Chiral Assembly Preferences and Directing Effects in Supramolecular Two-Component Organogels


Abstract
1. Introduction
2. Results and Discussion
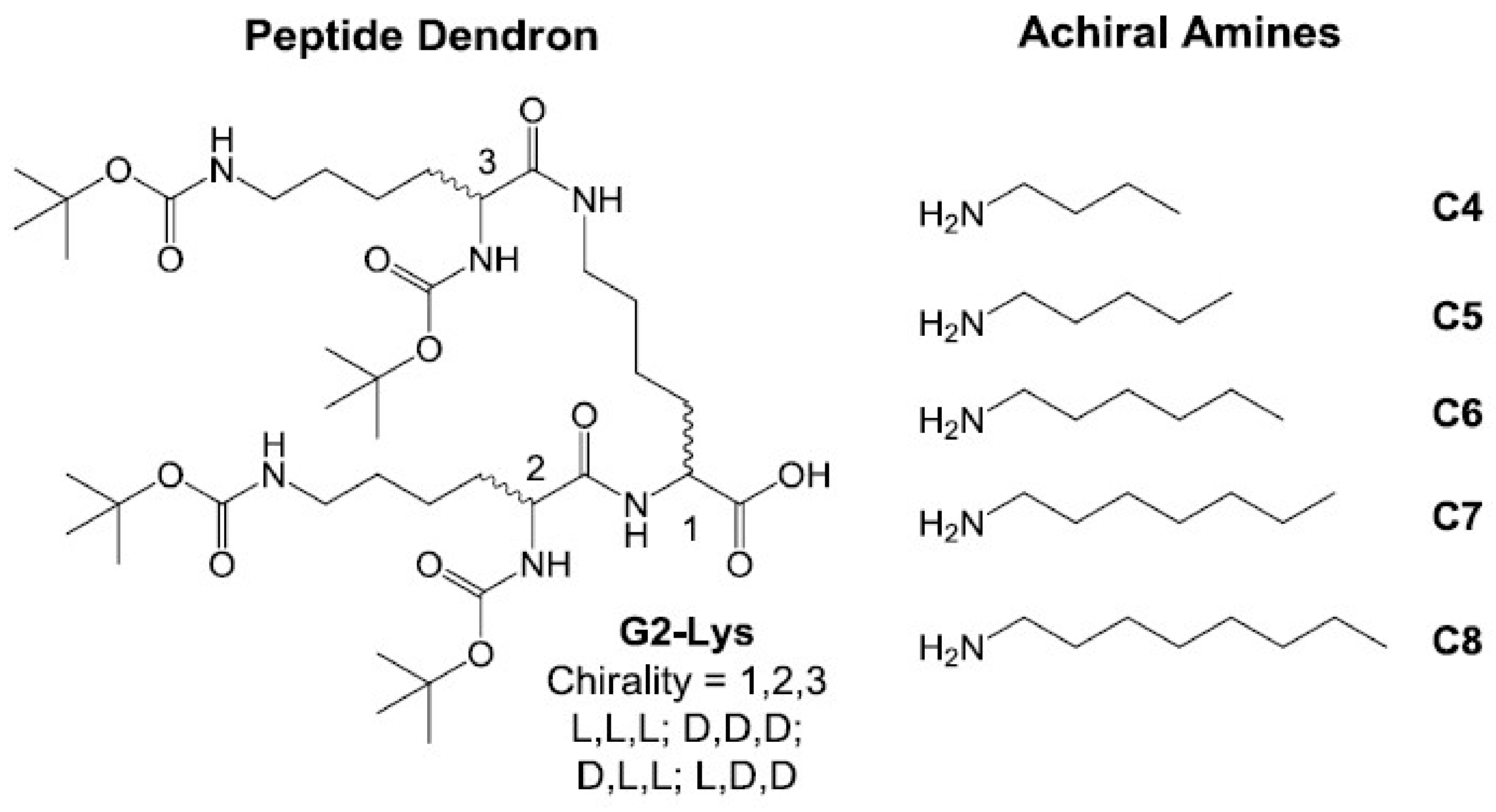
2.1. Synthesis and Characterisation
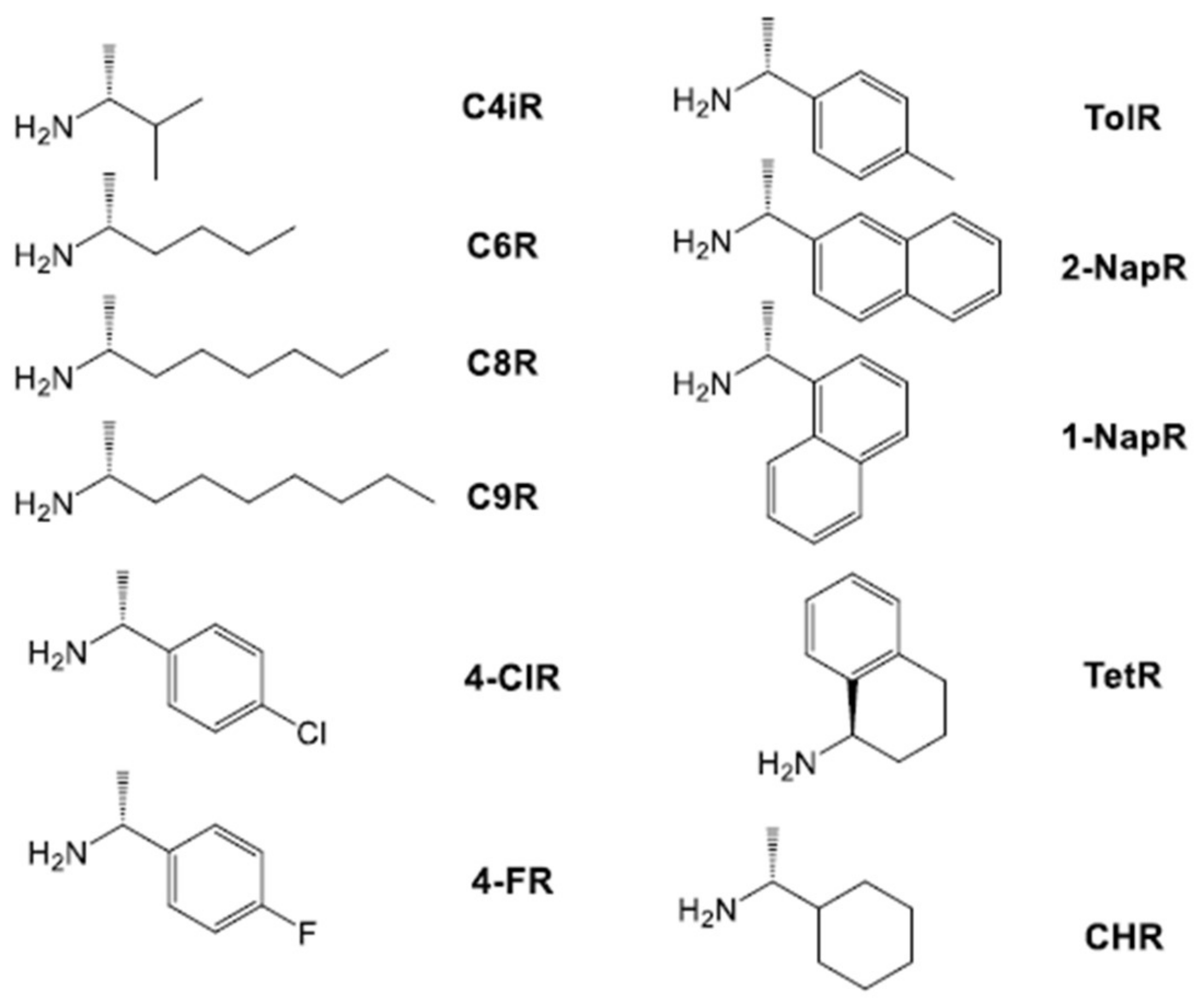
2.2. Investigation of Gel Formation with Simple Achiral Amines
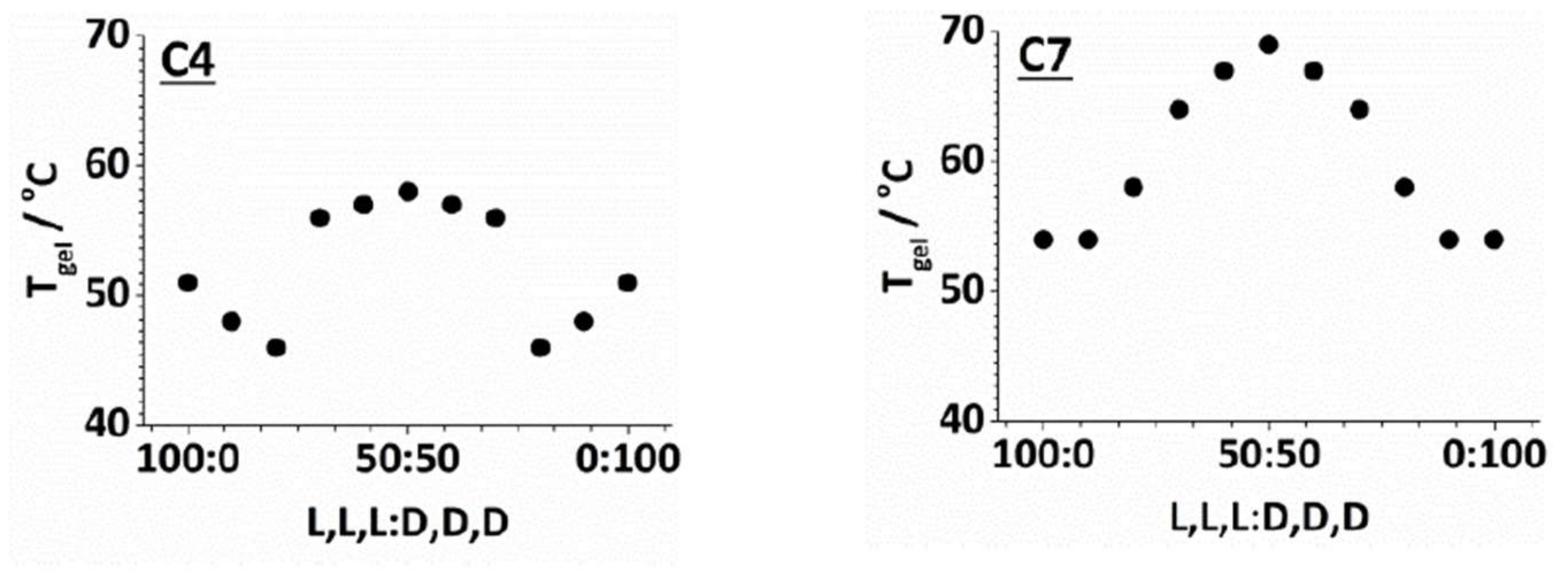
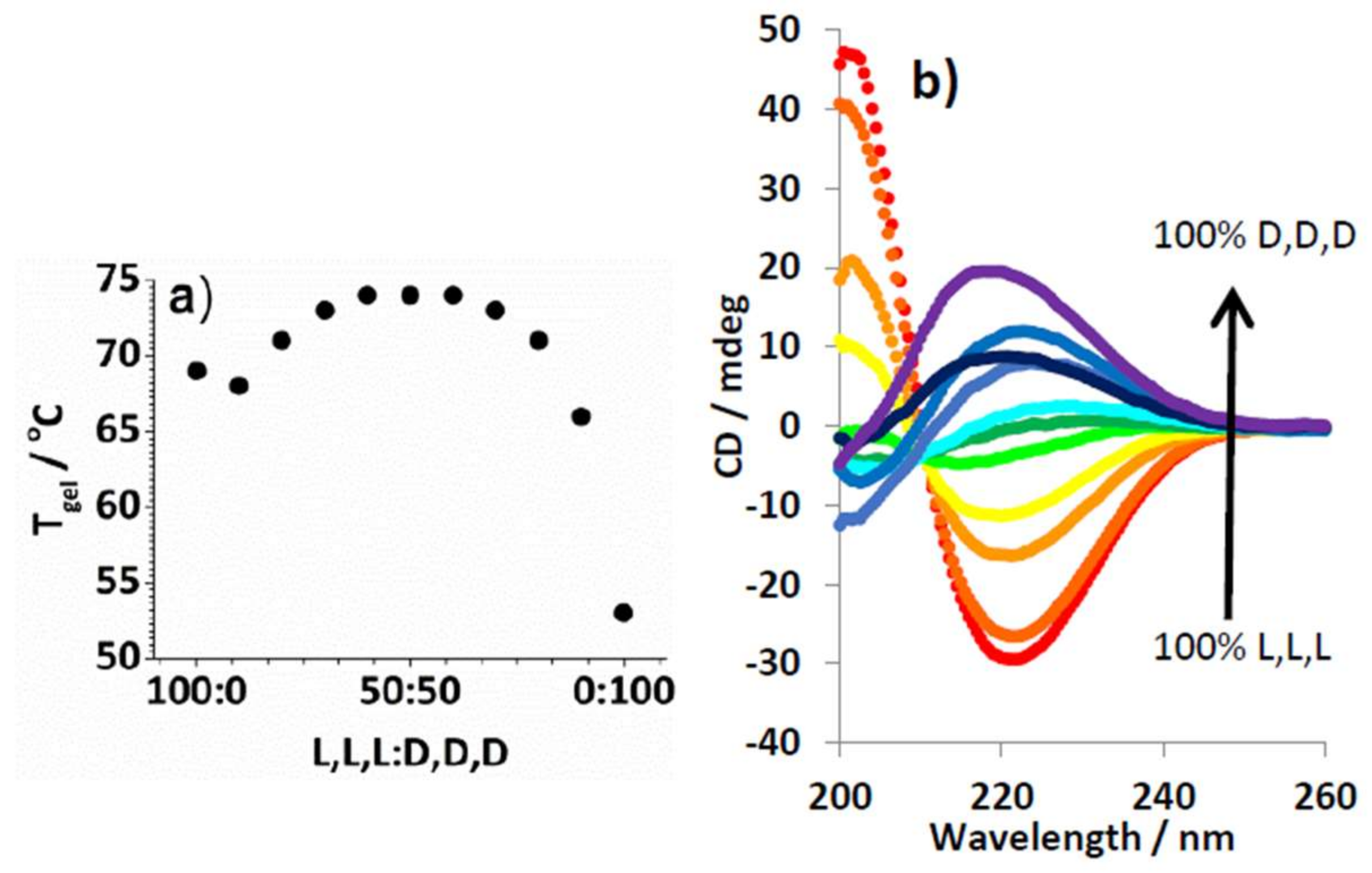
2.3. Effect of Enantiomeric Mixing on Gelation with Achiral Amines
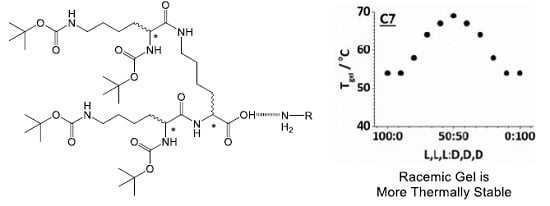
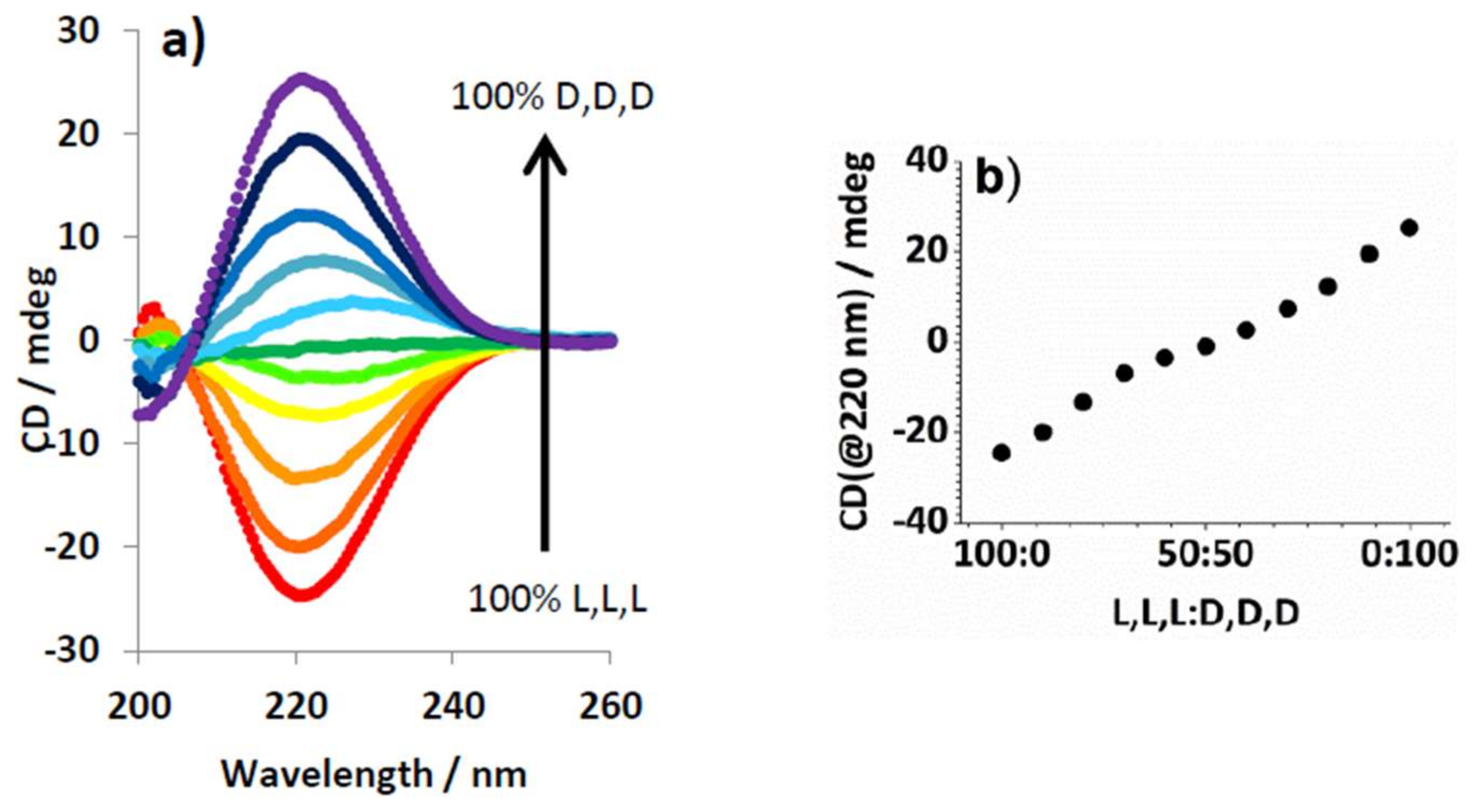
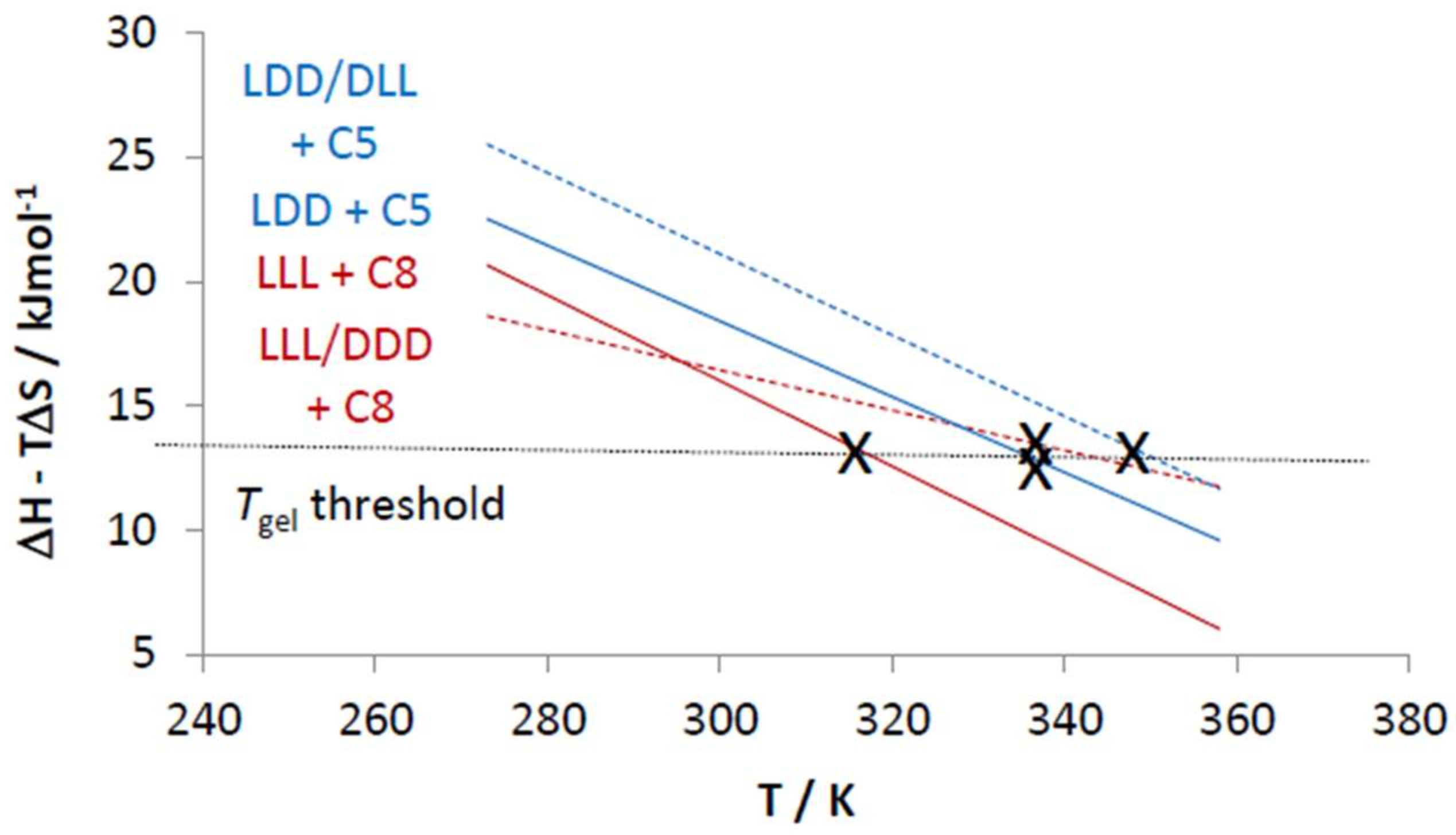
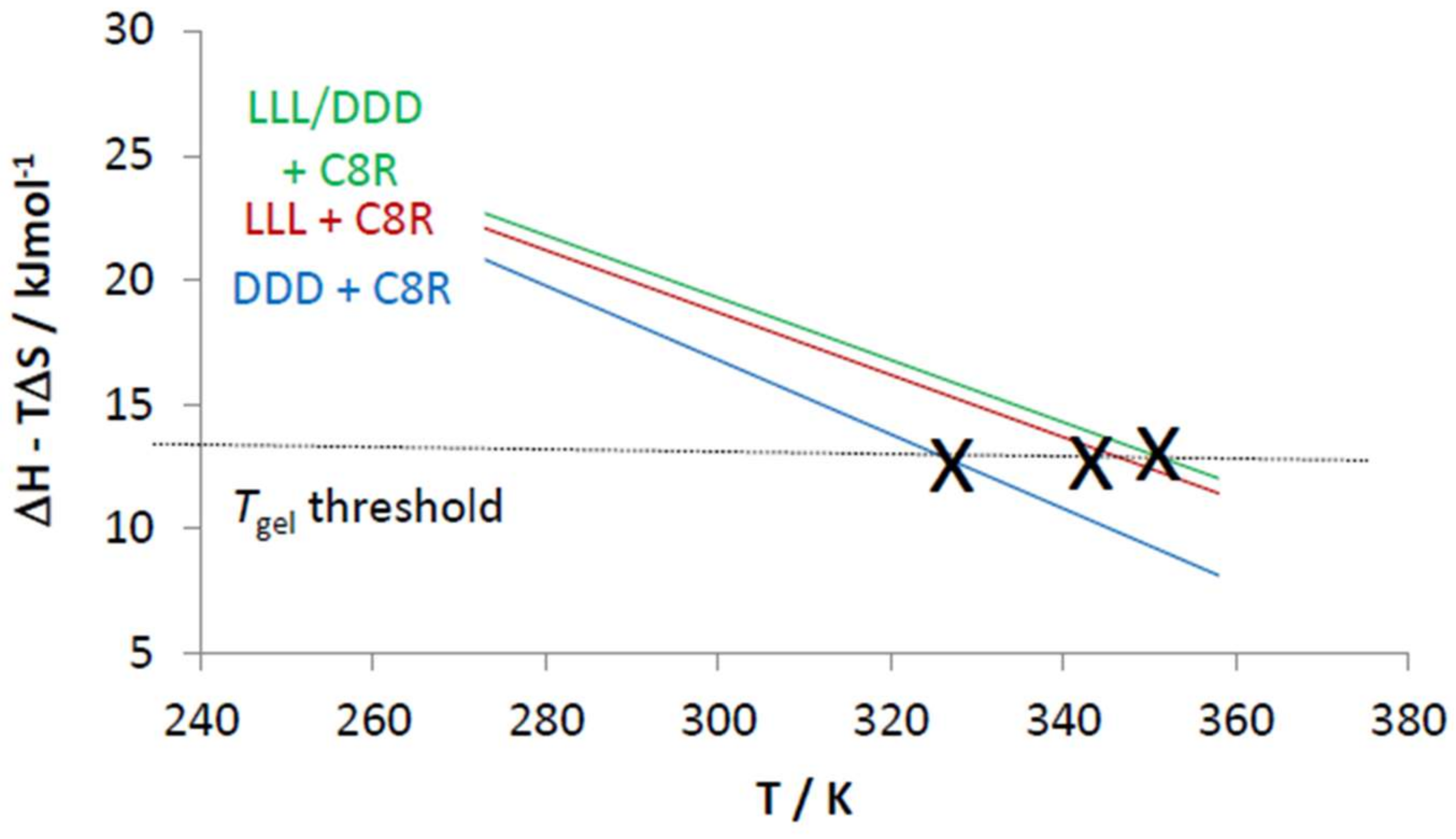
2.4. Chiral Dendron and Chiral Amines
3. Conclusions
4. Materials and Methods
4.1. Synthesis and Characterisation
4.2. Experimental Methods
4.2.1. Gel Formation
4.2.2. Tgel Measurements
4.2.3. Field Emission Gun Scanning Electron Microscopy (FEG-SEM)
4.2.4. Circular Dichroism (CD)
4.2.5. NMR Studies
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hirst, A.R.; Escuder, B.; Miravet, J.F.; Smith, D.K. High-Tech Applications of Self-Assembling Supramolecular Nanostructured Gel-Phase Materials: From Regenerative Medicine to Electronic Devices. Angew. Chem. Int. Ed. 2008, 47, 8002–8018. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, J.H. We Can Design Molecular Gelators, But Do We Understand Them? Langmuir 2009, 25, 8392–8394. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Das, R.K.; Maitra, U. Supramolecular Gels ‘In Action’. J. Mater. Chem. 2009, 19, 6649–6687. [Google Scholar] [CrossRef]
- Steed, J.W. Supramolecular Gel Chemistry: Developments Over the Last Decade. Chem. Commun. 2011, 47, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Dawn, A.; Shiraki, T.; Haraguchi, S.; Tamaru, S.-I.; Shinkai, S. What Kind of ‘Soft Materials’ Can We Design from Molecular Gels. Chem. Asian J. 2011, 6, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G. The Past, Present, and Future of Molecular Gels. J. Am. Chem. Soc. 2014, 136, 7519–7530. [Google Scholar] [CrossRef] [PubMed]
