
Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems

 ,
,  , ,
, ,  ,
,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
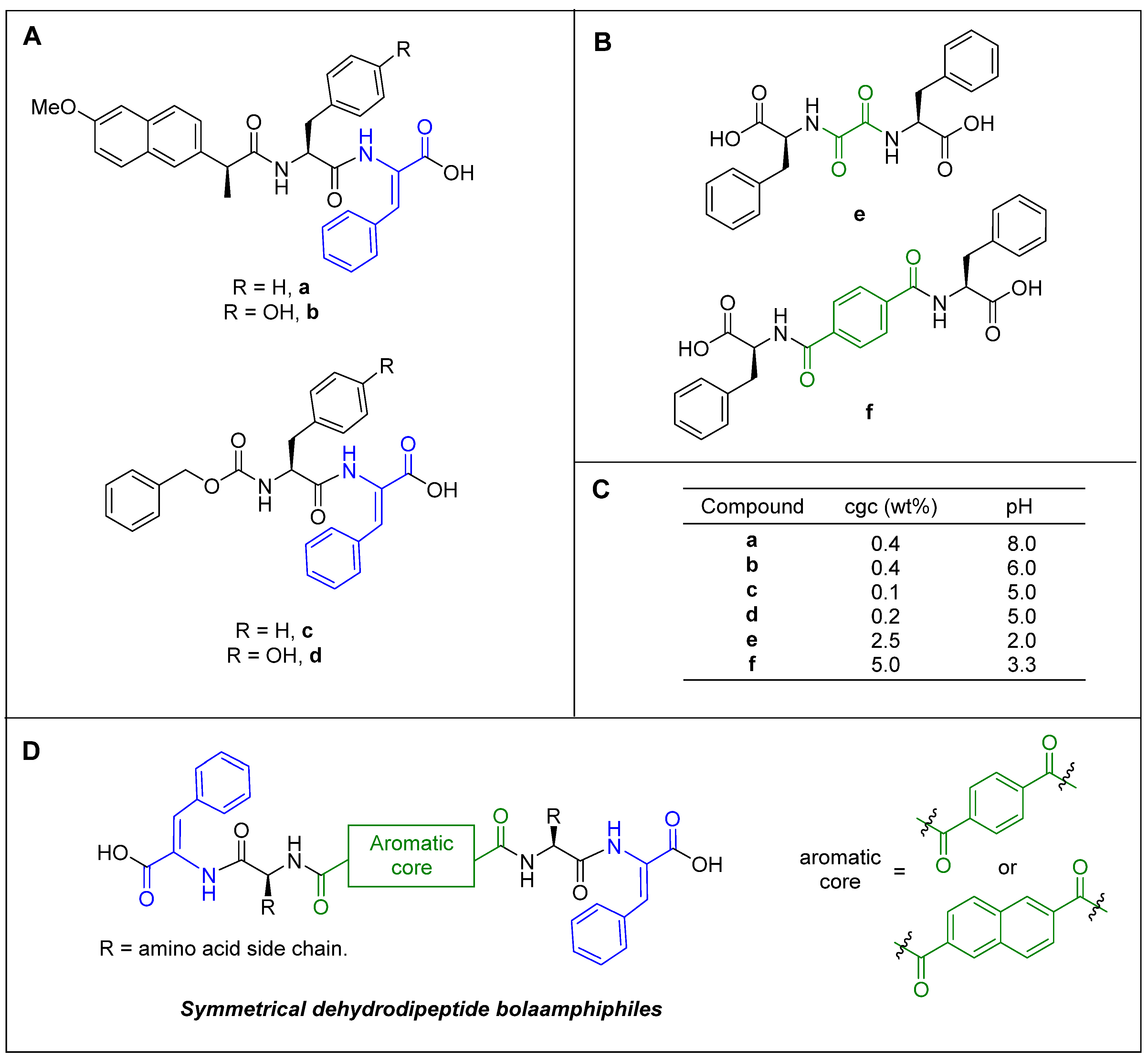
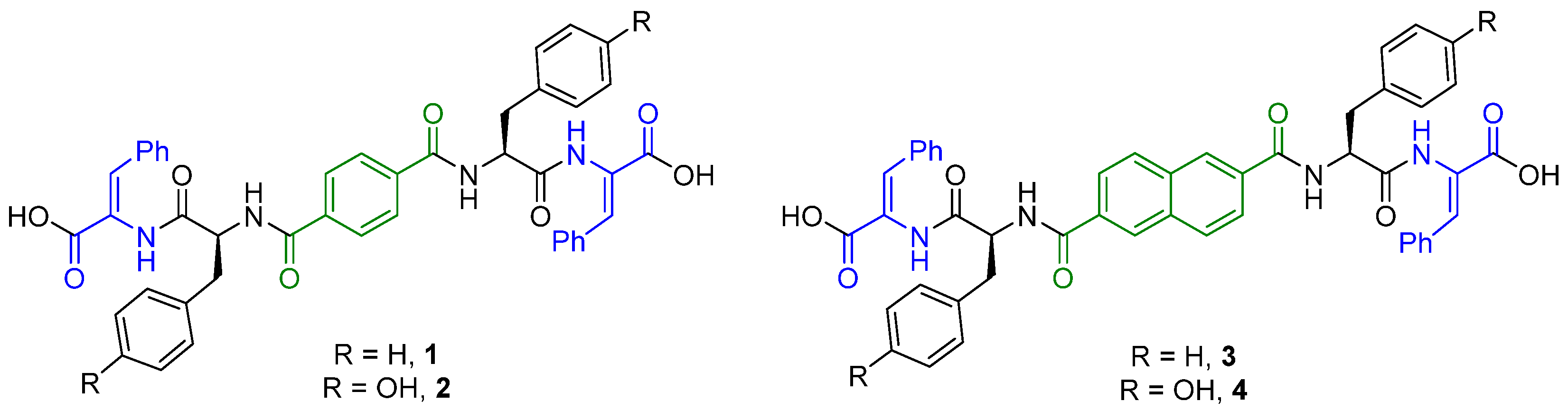
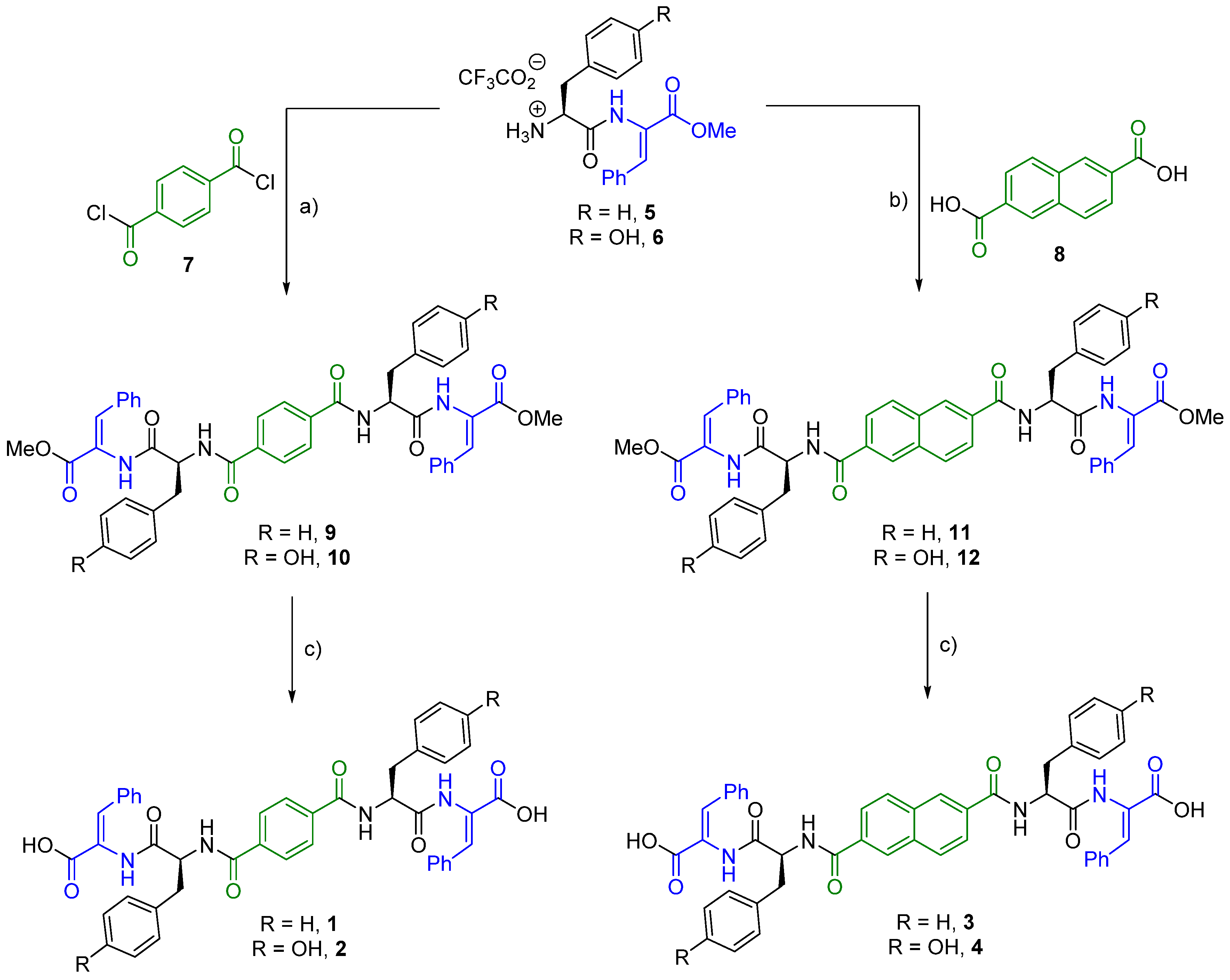
2.1. Synthesis
2.2. Gelation Study
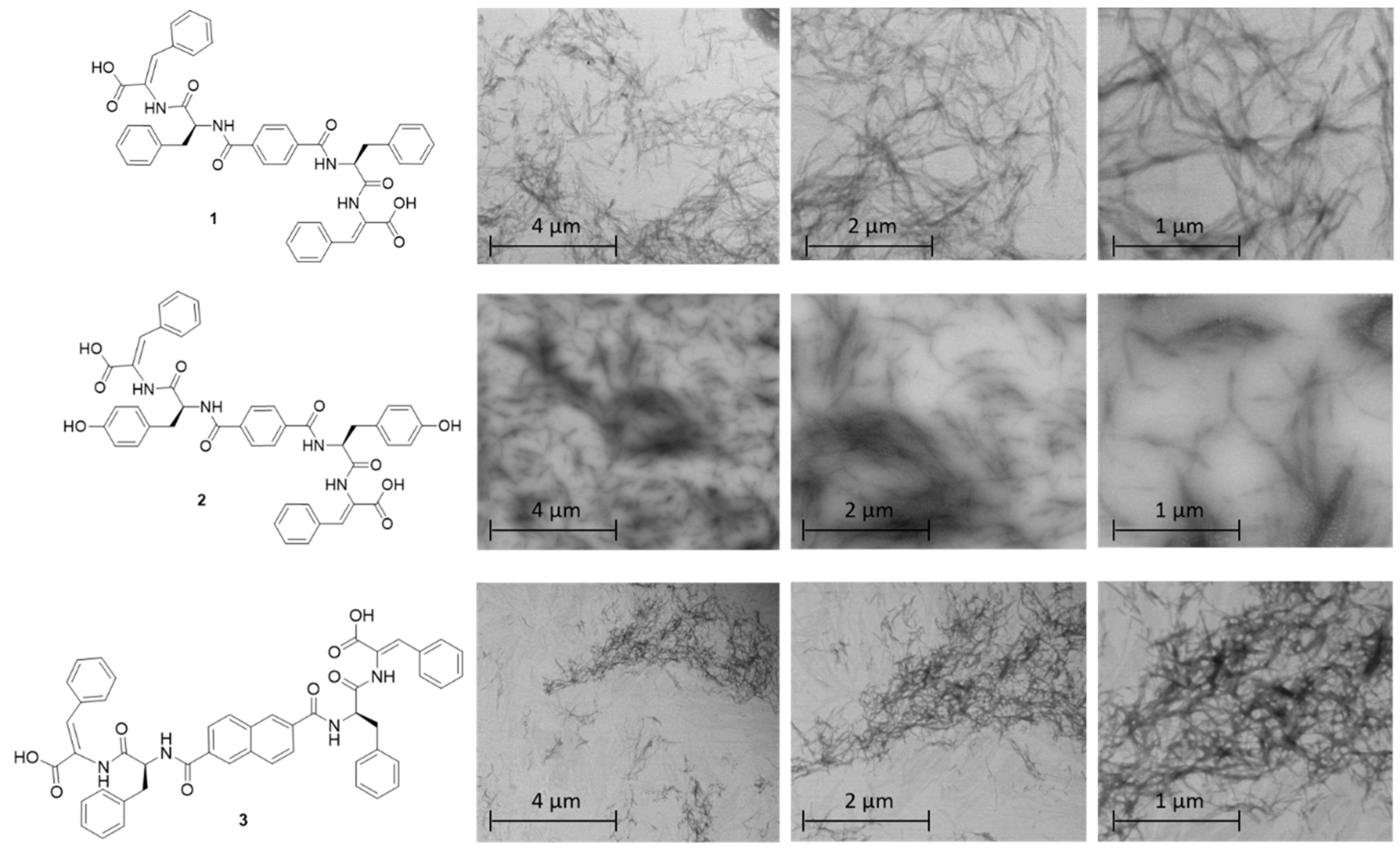
2.3. STEM Studies
2.4. Circular Dichroism (CD) Spectroscopy
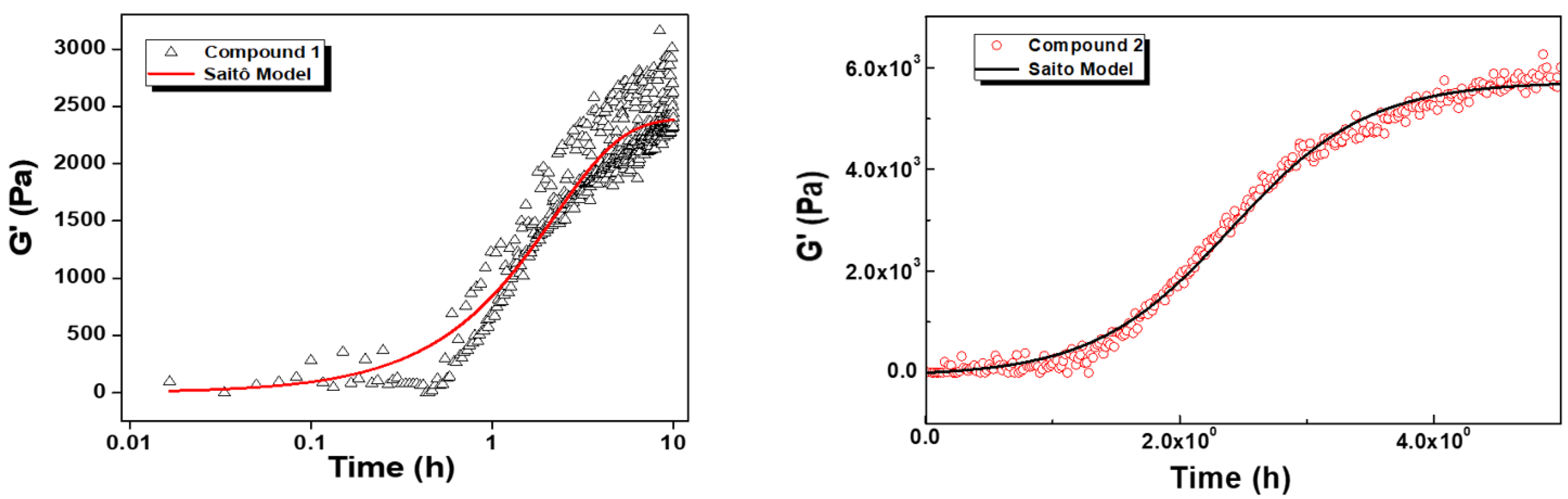
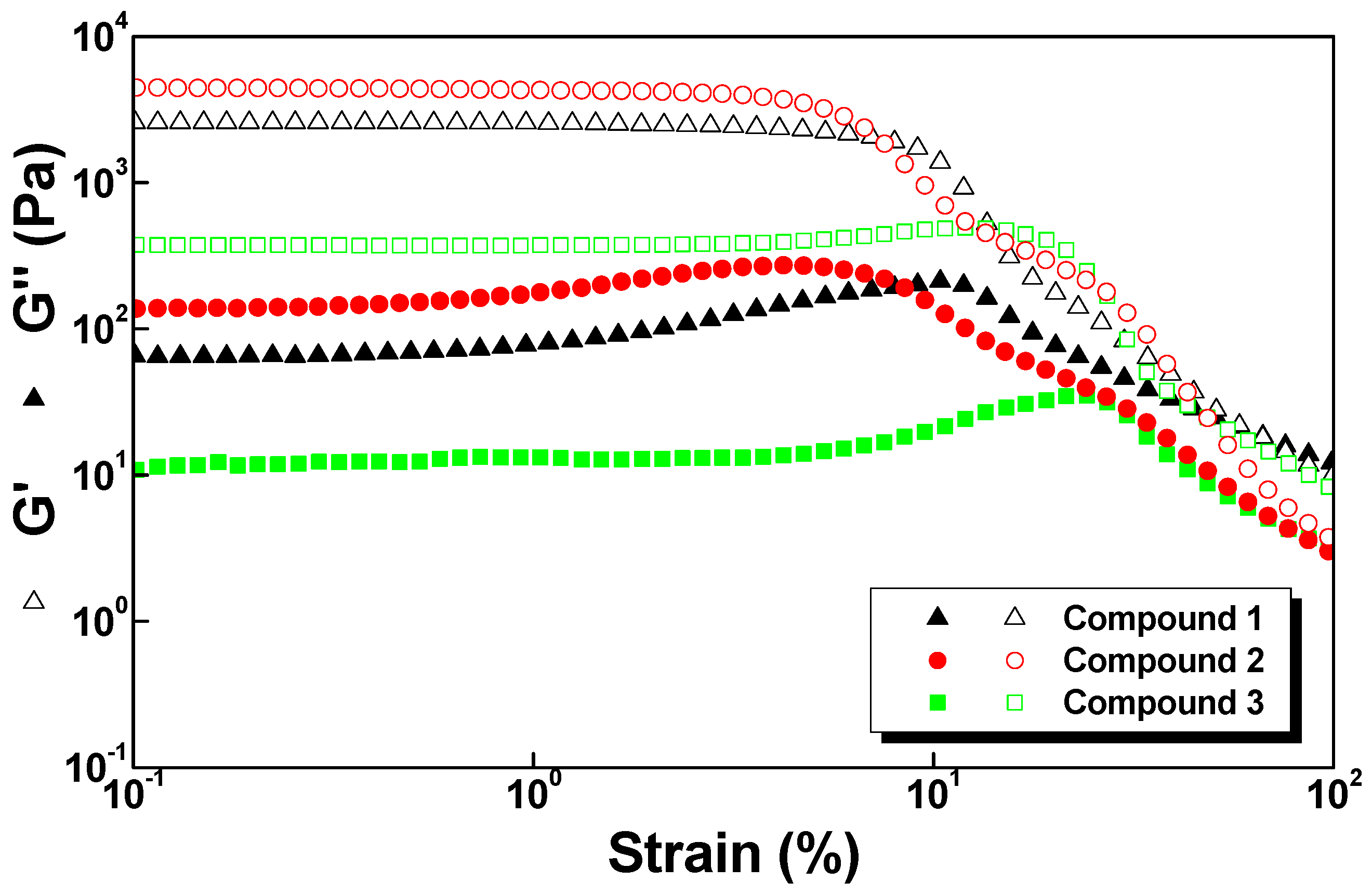
2.5. Rheology
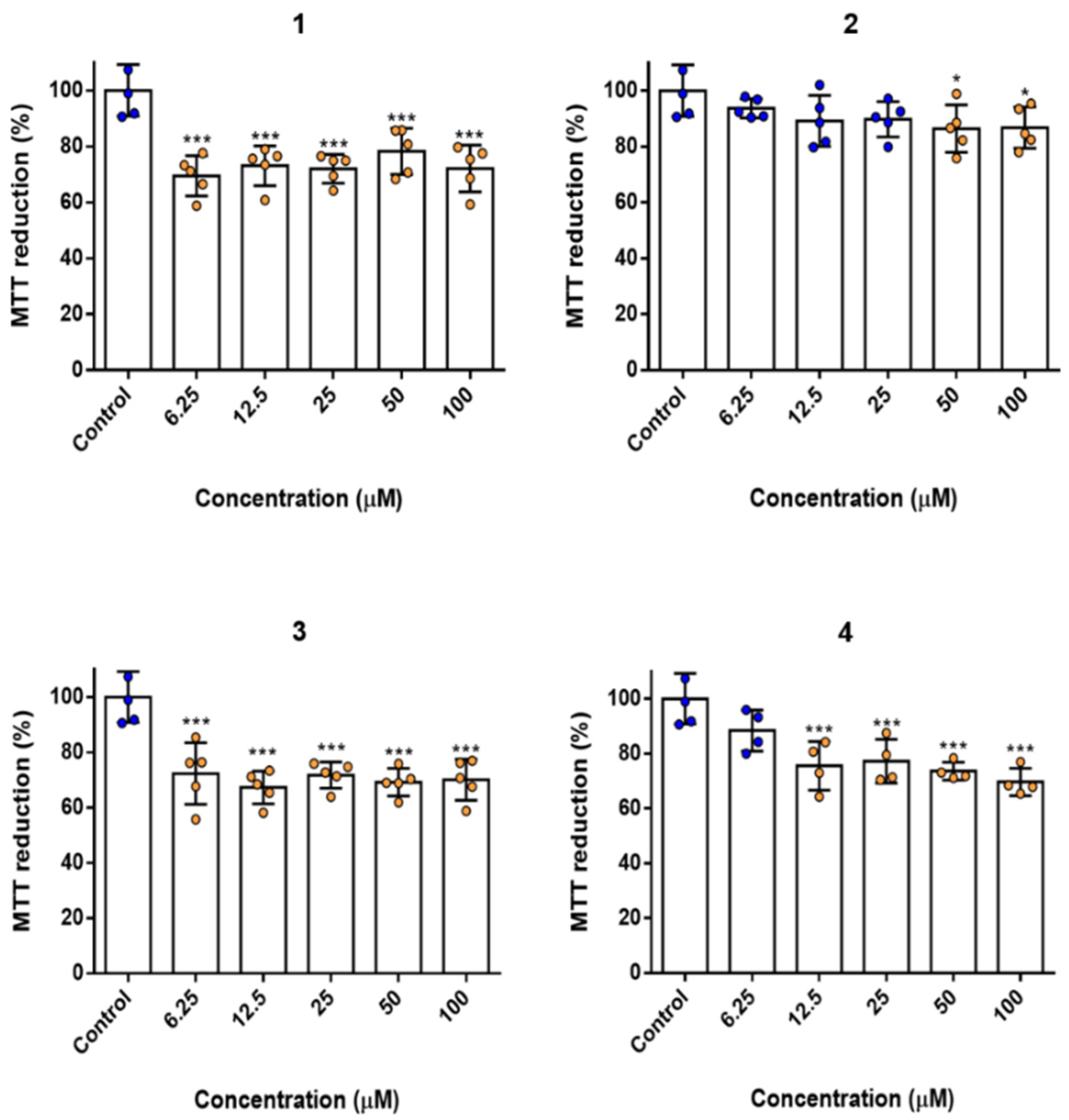
2.6. Biocompatibility and Cytotoxicity Studies
2.7. Drug Release Studies
3. Conclusions
4. Materials and Methods
4.1. Synthesis
4.1.1. Compound 9
4.1.2. Compound 1
4.1.3. Compound 10
4.1.4. Compound 2
4.1.5. Compound 11
4.1.6. Compound 3
4.1.7. Compound 12
4.1.8. Compound 4
4.2. Hydrogel Preparation
4.3. Rheology
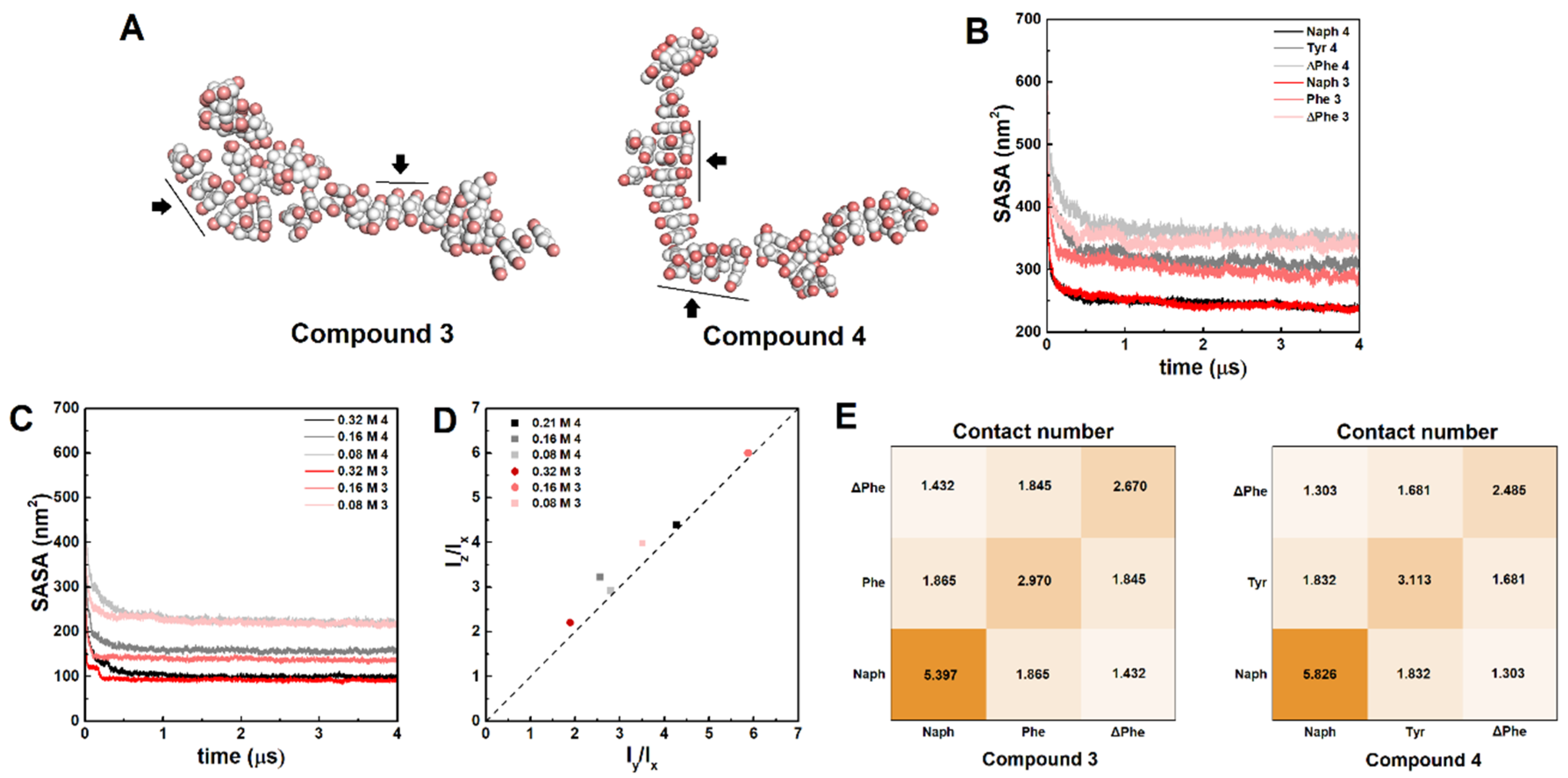
4.4. Molecular Dynamics
4.5. CD Spectroscopy
4.6. Scanning Transmission Electron Microscopy (STEM)
4.7. Sustained Release Assays
4.8. Cell Culture
4.9. MTT Assay/LDH Leakage
4.10. DNA/Protein Quantification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abraham, B.L.; Toriki, E.S.; Tucker, N.D.J.; Nilsson, B.L. Electrostatic interactions regulate the release of small molecules from supramolecular hydrogels. J. Mater. Chem. B 2020, 8, 6366–6377. [Google Scholar] [CrossRef]
