On the Race for More Stretchable and Tough Hydrogels




Abstract

{kind=link}
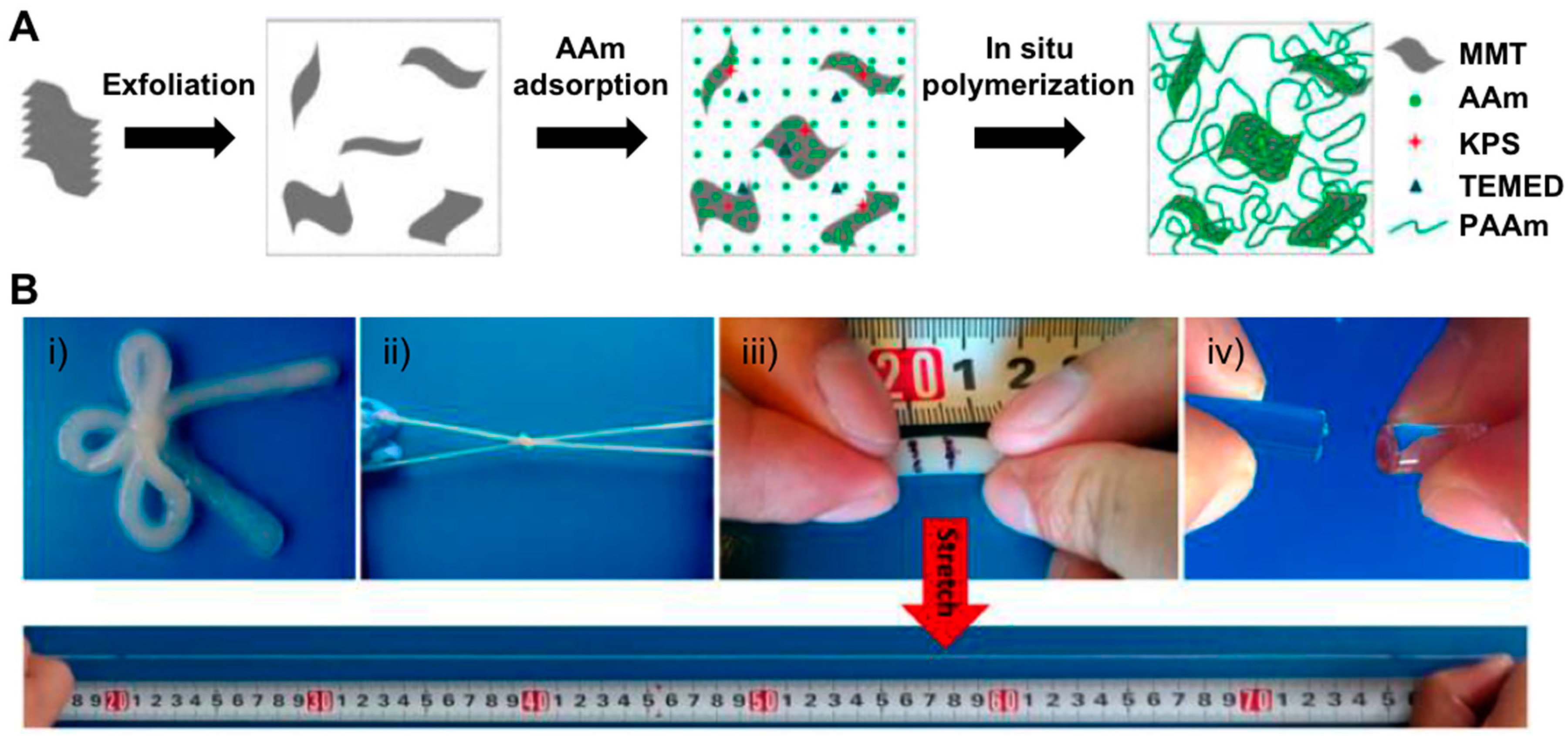{kind=link}
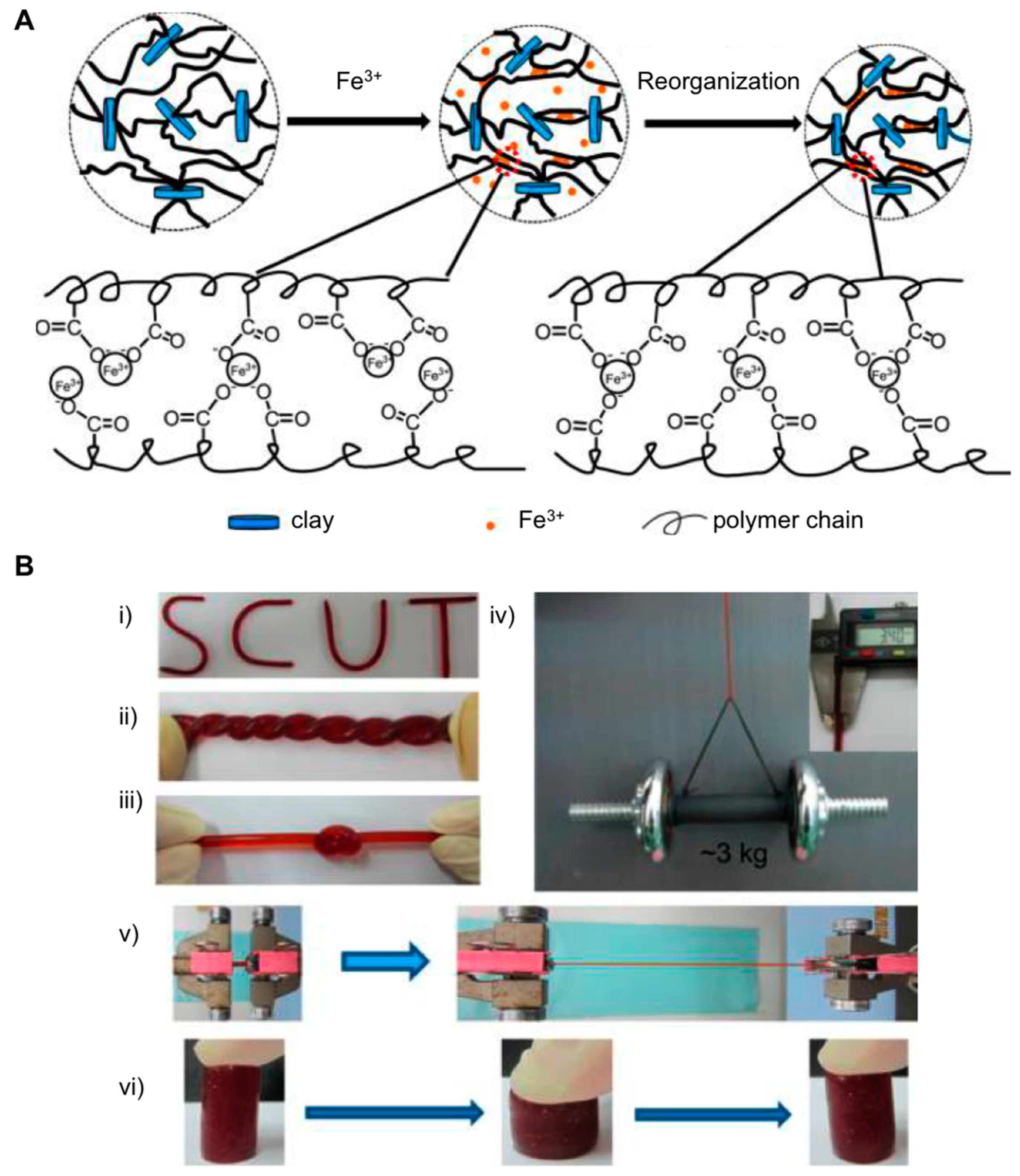{kind=link}
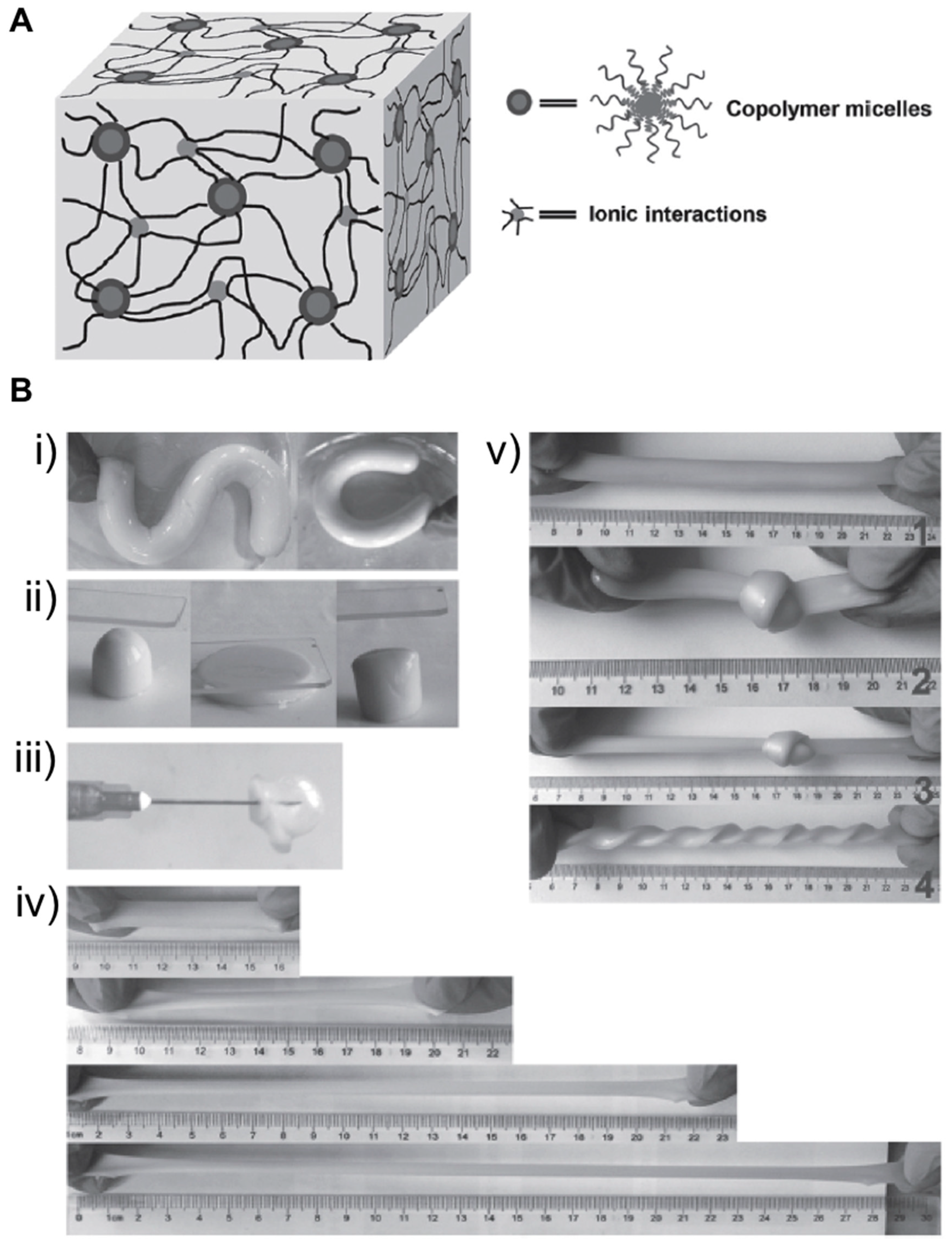{kind=link}
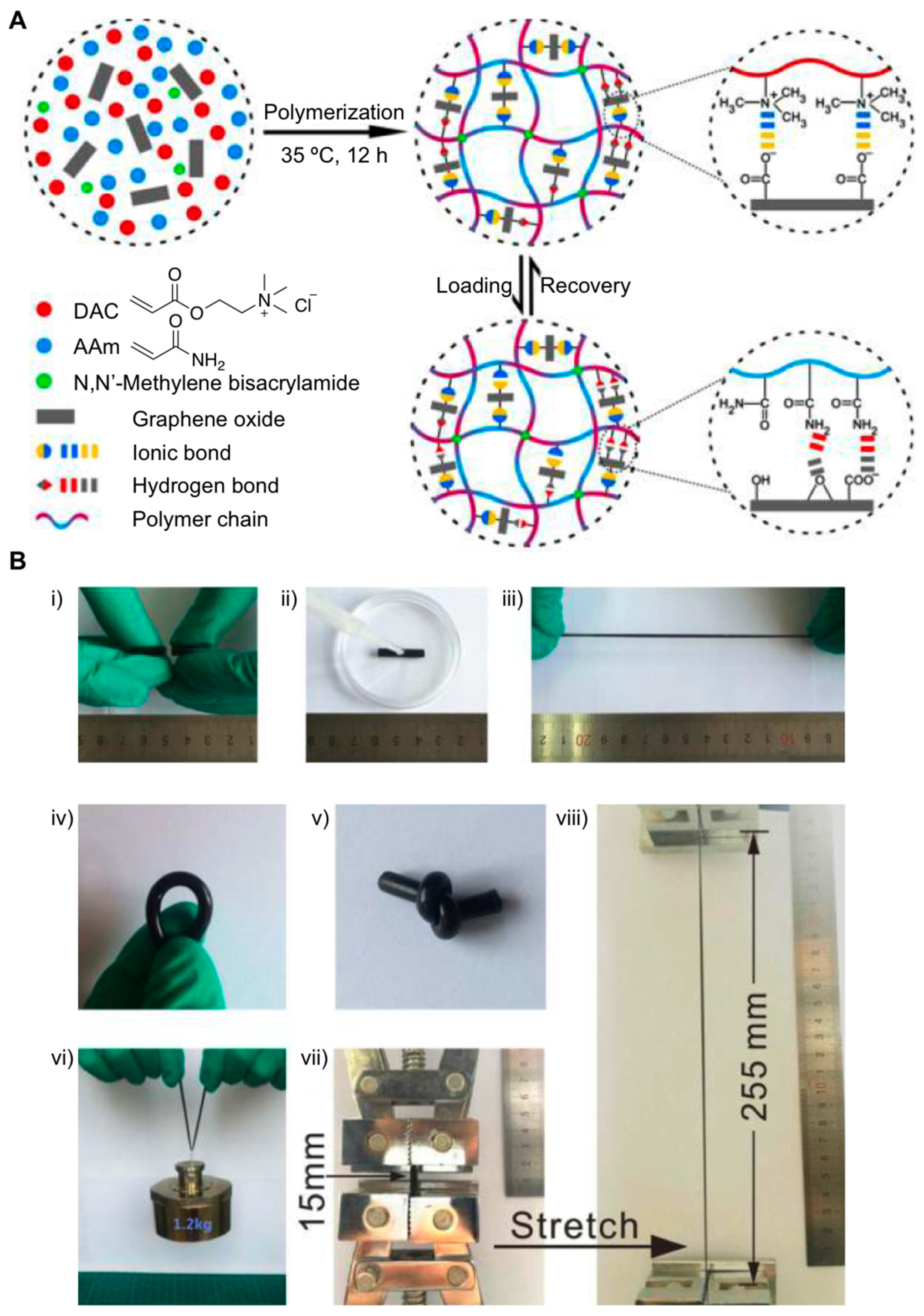{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Stretchable and Tough Hydrogels from Synthetic Polymers
2.1. Nanocomposite Hydrogels
2.2. Supramolecular Hydrogels
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAc | acrylic acid |
| AAm | acrylamide |
| AIBN | 2,2′-azobis(2-methylpropionitrile) |
| BA | butyl acrylate |
| BHT | butyl hydroxyl toluene |
| CaGP | 2-glycerol phosphate |
| CB[8] | cucurbit[8]uril |
| α-CD | α-cyclodextrin |
| 3D | tridimensional |
| DAC | 2-(dimethylamino)ethylacrylatemethyl chloride |
| DC | double cross-linked (DBTDL: dibutyltin dilaurate |
| DCPA | dicyclopentyl acrylate |
| D-Gel | dual-cross-linked hydrogels |
| DLS | dynamic light scattering |
| DMBA | dimethylbutanoic acid |
| DN | double-network |
| DPC | dual physically cross-linked |
| EDPOA | ethyl-2-[4-(dihydroxyphosphoryl)-2-oxabutyl]-acrylate |
| G-6-P | glucose-6-phosphate |
| HMA | hexadecyl methacrylate |
| HPR | hydropropylated polyrotaxane |
| IPDI | isophorone diisocyanate |
| IPN | interpenetrating polymer network |
| KPS | potassium peroxodisulfate |
| MBAAm | N,N′-methylene-bis(acrylamide) |
| M-Gel | mono-cross-linked hydrogels |
| MMSPH | macromolecular microspheres |
| MPS | 3-methacryloxypropyltrimethoxysilane |
| NaAAc | sodium acrylic acid |
| NaMMT | sodium montmorillonite |
| NCHs | nanocomposite hydrogels |
| NIPA | N-isopropylacrylamide |
| LMWG | low molecular weight gelators |
| PAAc | polyacrylic acid |
| P(AAm-co-AAc) | poly(acrylamide-co-acrylic acid) |
| P(AAm-co-LMA) | poly(acrylamide-co-laurylmethacrylate) |
| PAAm-l-MBAm | Polyacrylamide-l-N,N′-methylenebis-(acrylamide) |
| PDMA-l-TEG | poly-N,N-dimethyl acrylamide-l-TEG |
| PEG | polyethylene glycol |
| PF127 | pluronic F127 |
| PHEA-l-TEG | triethylene glycol dimethacrylate |
| PNIPAm | poly(N-isopropyl acrylamide) |
| PUU | polyurethane-urea |
| Q-CNCs | quaternized cellulose nanocrystals |
| TEMED | N,N,N,N-tetramethylethylenediamine |
| SEM | scanning electron microscopy |
