, P.; Scott, G.G.; Abul-Haija, Y.M.; Kalafatovic, D.; Pappas, C.G.; Javid, N.; Hunt, N.T.; Ulijn, R.V.; Tuttle, T. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 2015, 7, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, J.K.; Adams, D.J.; Berry, N.G. Will it gel? Successful computational prediction of peptide gelators using physicochemical properties and molecular fingerprints. Chem. Sci. 2016, 7, 4713–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Han, J.; Cao, T.; Lam, W.; Fan, B.; Tang, W.; Chen, S.; Fok, E.; Li, L. Design of self-assembly dipeptide hydrogels and machine learning via their chemical features. Proc. Natl. Acad. Sci. USA 2019, 116, 11259–11264. [Google Scholar] [CrossRef] [Green Version]
- Van Lommel, R.; Zhao, J.; De Borggraeve, W.M.; De Proft, F.; Alonso, M. Molecular dynamics based descriptors for predicting supramolecular gelation. Chem. Sci. 2020, 11, 4226–4238. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Schreiner, P.R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem. Int. Ed. 2018, 57, 4170–4176. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, T.; Ju, W.; Shi, S. Materials discovery and design using machine learning. J. Mater. 2017, 3, 159–177. [Google Scholar] [CrossRef]
- Schmidt, J.; Marques, M.R.; Botti, S.; Marques, M.A. Recent Advances and Applications of Machine Learning in Solid-State Materials Science. NPJ Comput. Mater. 2019, 5, 83. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef] [Green Version]
- van Teijlingen, A.; Tuttle, T. Beyond Tripeptides Two-Step Active Machine Learning for Very Large Data sets. J. Chem. Theory Comput. 2021, 17, 3221–3232. [Google Scholar] [CrossRef] [PubMed]
- Zanganeh, S.; Firoozpour, L.; Sardari, S.; Afgar, A.; Cohan, R.A.; Mohajel, N. Novel Descriptors Derived from the Aggregation Propensity of Di- and Tripeptides Can Predict the Critical Aggregation Concentration of Longer Peptides. ACS Omega 2021, 6, 13331–13340. [Google Scholar] [CrossRef] [PubMed]



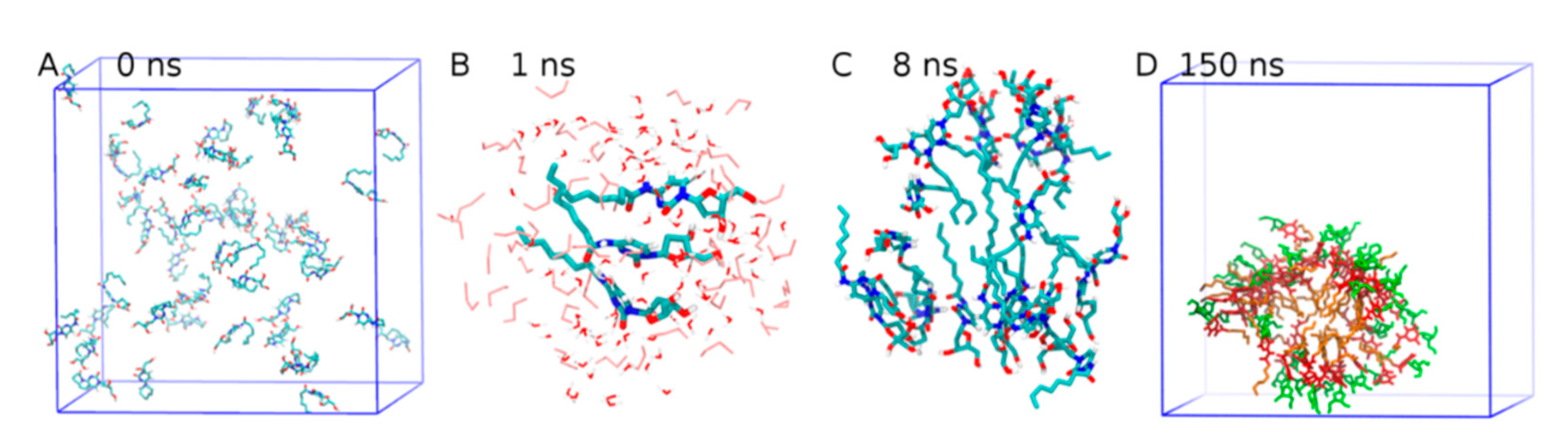
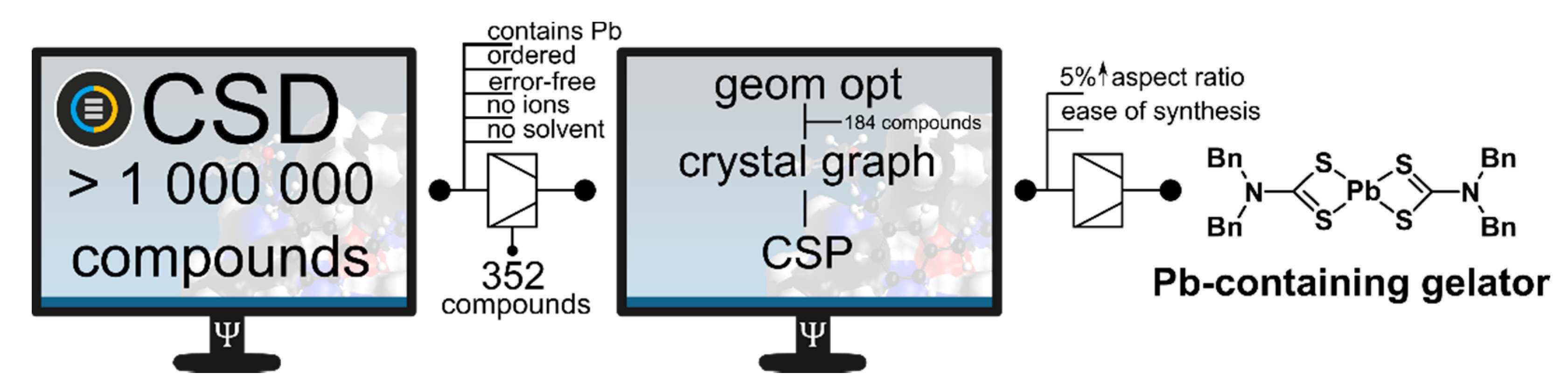