- Song, Y.; Gao, J.; Xu, X.; Zhao, H.; Xue, R.; Zhou, J.; Hong, W.; Qiu, H. Fabrication of thermal sensitive folic acid based supramolecular hybrid gels for injectable drug release gels. Mater. Sci. Eng. C 2017, 75, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Jayawarna, V.; Richardson, S.M.; Hirst, A.R.; Hodson, N.W.; Saiani, A.; Gough, J.E.; Ulijn, R.V. Introducing chemical functionality in Fmoc-peptide gels for cell culture. Acta Biomater. 2009, 5, 934–943. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, M.; Zhang, Y.; Yin, J.; Pei, R. Nanocomposite hydrogels for tissue engineering applications. Nanoscale 2020, 12, 14976–14995. [Google Scholar] [CrossRef]
- Yadav, N.; Chauhan, M.K.; Chauhan, V.S. Short to ultrashort peptide-based hydrogels as a platform for biomedical applications. Biomater. Sci. 2020, 8, 84–100. [Google Scholar] [CrossRef]
- Adams, D.J. Dipeptide and Tripeptide Conjugates as Low-Molecular-Weight Hydrogelators. Macromol. Biosci. 2011, 11, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Jervis, P.J.; Amorim, C.; Pereira, T.; Martins, J.A.; Ferreira, P.M.T. Exploring the properties and potential biomedical applications of NSAID-capped peptide hydrogels. Soft Matter 2020, 16, 10001–10012. [Google Scholar] [CrossRef]
- Arokianathan, J.F.; Ramya, K.A.; Janeena, A.; Deshpande, A.P.; Ayyadurai, N.; Leemarose, A.; Shanmugam, G. Non-proteinogenic amino acid based supramolecular hydrogel material for enhanced cell proliferation. Colloids Surf. B Biointerfaces 2020, 185, 110581. [Google Scholar] [CrossRef]
- Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Metal- and Anion-Binding Supramolecular Gels. Chem. Rev. 2010, 110, 1960–2004. [Google Scholar] [CrossRef]