| SDS | sodium dodecyl sulfate |
| SMA | stearyl methacrylate |
| SPBs | spherical polymer brushes |
| TEM | transmission electron microscopy |
References
- Ullah, F.; Othman, M.B.H.; Javed, F.; Ahmad, Z.; Akil, H.M. Classification, processing and application of hydrogels: A review. Mater. Sci. Eng. C 2015, 57, 414–433. [Google Scholar] [CrossRef]
- Richter, A.; Paschew, G.; Klatt, S.; Lienig, J.; Arndt, K.-F.; Adler, H.-J.P. Review on Hydrogel-based pH Sensors and Microsensors. Sensors 2008, 8, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, S.; Dubruel, P.; Schacht, E. Biopolymer-based hydrogels as scaffolds for tissue engineering applications: A review. Biomacromolecules 2011, 12, 1387–1408. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Varaprasad, K.; Raghavendra, G.M.; Jayaramudu, T.; Yallapu, M.M.; Sadiku, R. A mini review on hydrogels classification and recent developments in miscellaneous applications. Mater. Sci. Eng. C 2017, 79, 958–971. [Google Scholar] [CrossRef]
- Sood, N.; Bhardwaj, A.; Mehta, S.; Mehta, A. Stimuli-responsive hydrogels in drug delivery and tissue engineering. Drug Deliv. 2016, 23, 758–780. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Coviello, T.; Matricardi, P.; Marianecci, C.; Alhaique, F. Polysaccharide hydrogels for modified release formulations. J. Control. Release 2007, 119, 5–24. [Google Scholar] [CrossRef]
- Anseth, K.S.; Bowman, C.N.; Brannon-Peppas, L. Mechanical properties of hydrogels and their experimental determination. Biomaterials 1996, 17, 1647–1657. [Google Scholar] [CrossRef]
- Pushpamalar, J.; Veeramachineni, A.K.; Owh, C.; Loh, X.J. Biodegradable Polysaccharides for Controlled Drug Delivery. Chempluschem 2016, 81, 504–514. [Google Scholar] [CrossRef]
- Grijalvo, S.; Mayr, J.; Eritja, R.; Díaz, D.D. Biodegradable liposome-encapsulated hydrogels for biomedical applications: A marriage of convenience. Biomater. Sci. 2016, 555–574. [Google Scholar] [CrossRef]
- Marsich, E.; Travan, A.; Donati, I.; Di Luca, A.; Benincasa, M.; Crosera, M.; Paoletti, S. Biological response of hydrogels embedding gold nanoparticles. Colloids Surf. B Biointerfaces 2011, 83, 331–339. [Google Scholar] [CrossRef]