- Draper, E.R.; Adams, D.J. Low-Molecular-Weight Gels: The State of the Art. Chem 2017, 3, 390–410. [Google Scholar] [CrossRef]
- Amabilino, D.J.; Smith, D.K.; Steed, J.W. Supramolecular Materials. Chem. Soc. Rev. 2017, 46, 2404–2420. [Google Scholar] [CrossRef] [PubMed]
- Brizard, A.; Oda, R.; Huc, I. Chirality Effects in Self-Assembled Fibrillar Networks. Top. Curr. Chem. 2005, 256, 167–218. [Google Scholar] [PubMed]
- Smith, D.K. Lost in Translation? Chirality Effects in the Self-Assembly of Nanostructured Gel-Phase Materials. Chem. Soc. Rev. 2009, 38, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Cai, H.; Zhang, L.; Liu, M. Gelation Induced Supramolecular Chirality: Chirality Transfer, Amplification and Application. Soft Matter 2014, 10, 5428–5448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, Q.; Liu, M. Enantioselective Recognition by Chiral Supramolecular Gels. Chem. Asian J. 2016, 11, 2642–2649. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Wang, Y.; Chen, H. Emerging Chirality in Nanoscience. Chem. Soc. Rev. 2013, 42, 2930–2962. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; George, S.J. Homotropic and Heterotropic Allosteric Regulation of Supramolecular Chirality. Chem. Sci. 2014, 5, 3025–3030. [Google Scholar] [CrossRef]
- Yashima, E.; Ousaka, N.; Taura, D.; Shimomura, K.; Ikai, T.; Maeda, K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification of Their Functions. Chem. Rev. 2016, 116, 13752–13990. [Google Scholar] [CrossRef] [PubMed]
- Jędrzejewska, H.; Szumna, A. Making a Right or Left Choice: Chiral Self-Sorting as a Tool for the Formation of Discrete Complex Structures. Chem. Rev. 2017, 117, 4863–4899. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Smith, D.K. Two-Component Gel-Phase Materials—Highly Tunable Self-Assembling Systems. Chem. Eur. J. 2005, 11, 5496–5504. [Google Scholar] [CrossRef] [PubMed]