- Draper, E.R.; Adams, D.J. Controlling the Assembly and Properties of Low-Molecular-Weight Hydrogelators. Langmuir 2019, 35, 6506–6521. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, G.; Ma, M.; Gao, Y.; Xu, B. Conjugates of naphthalene and dipeptides produce molecular hydrogelators with high efficiency of hydrogelation and superhelical nanofibers. J. Mater. Chem. 2007, 17, 850–854. [Google Scholar] [CrossRef]
- Vilaça, H.; Hortelão, A.C.L.; Castanheira, E.M.S.; Queiroz, M.-J.R.P.; Hilliou, L.; Hamley, I.W.; Martins, J.A.; Ferreira, P.M.T. Dehydrodipeptide Hydrogelators Containing Naproxen N-Capped Tryptophan: Self-Assembly, Hydrogel Characterization, and Evaluation as Potential Drug Nanocarriers. Biomacromolecules 2015, 16, 3562–3573. [Google Scholar] [CrossRef]
- Vilaça, H.; Pereira, G.; Castro, T.G.; Hermenegildo, B.F.; Shi, J.; Faria, T.Q.; Micaêlo, N.; Brito, R.M.M.; Xu, B.; Castanheira, E.M.S.; et al. New self-assembled supramolecular hydrogels based on dehydropeptides. J. Mater. Chem. B 2015, 3, 6355–6367. [Google Scholar] [CrossRef]
- Carvalho, A.; Gallo, J.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Hilliou, L.; Ferreira, P.M.T.; Bañobre-López, M.; Martins, J.A. Magnetic Dehydrodipeptide-Based Self-Assembled Hydrogels for Theragnostic Applications. Nanomaterials 2019, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Veloso, S.R.S.; Magalhães, C.A.B.; Rodrigues, A.R.O.; Vilaça, H.; Queiroz, M.-J.R.P.; Martins, J.A.; Coutinho, P.J.G.; Ferreira, P.M.T.; Castanheira, E.M.S. Novel dehydropeptide-based magnetogels containing manganese ferrite nanoparticles as antitumor drug nanocarriers. Phys. Chem. Chem. Phys. 2019, 21, 10377–10390. [Google Scholar] [CrossRef] [PubMed]