- Kopeĉek, J. Hydrogels: From soft contact lenses and implants to self-assembled nanomaterials. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 5929–5946. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, J.; Yan, H.; Wang, Y.; Zhao, Y.; Feng, B.; Duan, K.; Weng, J. An injectable supramolecular self-healing bio-hydrogel with high stretchability, extensibility and ductility, and a high swelling ratio. J. Mater. Chem. B 2017, 5, 7021–7034. [Google Scholar] [CrossRef]
- Sun, G.; Li, Z.; Liang, R.; Weng, L.T.; Zhang, L. Super stretchable hydrogel achieved by non-aggregated spherulites with diameters <5 nm. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Le, X.; Lu, W.; Zheng, J.; Tong, D.; Zhao, N.; Ma, C.; Xiao, H.; Zhang, J.; Huang, Y.; Chen, T. Stretchable supramolecular hydrogels with triple shape memory effect. Chem. Sci. 2016, 7, 6715–6720. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Simon, J.R.; Ghoorchian, A.; Scholl, Z.; Lin, S.; Rubinstein, M.; Marszalek, P.; Chilkoti, A.; López, G.P.; Zhao, X. Strong, Tough, Stretchable, and Self-Adhesive Hydrogels from Intrinsically Unstructured Proteins. Adv. Mater. 2017, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, J.P.; Kholiya, F.; Vadodariya, N.; Budheliya, V.M.; Gogda, A.; Meena, R. Carboxymethylagarose-based multifunctional hydrogel with super stretchable, self-healable having film and fiber forming properties. Arab. J. Chem. 2018. [Google Scholar] [CrossRef]
- Costa, A.M.S.; Mano, J.F. Extremely strong and tough hydrogels as prospective candidates for tissue repair—A review. Eur. Polym. J. 2015, 72, 344–364. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Peak, C.W.; Wilker, J.J.; Schmidt, G. A review on tough and sticky hydrogels. Colloid Polym. Sci. 2013, 291, 2031–2047. [Google Scholar] [CrossRef]
- Dragan, E.S. Advances in interpenetrating polymer network hydrogels and their applications. Pure Appl. Chem. 2014, 86, 1707–1721. [Google Scholar] [CrossRef]
- Haque, M.A.; Kurokawa, T.; Gong, J.P. Super tough double network hydrogels and their application as biomaterials. Polymer (Guildf) 2012, 53, 1805–1822. [Google Scholar] [CrossRef]
- Gong, J.P. Why are double network hydrogels so tough? Soft Matter 2010, 6, 2583–2590. [Google Scholar] [CrossRef]