- Buerkle, L.E.; Rowan, S.J. Supramolecular Gels Formed from Multi-Component Low Molecular Weight Species. Chem. Soc. Rev. 2012, 41, 6089–6102. [Google Scholar] [CrossRef] [PubMed]
- Raeburn, J.; Adams, D.J. Multicomponent Low Molecular Weight Gelators. Chem. Commun. 2015, 51, 5170–5180. [Google Scholar] [CrossRef] [PubMed]
- Draper, E.R.; Adams, D.J. How Should Multicomponent Supramolecular Gels be Characterised. Chem. Soc. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hanabusa, K.; Miki, T.; Taguchi, Y.; Koyama, T.; Shirai, H. Two-Component, Small Molecule Gelling Agents. J. Chem. Soc. Chem. Commun. 1993, 1382–1384. [Google Scholar] [CrossRef]
- Inoue, K.; Ono, Y.; Kanekiyo, Y.; Ishi-i, T.; Yoshihara, K.; Shinkai, S. Design of New Organic Gelators Stabilised by a Host-Guest Interaction. J. Org. Chem. 1999, 64, 2933–2937. [Google Scholar] [CrossRef] [PubMed]
- Partridge, K.S.; Smith, D.K.; Dykes, G.M.; McGrail, P.T. Supramolecular Dendritic Two-Component Gel. Chem. Commun. 2001, 319–320. [Google Scholar] [CrossRef]
- Sreenivasachary, N.; Lehn, J.-M. Gelation-Driven Component Selection in the Generation of Constitutional Dynamic Hydrogels Based on Guanine-Quartet Formation. Proc. Natl. Acad. Sci. USA 2005, 102, 5938–5943. [Google Scholar] [CrossRef] [PubMed]
- Buhler, E.; Sreenivasachary, N.; Candau, D.-J.; Lehn, J.-M. Modulation of the Supramolecular Structure of G-Quartet Assemblies by Dynamic Covalent Decoration. J. Am. Chem. Soc. 2007, 129, 10058–10059. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Miravet, J.F.; Escuder, B.; Noirez, L.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Self-Assembly of Two-Component Gels: Stoichiometric Control and Component Selection. Chem. Eur. J. 2009, 15, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Buchs, B.; Fieber, W.; Vigoroux-Elie, F.; Sreenivasachary, N.; Lehn, J.-M.; Herrmann, A. Release of Bioactive Volatiles from Supramolecular Hydrogels: Influence of Reversible Acylhydrazone Formation on Gel Stability and Volatile Compound Evaporation. Org. Biomol. Chem. 2011, 9, 2906–2919. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M.; Edwards, W.; Smith, D.K. Self-Organisation Effects in Dynamic Nanoscale Gels Self-Assembled from Simple Mixtures of Commercially Available Molecular-Scale Components. Chem. Sci. 2013, 4, 671–676. [Google Scholar] [CrossRef]