- Vilaça, H.; Castro, T.; Costa, F.M.G.; Melle-Franco, M.; Hilliou, L.; Hamley, I.W.; Castanheira, E.M.S.; Martins, J.A.; Ferreira, P.M.T. Self-assembled RGD dehydropeptide hydrogels for drug delivery applications. J. Mater. Chem. B 2017, 5, 8607–8617. [Google Scholar] [CrossRef] [PubMed]
- Veloso, S.R.S.; Jervis, P.J.; Silva, J.F.G.; Hilliou, L.; Moura, C.; Pereira, D.M.; Coutinho, P.J.G.; Martins, J.A.; Castanheira, E.M.S.; Ferreira, P.M.T. Supramolecular ultra-short carboxybenzyl-protected dehydropeptide-based hydrogels for drug delivery. Mater. Sci. Eng. C 2021, 122, 111869. [Google Scholar] [CrossRef]
- Hughes, J.R.; Miller, A.S.; Wallace, C.E.; Vemuri, G.N.; Lovine, P.M. Biomedically Relevant Applications of Bolaamphiphiles and Bolaamphiphile-Containing Materials. Med. Pharm. Chem. 2021, 8, 1269. [Google Scholar]
- Qiu, F.; Chen, Y.; Tang, C.; Zhou, Q.; Wang, C.; Shi, Y.K.; Zhao, X. De Novo Design of a Bolaamphiphilic Peptide with only natural amino acids. Macromol. Biosci. 2008, 8, 1053–1059. [Google Scholar]
- Estroff, L.A.; Hamilton, A.D. Water Gelation by Small Organic Molecules. Chem. Rev. 2004, 104, 1201–1217. [Google Scholar]
- Dou, X.; Li, P.; Zhang, D.; Feng, C.L. C2-symmetric benzene-based hydrogels with unique layered structures for controllable organic dye adsorption. Soft Matter 2012, 8, 3231–3238. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Tang, Y.; Yamamoto, T.; Yao, Y.; Guterman, T.; Zilberzwige-Tal, S.; Adadi, N.; Ji, W.; Dvir, T.; Ramamoorthy, A.; et al. Unusual two-step assembly of a minimalistic dipeptide-based functional hypergelator. Adv. Mater. 2020, 32, 1906043. [Google Scholar]