- Fares, M.M.; Shirzaei Sani, E.; Portillo Lara, R.; Oliveira, R.B.; Khademhosseini, A.; Annabi, N. Interpenetrating network gelatin methacryloyl (GelMA) and pectin-g-PCL hydrogels with tunable properties for tissue engineering. Biomater. Sci. 2018, 6, 2938–2950. [Google Scholar] [CrossRef] [PubMed]
- Rennerfeldt, D.A.; Renth, A.N.; Talata, Z.; Gehrke, S.H.; Detamore, M.S. Tuning mechanical performance of poly(ethylene glycol) and agarose interpenetrating network hydrogels for cartilage tissue engineering. Biomaterials 2013, 34, 8241–8257. [Google Scholar] [CrossRef]
- Truong, V.X.; Ablett, M.P.; Richardson, S.M.; Hoyland, J.A.; Dove, A.P. Simultaneous Orthogonal Dual-Click Approach to Tough, in-Situ-Forming Hydrogels for Cell Encapsulation. J. Am. Chem. Soc. 2015, 137, 1618–1622. [Google Scholar] [CrossRef]
- Gu, Z.; Huang, K.; Luo, Y.; Zhang, L.; Kuang, T.; Chen, Z.; Liao, G. Double network hydrogel for tissue engineering. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1520. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Olsen, B.D.; Khademhosseini, A. The mechanical properties and cytotoxicity of cell-laden double-network hydrogels based on photocrosslinkable gelatin and gellan gum biomacromolecules. Biomaterials 2012, 33, 3143–3152. [Google Scholar] [CrossRef]
- Rodell, C.B.; Dusaj, N.N.; Highley, C.B.; Burdick, J.A. Injectable and Cytocompatible Tough Double-Network Hydrogels through Tandem Supramolecular and Covalent Crosslinking. Adv. Mater. 2016, 28, 8419–8424. [Google Scholar] [CrossRef]
- Gaharwar, A.K.; Peppas, N.A.; Khademhosseini, A. Nanocomposite hydrogels for biomedical applications. Biotechnol. Bioeng. 2014, 111, 441–453. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Samanta, S.K. Soft-Nanocomposites of Nanoparticles and Nanocarbons with Supramolecular and Polymer Gels and Their Applications. Chem. Rev. 2016, 116, 11967–12028. [Google Scholar] [CrossRef] [PubMed]
- Rahman, C.V.; Saeed, A.; White, L.J.; Gould, T.W.A.; Kirby, G.T.S.; Sawkins, M.J.; Alexander, C.; Rose, F.R.A.J.; Shakesheff, K.M. Chemistry of polymer and ceramic-based injectable scaffolds and their applications in regenerative medicine. Chem. Mater. 2012, 24, 781–795. [Google Scholar] [CrossRef]