- Edwards, W.; Smith, D.K. Dynamic Evolving Two-Component Supramoleular Gels—Hierarchical Control over Component Selection in Complex Mixtures. J. Am. Chem. Soc. 2013, 135, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Messmore, B.W.; Sukerkar, P.A.; Stupp, S.I. Mirror Image Nanostructures. J. Am. Chem. Soc. 2005, 127, 7992–7993. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Huang, B.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Self-Organisation in the Assembly of Gels from Mixtures of Different Dendritic Peptide Building Blocks. Chem. Eur. J. 2007, 13, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Sugiyasu, K.; Kawano, S.I.; Fujita, N.; Shinkai, S. Self-Sorting Organogels with p-n Heterojunction Points. Chem. Mater. 2008, 20, 2863–2865. [Google Scholar] [CrossRef]
- Moffat, J.R.; Smith, D.K. Controlled Self-Sorting in the Assembly of ‘Multi-Gelator’ Gels. Chem. Commun. 2009, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.R.; Coates, I.A.; Leng, F.J.; Smith, D.K. Metathesis within Self-Assembled Gels: Transcribing Nanostructured Soft Materials into a More Robust Form. Langmuir 2009, 25, 8786–8793. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, S.; Ghini, G.; Lascialfari, L.; Brandi, A.; Betti, F.; Berti, D.; Baglioni, P.; Di Bari, L.; Pescitelli, G.; Mannini, M.; et al. Self-Sorting Chiral Organogels from a Long Chain Carbamate of 1-Benzyl-pyrrolidine-3,4-diol. Soft Matter 2010, 6, 1655–1661. [Google Scholar] [CrossRef]
- Smith, M.M.; Smith, D.K. Self-Sorting Multi-Gelator Gels: Mixing and Ageing Effects in Thermally Addressable Supramolecular Soft Nanomaterials. Soft Matter 2011, 7, 4856–4860. [Google Scholar] [CrossRef]