- Frederix, P.W.J.M.; Scott, G.G.; Abul-Haija, Y.M.; Kalafatovic, D.; Pappas, C.G.; Javid, N.; Hunt, N.T.; Ulijn, R.V.; Tuttle, T. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 2015, 7, 30–37. [Google Scholar]
- Moreira, I.P.; Scott, G.G.; Ulijn, R.V.; Tuttle, T. Computational prediction of tripeptide-dipeptide co-assembly. Mol. Phys. 2019, 117, 1151–1163. [Google Scholar]
- Guo, C.; Luo, Y.; Zhou, R.; Wei, G. Triphenylalanine peptides self-assemble into nanospheres and nanorods that are different from the nanovesicles and nanotubes formed by diphenylalanine peptides. Nanoscale 2014, 6, 2800–2811. [Google Scholar] [CrossRef]
- Frederix, P.W.J.M.; Ulijn, R.V.; Hunt, N.T.; Tuttle, T. Virtual screening for dipeptide aggregation: Toward predictive tools for peptide self-assembly. J. Phys. Chem. Lett. 2011, 2, 2380–2384. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, H.; Poh, P.S.P.; Machens, H.-G.; Schilling, A.F. Hydrogels for Engineering of Perfusable Vascular Networks. Int. J. Mol. Sci. 2015, 16, 15997–16016. [Google Scholar] [CrossRef]
- Adams, D.J.; Mullen, L.M.; Berta, M.; Chen, L.; Frith, W.J. Relationship between molecular structure, gelation behaviour and gel properties of Fmoc-dipeptides. Soft Matter 2010, 6, 1971–1980. [Google Scholar] [CrossRef]