- Zhao, L.Z.; Zhou, C.H.; Wang, J.; Tong, D.S.; Yu, W.H.; Wang, H. Recent advances in clay mineral-containing nanocomposite hydrogels. Soft Matter 2015, 11, 9229–9246. [Google Scholar] [CrossRef]
- Wahid, F.; Zhong, C.; Wang, H.-S.; Hu, X.-H.; Chu, L.-Q. Recent Advances in Antimicrobial Hydrogels Containing Metal Ions and Metals/Metal Oxide Nanoparticles. Polymers 2017, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Takehisa, T.; Fan, S. Effects of clay content on the properties of nanocomposite hydrogels composed of poly(N-isopropylacrylamide) and clay. Macromolecules 2002, 35, 10162–10171. [Google Scholar] [CrossRef]
- Song, F.; Li, X.; Wang, Q.; Liao, L.; Zhang, C. Nanocomposite hydrogels and their applications in drug delivery and tissue engineering. J. Biomed. Nanotechnol. 2015, 11, 40–52. [Google Scholar] [CrossRef]
- Gao, G.; Du, G.; Sun, Y.; Fu, J. Self-healable, tough, and ultrastretchable nanocomposite hydrogels based on reversible polyacrylamide/montmorillonite adsorption. ACS Appl. Mater. Interfaces 2015, 7, 5029–5037. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Hu, Y.; Du, Z.; Deng, X.; Wang, T.; Yang, Z.; Zhou, W.; Wang, C. Dual Physically Cross-Linked Hydrogels with High Stretchability, Toughness, and Good Self-Recoverability. Macromolecules 2016, 49, 5660–5668. [Google Scholar] [CrossRef]
- Qin, Z.; Niu, R.; Tang, C.; Xia, J.; Ji, F.; Dong, D.; Zhang, H.; Zhang, S.; Li, J.; Yao, F. A Dual-Crosslinked Strategy to Construct Physical Hydrogels with High Strength, Toughness, Good Mechanical Recoverability, and Shape-Memory Ability. Macromol. Mater. Eng. 2018, 303, 1–12. [Google Scholar] [CrossRef]
- Zhang, T.; Zuo, T.; Hu, D.; Chang, C. Dual Physically Cross-Linked Nanocomposite Hydrogels Reinforced by Tunicate Cellulose Nanocrystals with High Toughness and Good Self-Recoverability. ACS Appl. Mater. Interfaces 2017, 9, 24230–24237. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, M.; Cao, J.; Yan, B.; Yang, W.; Jin, X.; Ma, A.; Chen, W.; Ding, X.; Zhang, G.; et al. Highly Flexible, Tough, and Self-Healable Hydrogels Enabled by Dual Cross-Linking of Triblock Copolymer Micelles and Ionic Interactions. Macromol. Mater. Eng. 2017, 302, 1–5. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.B.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–286. [Google Scholar] [CrossRef]