- Das, A.; Ghosh, S. A Generalized Supramolecular Strategy for Self-Sorted Assembly between Donor and Acceptor Gels. Chem. Commun. 2011, 47, 8922–8924. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, D.G.; Luque, R. Spontaneous Orthogonal Self-Assembly of a Synergetic Gelator System. Chem. Eur. J. 2011, 17, 3847–3849. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.L.; Chen, L.; Raeburn, J.; Sellick, O.R.; Cotanda, P.; Paul, A.; Griffiths, P.C.; King, S.M.; O’Reilly, R.K.; Serpell, L.C.; et al. Chemically Programmed Self-Sorting of Gelator Networks. Nat. Commun. 2013, 4, 1480. [Google Scholar] [CrossRef] [PubMed]
- Tena-Solsona, M.; Escuder, B.; Miravet, J.F.; Casttelleto, V.; Hamley, I.W.; Dehsorkhi, A. Themodynamic and Kinetic Study of the Fibrillization of a Family of Tetrapeptides and Its Application to Self-Sorting. What Takes So Long? Chem. Mater. 2015, 27, 3358–3365. [Google Scholar] [CrossRef]
- Cornwell, D.J.; Daubney, O.J.; Smith, D.K. Photopatterned Multidomain Gels: Multi-Component Self-Assembled Hydrogels Based on Partially Self-Sorting 1,3:2,4-Dibenzylidene-d-dorbitol Derivatives. J. Am. Chem. Soc. 2015, 137, 15486–15492. [Google Scholar] [CrossRef] [PubMed]
- Onogi, S.; Shigemitsu, H.; Yoshii, T.; Tanida, T.; Ikeda, M.; Kubota, R.; Hamachi, I. In Situ Real-Time Imaging of Self-Sorted Supramolecular Nanofibres. Nat. Chem. 2016, 8, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Shigemitsu, H.; Fujisaku, T.; Tanaka, W.; Kubota, R.; Minami, S.; Urayama, K.; Hamachi, I. An Adaptive Supramolecular Hydrogel Comprising Self-Sorting Double Nanofibre Networks. Nat. Nanotechnol. 2018, 13, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-L.; Matsumoto, S.; Tian, H.D.; Yamane, H.; Ojida, A.; Kiyonaka, S.; Hamachi, I. pH-Responsive Shrinkage/Swelling of a Supramolecular Hydrogel Composed of Two Small Amphiphilic Molecules. Chem. Eur. J. 2005, 11, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; Edkins, R.M.; Cameron, G.J.; Colgin, N.; Fucke, K.; Ridgeway, S.; Crawford, A.G.; Marder, T.B.; Beeby, A.; Cobb, S.L.; et