- Kamihira, M.; Naito, A.; Tuzi, S.; Saitô, H.; Nosaka, A.Y. Conformational transitions and fibrillation mechanism of human calcitonin as studied by high-resolution solid-state 13C NMR. Protein Sci. 2000, 9, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Kini, S.; Bahadur, D.; Panda, D. Mechanism of Anti-Cancer Activity of Benomyl Loaded Nanoparticles in Multidrug Resistant Cancer Cells. J. Biomed. Nanotechnol. 2015, 11, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Bagaria, A.; Mishra, A.; Mathur, P.; Basu, A.; Ramakumar, S.; Chauhan, V.S. Self-Assembly of a Dipeptide- Containing Conformationally Restricted Dehydrophenylalanine Residue to Form Ordered Nanotubes. Adv. Mater. 2007, 19, 858–861. [Google Scholar] [CrossRef]
- Mishra, A.; Panda, J.J.; Basu, A.; Chauhan, V.S. Nanovesicles Based on Self-Assembly of Conformationally Constrained Aromatic Residue Containing Amphiphilic Dipeptides. Langmuir 2008, 24, 4571–4576. [Google Scholar] [CrossRef] [PubMed]
- Parween, S.; Misra, A.; Ramakumar, S.; Chauhan, V.S. Self-assembled dipeptide nanotubes constituted by flexible β-phenylalanine and conformationally constrained α,β-dehydrophenylalanine residues as drug delivery system. J. Mater. Chem. B 2014, 2, 3096–3106. [Google Scholar] [CrossRef]
- Veloso, S.R.S.; Martins, J.A.; Hilliou, L.; Amorim, C.O.; Amaral, V.S.; Almeida, B.G.; Jervis, P.J.; Moreira, R.; Pereira, D.M.; Coutinho, P.J.G.; et al. Dehydropeptide-based plasmonic magnetogels: A supramolecular composite nanosystem for multimodal cancer therapy. J. Mater. Chem. B 2020, 8, 45–64. [Google Scholar] [CrossRef]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09, Revision, A.02; Fox, Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Huang, W.L.; Lin, Z.X.; van Gunsteren, W.F. Rapid sampling of folding equilibria of β-Peptides in methanol using a supramolecular solvent model. J. Chem. Theory Comput. 