- Lu, B.; Li, T.; Zhao, H.; Li, X.; Gao, C.; Zhang, S.; Xie, E. Graphene-based composite materials beneficial to wound healing. Nanoscale 2012, 4, 2978. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, D.; Liu, L.; Gai, G. Recent achievements of self-healing graphene/polymer composites. Polymers (Basel) 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Liu, L.; Chen, Q.; Zhang, Q.; Guo, G. Tough, Stretchable, Compressive Novel Polymer/Graphene Oxide Nanocomposite Hydrogels with Excellent Self-Healing Performance. ACS Appl. Mater. Interfaces 2017, 9, 38052–38061. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Liu, Y.-T.; Xie, X.-M. Self-healable, super tough graphene oxide–poly(acrylic acid) nanocomposite hydrogels facilitated by dual cross-linking effects through dynamic ionic interactions. J. Mater. Chem. B 2015, 3, 4001–4008. [Google Scholar] [CrossRef]
- Rauner, N.; Meuris, M.; Zoric, M.; Tiller, J.C. Enzymatic mineralization generates ultrastiff and tough hydrogels with tunable mechanics. Nature 2017, 543, 407–410. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1879. [Google Scholar] [CrossRef]
- Jiang, G.; Liu, C.; Liu, X.; Chen, Q.; Zhang, G.; Yang, M.; Liu, F. Network structure and compositional effects on tensile mechanical properties of hydrophobic association hydrogels with high mechanical strength. Polymer 2010, 51, 1507–1515. [Google Scholar] [CrossRef]
- Huang, T.; Xu, H.; Jiao, K.; Zhu, L.; Brown, H.R.; Wang, H. A novel hydrogel with high mechanical strength: A macromolecular microsphere composite hydrogel. Adv. Mater. 2007, 19, 1622–1626. [Google Scholar] [CrossRef]
- Jiang, F.; Huang, T.; He, C.; Brown, H.R.; Wang, H. Interactions affecting the mechanical properties of macromolecular microsphere composite hydrogels. J. Phys. Chem. B 2013, 117, 13679–13687. [Google Scholar] [CrossRef]
- Ren, X.Y.; Yu, Z.; Liu, B.; Liu, X.J.; Wang, Y.J.; Su, Q.; Gao, G.H. Highly tough and puncture resistant hydrogels driven by macromolecular microspheres. RSC Adv. 2016, 6, 8956–8963. [Google Scholar] [CrossRef]
- Shibayama, M. Structure-mechanical property relationship of tough hydrogels. Soft Matter 2012, 8, 8030. [Google Scholar] [CrossRef]