al. Blending Gelators to Tunde Gel Structure and Probe Anion-Induced Disassembly. Chem. Eur. J. 2014, 20, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, C.; Draper, E.R.; Eden, E.G.B.; Cattoz, B.N.; Morris, K.L.; McDonald, T.O.; Terry, A.E.; Griffiths, P.C.; Serpell, L.C.; Adams, D.J. The Effect of Self-Sorting and Co-Assembly on the Mechanical Properties of Low Molecular Weight Hydrogels. Nanoscale 2014, 6, 13719–13725. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, W.; Nilsson, B.L. Substituent Effects on the Self-Assembly/Coassembly and Hydrogelation of Phenylalanine Derivatives. Langmuir 2016, 32, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Heeres, A.; van der Pol, C.; Stuart, M.C.A.; Friggeri, A.; Feringa, B.L.; van Esch, J. Orthogonal Self-Assembly of Low Molecular Weight Hydrogelators and Surfactants. J. Am. Chem. Soc. 2003, 125, 14252–14253. [Google Scholar] [CrossRef] [PubMed]
- Brizard, A.M.; Stuart, M.C.A.; van Bommel, K.; Friggeri, A.; de Jong, M.; van Esch, J.H. Preparation of Nanostructures by Orthogonal Self-Assembly of Hydrogelators and Surfactants. Angew. Chem. Int. Ed. 2008, 47, 2063–2066. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Liu, X.Y.; Strom, C.S. Producing Supramolecular Functional Materials Based on Fiber Network Reconstruction. Adv. Funct. Mater. 2009, 19, 2252–2259. [Google Scholar] [CrossRef]
- Buerkle, L.E.; Galleguillos, R.; Rowan, S.J. Nonionic Surfactant-Induced Stabilization and Tailorability of Sugar-Amphiphile Hydrogels. Soft Matter 2011, 7, 6984–6990. [Google Scholar] [CrossRef]
- Adhia, Y.J.; Schloemer, T.H.; Perez, M.T.; McNeil, A.J. Using Polymeric Additives to Enhance Molecular Gelation: Impact of Poly(acrylic acid) on Pyridine-Based Gelators. Soft Matter 2012, 8, 430–434. [Google Scholar] [CrossRef]
- Himmelein, S.; Lewe, V.; Stuart, M.C.A.; Ravoo, B.J. A Carbohydrate-Based Hydrogel Containing Vesicles as Responsive Non-Covalent Cross-Linkers. Chem. Sci. 2014, 5, 1054–1058. [Google Scholar] [CrossRef]