2011, 5, 1237–1243. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. Biophys. Lett. 2011, 40, 843. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Abraham, M.J.; van der Spoel, D.; Lindahl, E.; Hess, B. The GROMACS Development Team. GROMACS User Manual Version 5.1.4. 2016. Available online: www.gromacs.org (accessed on 15 March 2021).
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R.J. Molecular dynamics with coupling to an external bath. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Szała, B.; Molski, A. Aggregation kinetics of short peptides: All-atom and coarse-grained molecular dynamics study. Biophys. Chem. 2019, 53, 106219. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Critical Gelation Concentration (cgc) | GDL Concentration (wt %) | pH | cLogP * | |
|---|---|---|---|---|---|
| wt % | mM | ||||
| 1 | 0.3 | 4.0 | 0.4 | 5.10 | 5.82 |
| 2 | 0.4 | 5.1 | 0.4 | 4.15 | 4.49 |
| 3 | 0.3 | 3.7 | 0.4 | 5.30 | 6.69 |
| 4 | No hydrogel | 5.55 | |||
| Compound | G0 (Pa) | dG (Pa) | kn (h−1) | k (h−1) | R2 |
|---|---|---|---|---|---|
| 1 | 0 ± 57 | 2392 ± 66 | 0.40 ± 0.04 | 0.18 ± 0.10 | 0.89 |
| 2 | 0 ± 25 | 5722 ± 39 | 0.02 ± 0.001 | 1.84 ± 0.04 | 0.99 |
| Hydrogel | G′ (Pa) | G″ (Pa) |
|---|---|---|
| 1 | 2.5 × 103 | 6.5 × 101 |
| 2 | 4.5 × 103 | 1.4 × 102 |
| 3 | 3.7 × 102 | 1.3 × 101 |