- Haraguchi, K. Nanocomposite hydrogels. Curr. Opin. Solid State Mater. Sci. 2007, 11, 47–54. [Google Scholar] [CrossRef]
- Ye, E.; Loh, X.J. Polymeric hydrogels and nanoparticles: A merging and emerging field. Aust. J. Chem. 2013, 66, 997–1007. [Google Scholar] [CrossRef]
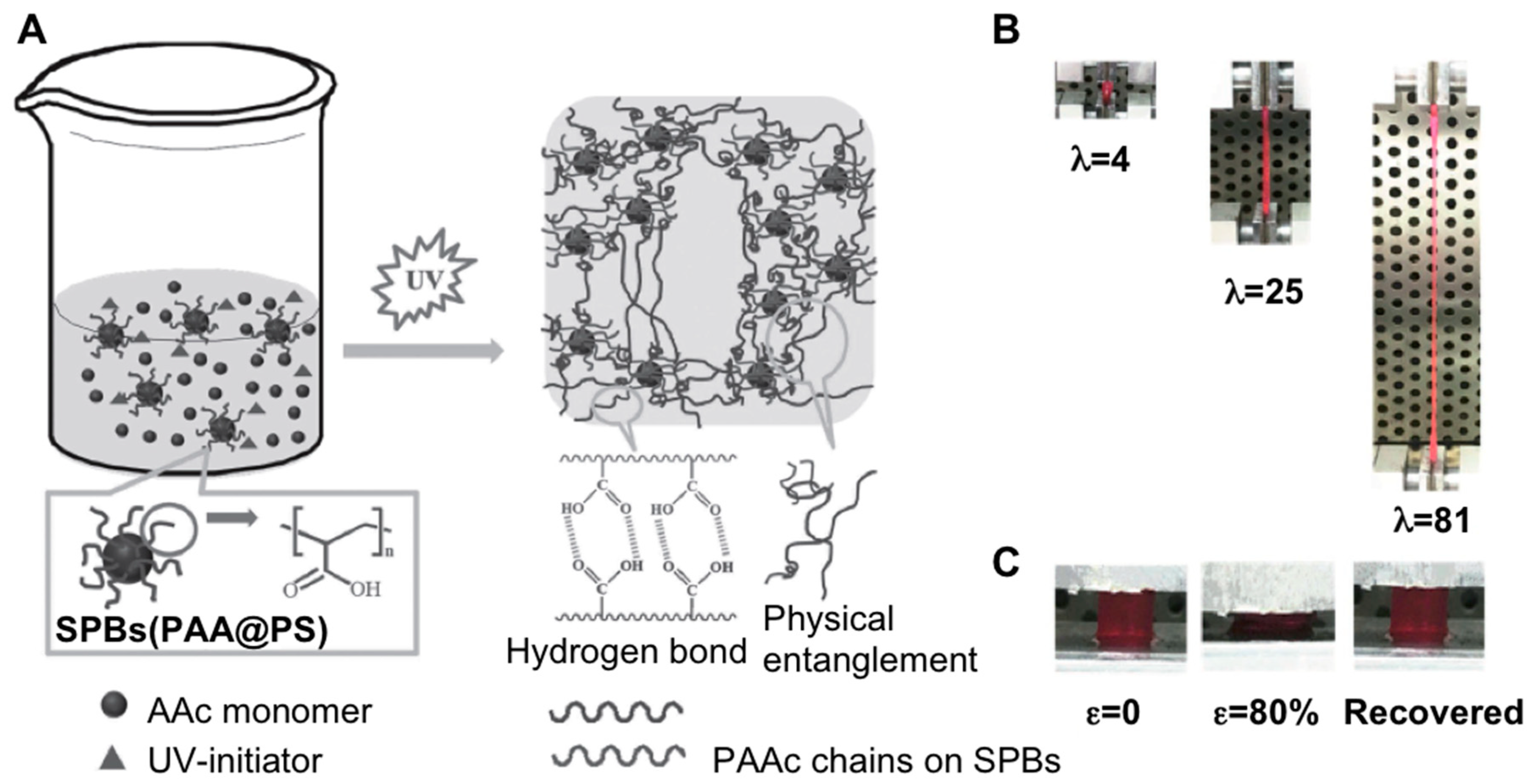
- Xia, S.; Song, S.; Ren, X.; Gao, G. Highly tough, anti-fatigue and rapidly self-recoverable hydrogels reinforced with core–shell inorganic–organic hybrid latex particles. Soft Matter 2017, 13, 6059–6067. [Google Scholar] [CrossRef] [PubMed]
- Reza Saboktakin, M.; Tabatabaei, R.M. Supramolecular hydrogels as drug delivery systems. Int. J. Biol. Macromol. 2015, 75, 426–436. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, N.; Liu, W.; Zhao, X.; Lu, W. Intermolecular hydrogen bonding strategy to fabricate mechanically strong hydrogels with high elasticity and fatigue resistance. Soft Matter 2013, 9, 6331. [Google Scholar] [CrossRef]
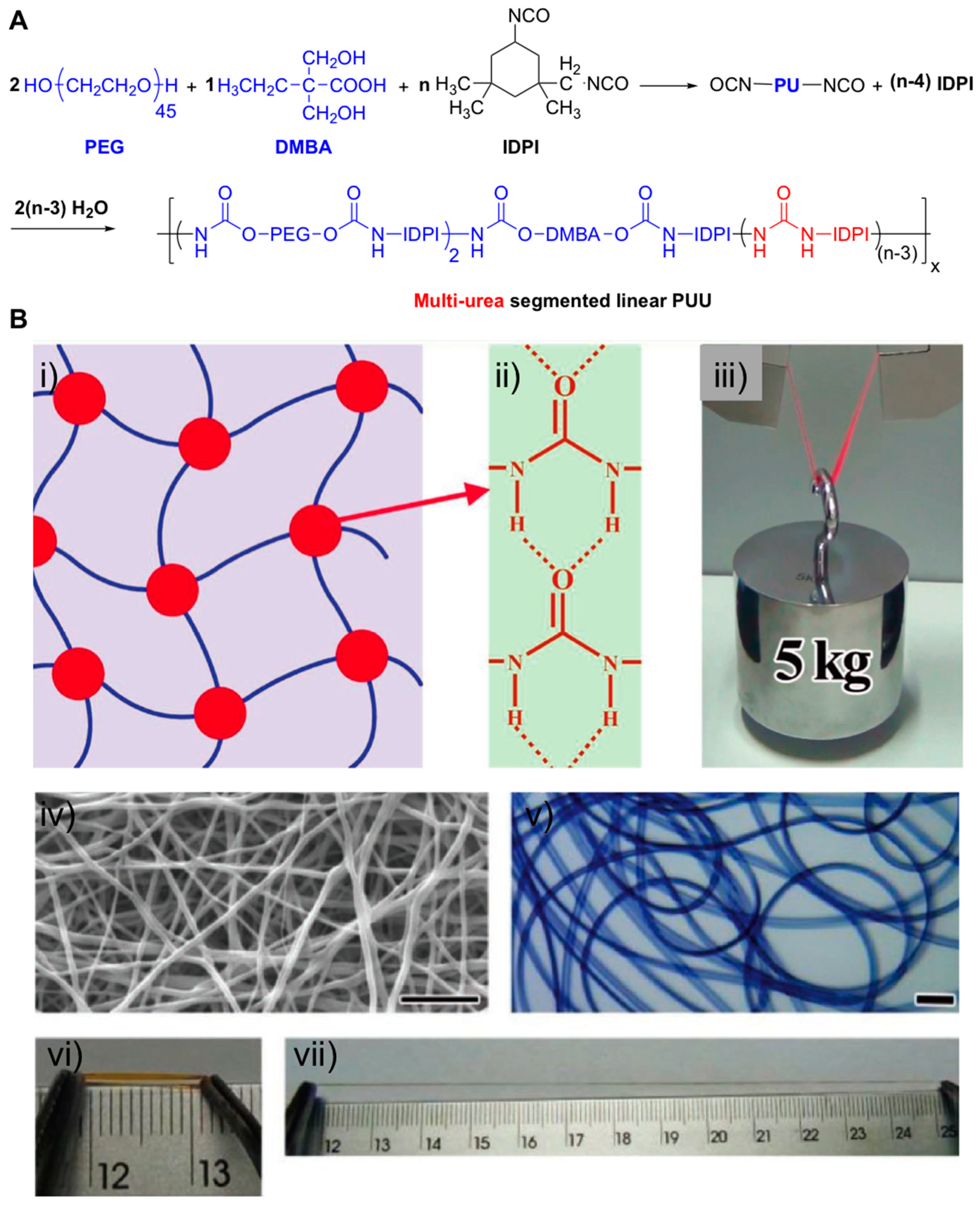
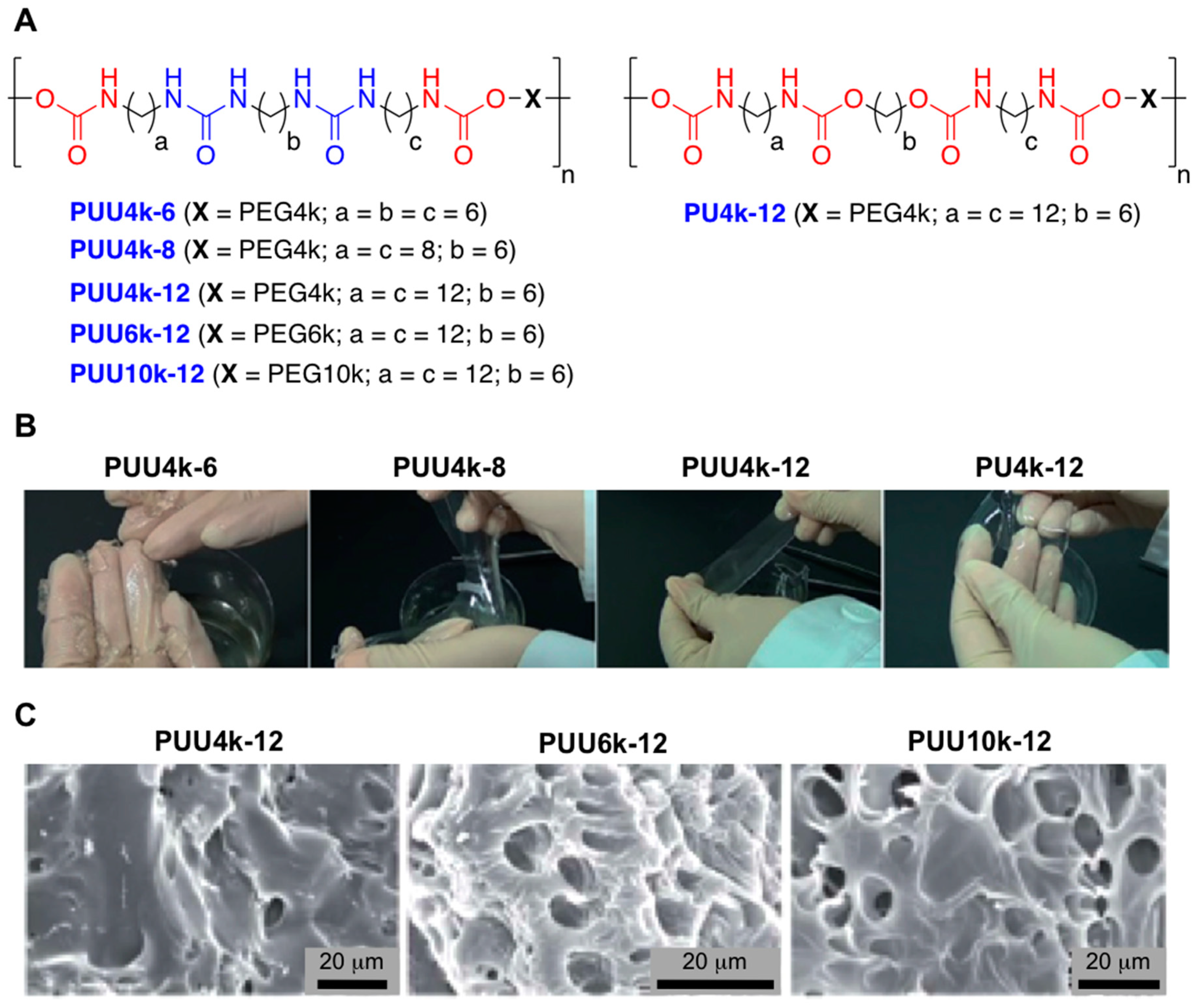
- Yang, N.; Yang, H.; Shao, Z.; Guo, M. Ultrastrong and Tough Supramolecular Hydrogels from Multiurea Linkage Segmented Copolymers with Tractable Processablity and Recyclability. Macromol. Rapid Commun. 2017, 38, 1–6. [Google Scholar] [CrossRef]
- Cui, Y.; Tan, M.; Zhu, A.; Guo, M. Non-covalent interaction cooperatively induced stretchy, tough and stimuli-responsive polyurethane–urea supramolecular (PUUS) hydrogels. J. Mater. Chem. B 2015, 3, 2834–2841. [Google Scholar] [CrossRef]
- Jeon, I.; Cui, J.; Illeperuma, W.R.K.; Aizenberg, J.; Vlassak, J.J. Extremely Stretchable and Fast Self-Healing Hydrogels. Adv. Mater. 2016, 4678–4683. [Google Scholar] [CrossRef] [PubMed]
- Förster, S.; Wenz, E.; Lindner, P. Density profile of spherical polymer brushes. Phys. Rev. Lett. 1996, 77, 95–98. [Google Scholar] [CrossRef]
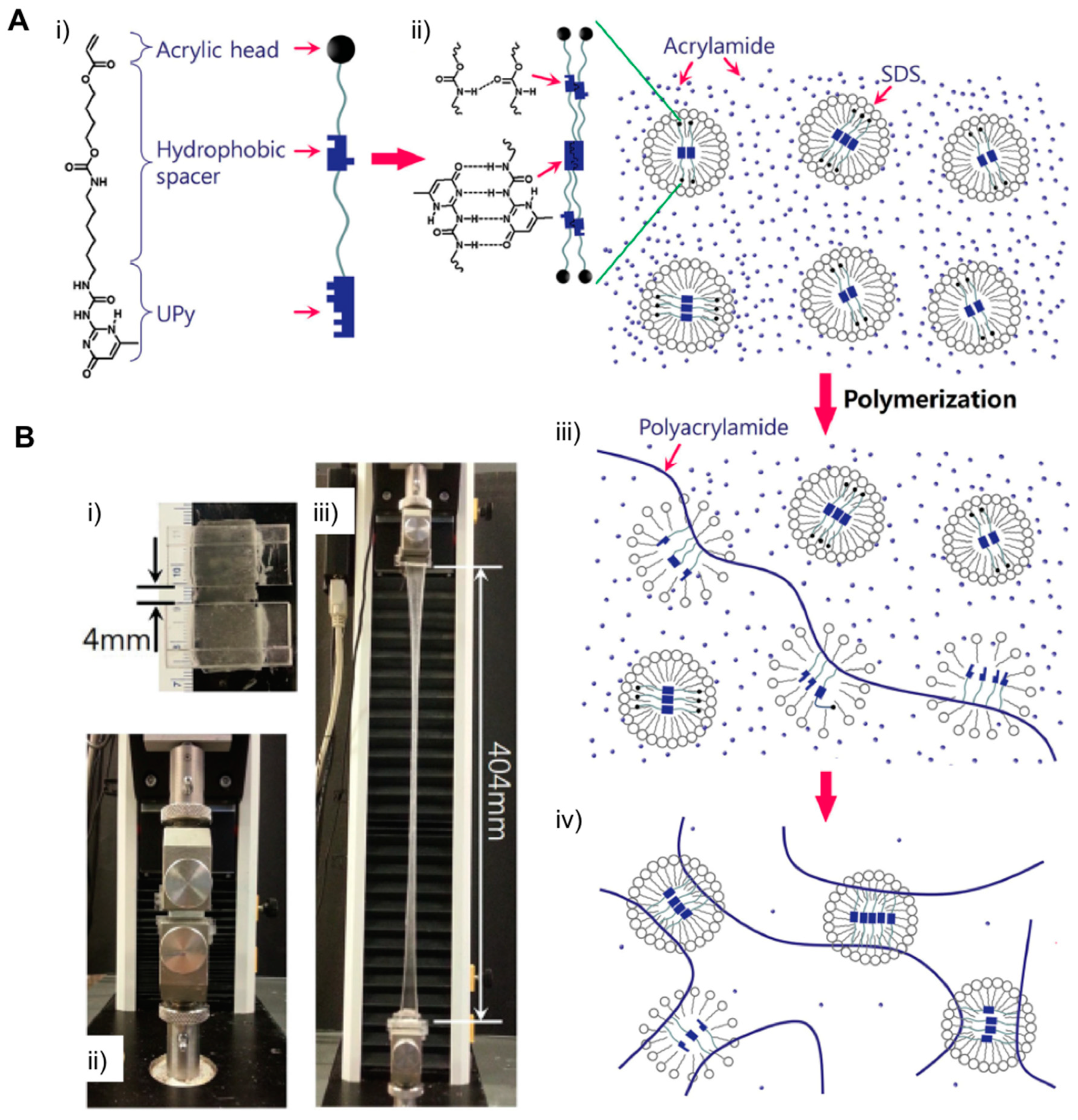
- Zhang, R.; Wang, L.; Shen, Z.; Li, M.; Guo, X.; Yao, Y. Ultrastretchable, Tough, and Notch-Insensitive Hydrogels Formed with Spherical Polymer Brush Crosslinker. Macromol. Rapid Commun. 2017, 38, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; Loh, X.J.; Jones, S.T.; Dreiss, C.A.; Scherman, O.A. Sustained release of proteins from high water content supramolecular polymer hydrogels. Biomaterials 2012, 33, 4646–4652. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, M.; Takashima, Y.; Harada, A. Highly Flexible, Tough, and Self-Healing Supramolecular Polymeric Materials Using Host-Guest Interaction. Macromol. Rapid Commun. 2016, 37, 86–92. [Google Scholar] [CrossRef]