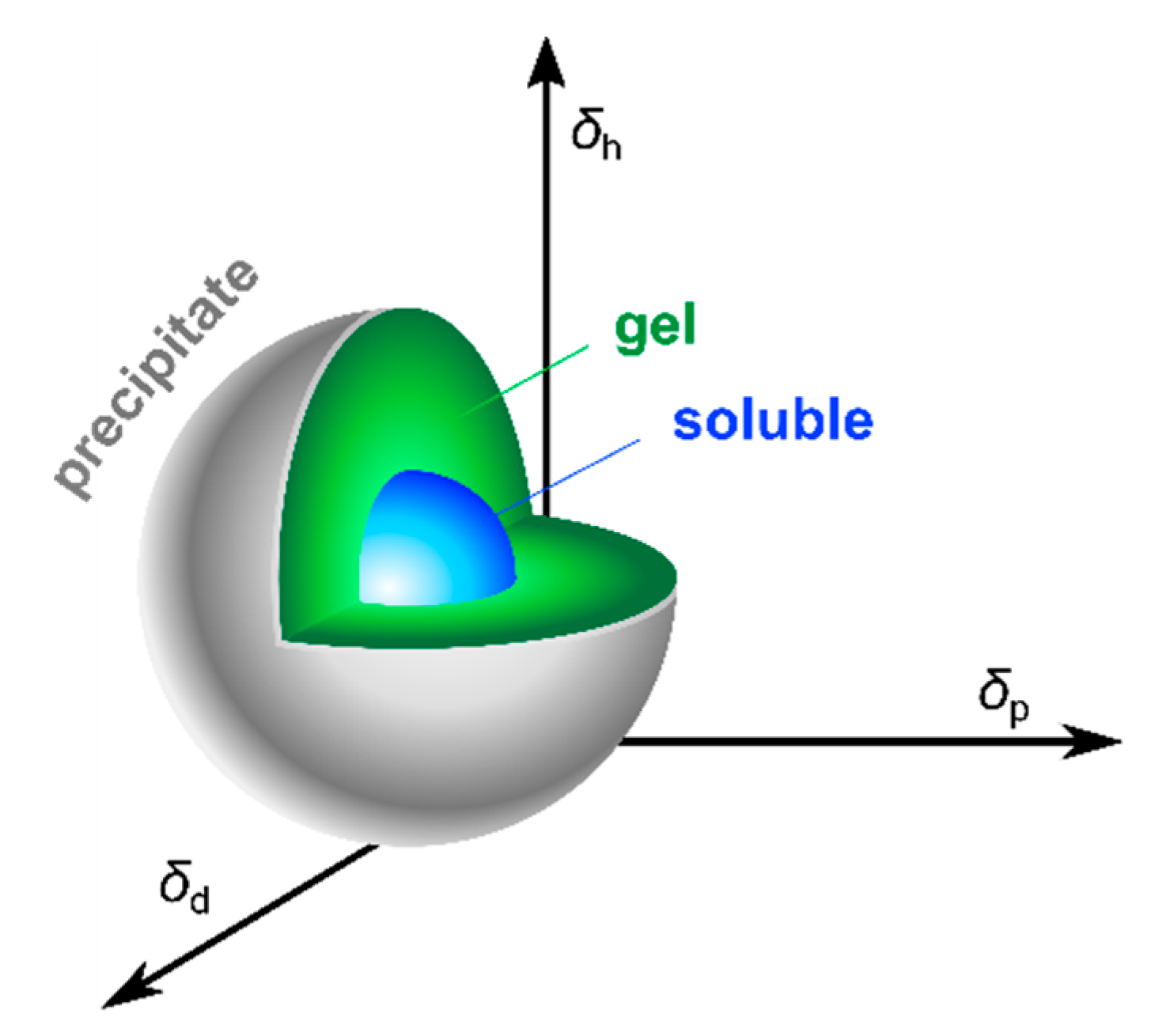
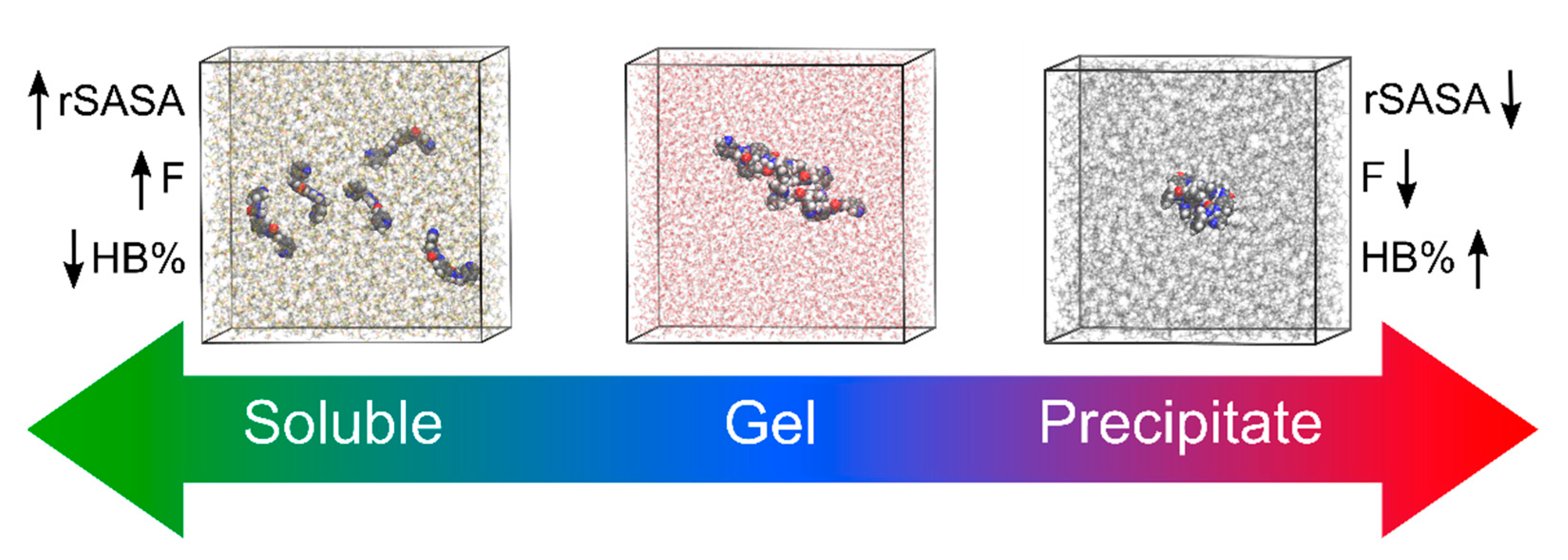
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Lommel, R.; De Borggraeve, W.M.; De Proft, F.; Alonso, M. Computational Tools to Rationalize and Predict the Self-Assembly Behavior of Supramolecular Gels. Gels 2021, 7, 87. https://doi.org/10.3390/gels7030087
Van Lommel R, De Borggraeve WM, De Proft F, Alonso M. Computational Tools to Rationalize and Predict the Self-Assembly Behavior of Supramolecular Gels. Gels. 2021; 7(3):87. https://doi.org/10.3390/gels7030087
Chicago/Turabian StyleVan Lommel, Ruben, Wim M. De Borggraeve, Frank De Proft, and Mercedes Alonso. 2021. "Computational Tools to Rationalize and Predict the Self-Assembly Behavior of Supramolecular Gels" Gels 7, no. 3: 87. https://doi.org/10.3390/gels7030087
APA StyleVan Lommel, R., De Borggraeve, W. M., De Proft, F., & Alonso, M. (2021). Computational Tools to Rationalize and Predict the Self-Assembly Behavior of Supramolecular Gels. Gels, 7(3), 87. https://doi.org/10.3390/gels7030087