| Hydrogelator | MB Released (%) | MO Released (%) | Ciprofloxacin Released (%) |
|---|---|---|---|
| 1 | 12.1 | 60.9 | 32.3 |
| 2 | 7.63 | 96.2 | 58.8 |
| 3 | 9.55 | 60.3 | 20.3 |
| Cargo | Hydrogel | K | n | R2 |
|---|---|---|---|---|
| Methyl Orange | 1 | 0.0200 | 0.4000 | 0.9509 |
| 2 | 0.0900 | 0.2700 | 0.9785 | |
| 3 | 0.0400 | 0.3300 | 0.9497 | |
| Ciprofloxacin | 1 | 0.0200 | 0.3300 | 0.8677 |
| 2 | 0.0030 | 0.5800 | 0.8956 | |
| 3 | 0.0001 | 0.6300 | 0.9406 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amorim, C.; Veloso, S.R.S.; Castanheira, E.M.S.; Hilliou, L.; Pereira, R.B.; Pereira, D.M.; Martins, J.A.; Jervis, P.J.; Ferreira, P.M.T. Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems. Gels 2021, 7, 52. https://doi.org/10.3390/gels7020052
Amorim C, Veloso SRS, Castanheira EMS, Hilliou L, Pereira RB, Pereira DM, Martins JA, Jervis PJ, Ferreira PMT. Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems. Gels. 2021; 7(2):52. https://doi.org/10.3390/gels7020052
Chicago/Turabian StyleAmorim, Carolina, Sérgio R. S. Veloso, Elisabete M. S. Castanheira, Loic Hilliou, Renato B. Pereira, David M. Pereira, José A. Martins, Peter J. Jervis, and Paula M. T. Ferreira. 2021. "Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems" Gels 7, no. 2: 52. https://doi.org/10.3390/gels7020052
APA StyleAmorim, C., Veloso, S. R. S., Castanheira, E. M. S., Hilliou, L., Pereira, R. B., Pereira, D. M., Martins, J. A., Jervis, P. J., & Ferreira, P. M. T. (2021). Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems. Gels, 7(2), 52. https://doi.org/10.3390/gels7020052