- Mealy, J.E.; Rodell, C.B.; Burdick, J.A. Sustained small molecule delivery from injectable hyaluronic acid hydrogels through host-guest mediated retention. J. Mater. Chem. B 2015, 3, 8010–8019. [Google Scholar] [CrossRef]
- Rodell, C.B.; Kaminski, A.L.; Burdick, J.A. Rational design of network properties in guest-host assembled and shear-thinning hyaluronic acid hydrogels. Biomacromolecules 2013, 14, 4125–4134. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J. Supramolecular host–guest polymeric materials for biomedical applications. Mater. Horiz. 2014, 1, 185–195. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, Y. Biomedical Applications of Supramolecular Systems Based on Host-Guest Interactions. Chem. Rev. 2015, 115, 7794–7839. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, C.S.Y.; Yu, Z.; Li, N.; Abell, C.; Scherman, O.A. Tough Supramolecular Polymer Networks with Extreme Stretchability and Fast Room-Temperature Self-Healing. Adv. Mater. 2017, 29, 1–7. [Google Scholar] [CrossRef]
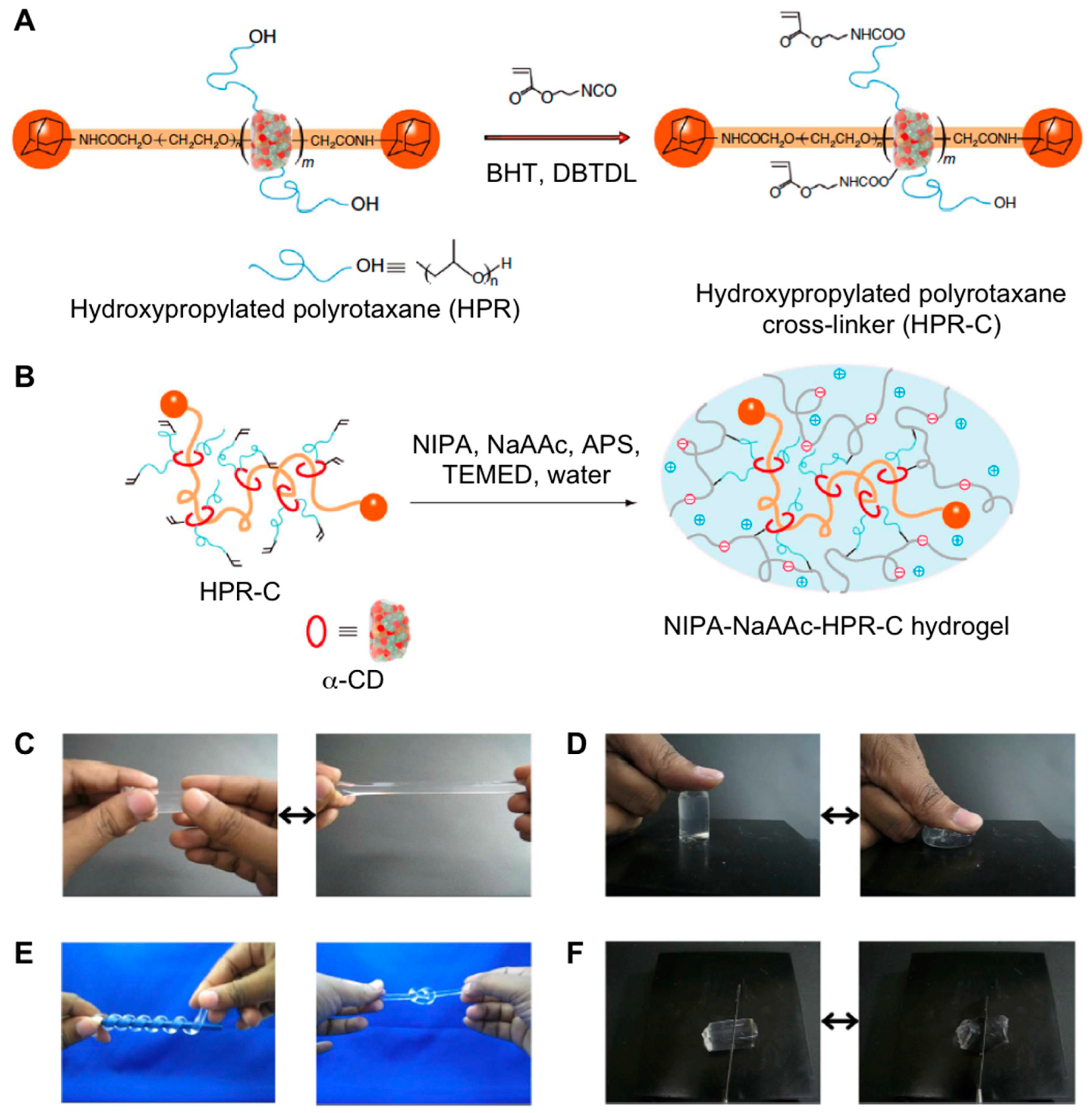
- Bin Imran, A.; Esaki, K.; Gotoh, H.; Seki, T.; Ito, K.; Sakai, Y.; Takeoka, Y. Extremely stretchable thermosensitive hydrogels by introducing slide-ring polyrotaxane cross-linkers and ionic groups into the polymer network. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grijalvo, S.; Eritja, R.; Díaz Díaz, D. On the Race for More Stretchable and Tough Hydrogels. Gels 2019, 5, 24. https://doi.org/10.3390/gels5020024
Grijalvo S, Eritja R, Díaz Díaz D. On the Race for More Stretchable and Tough Hydrogels. Gels. 2019; 5(2):24. https://doi.org/10.3390/gels5020024
Chicago/Turabian StyleGrijalvo, Santiago, Ramon Eritja, and David Díaz Díaz. 2019. "On the Race for More Stretchable and Tough Hydrogels" Gels 5, no. 2: 24. https://doi.org/10.3390/gels5020024
APA StyleGrijalvo, S., Eritja, R., & Díaz Díaz, D. (2019). On the Race for More Stretchable and Tough Hydrogels. Gels, 5(2), 24. https://doi.org/10.3390/gels5020024