- Stubenrauch, C.; Giesselmann, F. Gelled Complex Fluids: Combining Unique Structures with Mechanical Stability. Angew. Chem. Int. Ed. 2016, 55, 3268–3275. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.M.P.; Hay, L.L.; Smith, D.K. Multi-Component Hybrid Hydrogels—Understanding the Extent of Orthogonal assembly and its Impact on Controlled Release. Chem. Sci. 2017, 8, 6981–6990. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, B.; Liang, Y. Self-Assembled Organogels Formed by Mono-Chain l-Alanine Derivatives. Chem. Commun. 2001, 1556–1557. [Google Scholar] [CrossRef]
- Koga, T.; Matsuoka, M.; Higashi, N. Structural Control of Self-Assembled Nanofibers by Artificial β-Sheet Peptides Composed of d- or l-isomer. J. Am. Chem. Soc. 2005, 127, 17596–17597. [Google Scholar] [CrossRef] [PubMed]
- Puigmartí-Luis, J.; Laukhin, V.; Pérez del Pino, A.; Vidal-Gancedo, J.; Rovira, C.; Laukhina, E.; Amabilino, D.B. Supramolecular Conducting Nanowires from Organogels. Angew. Chem. Int. Ed. 2007, 46, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Makarević, J.; Jokić, M.; Raza, Z.; Štefanić, Z.; Kojić-Prodić, B.; Žinić, M. Chiral Bis(amino alcohol)oxalamide Gelators—Gelation Properties and Supramolecular Organization: Racemate versus Pure Enantiomer Gelation. Chem. Eur. J. 2003, 9, 5567–5580. [Google Scholar] [CrossRef] [PubMed]
- Čaplar, V.; Frkanec, L.; Vujičić, N.S.; Žinić, M. Positionally Isomeric Organic Gelators: Structure-Gelation Study, Racemic versus Enantiomeric Gelators and Solvation Effects. Chem. Eur. J. 2010, 16, 3066–3082. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, R.; Mizutani, M.; Yamaguchi, M. Two-Component Gel Formation by Pseudoenantiomeric Ethynylhelicene Oligomers. Angew. Chem. Int. Ed. 2010, 49, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Nagy, K.J.; Giano, M.C.; Jin, A.; Pochan, D.J.; Schneider, J.P. Enhanced Mechanical Rigidity of Hydrogels formed from Enantiomeric Peptide Assemblies. J. Am. Chem. Soc. 2011, 133, 14975–14977. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Guo, Z.; Plas, J.; Amabilino, D.B.; De Feyter, S.; Schenning, A.P.H.J. Homochiral and Heterochiral Assembly Preferences at Different Length Scales—Conglomerates and Racemates in the Same Assemblies. Chem. Commun. 2013, 49, 9320–9322. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wang, T.; Liu, M. Tuning the Gelation Ability of Racemic Mixture by Melamine: Enhanced Mechanical Rigidity and Tunable Nanoscale Chirality. Langmuir 2014, 30, 10772–10778. [Google Scholar] [CrossRef] [PubMed]
- Caumes, X.; Baldi, A.; Gontard, G.; Brocorens, P.; Lazzaroni, R.; Vanthuyne, N.; Troufflard, C.; Raynal, M.; Bouteiller, L. Tuning the Structure of 1,3,5-Benzenetricarboxamide Self-Assemblies through Stereochemistry. Chem. Commun. 2016, 52, 13369–13372. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, X.; Zhang, A.-Y.; Bezuidenhout, D.; Feng, Z.-G. Rapidly Recoverable Thixotropic Hydrogels from the Racemate of Chiral OFm Monosubstituted Cyclo(Glu-Glu) Derivatives. Langmuir 2017, 33, 13821–13827. [Google Scholar] [CrossRef] [PubMed]
- Brunsveld, L.; Vekemans, J.A.J.M.; Hirschberg, J.H.K.K.; Sjibesma, R.P.; Meijer, E.W. Hierarchical Formation of Helical Supramolecular Polymers via Stacking of Hydrogen-Bonded Pairs in Water. Proc. Natl. Acad. Sci. USA 2002, 99, 4977–4982. [Google Scholar] [CrossRef] [PubMed]
- Ajayaghosh, A.; Varghese, R.; George, S.J.; Vijayakumar, C. Transcription and Amplification of Molecular Chirality to Oppositely Biased Supramolecular π-Helices. Angew. Chem. Int. Ed. 2006, 45, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wang, G.-T.; Du, P.; Wang, R.-X.; Jiang, X.-K.; Li, Z.-T. Foldamer Organogels: A Circular Dichroism Study of Glucose-Mediated Dynamic Helicity Induction and Amplification. J. Am. Chem. Soc. 2008, 130, 13450–13459. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.R.; Lee, H.Y.; Hong, J.-L. Control of Macroscopic Helicity by Using the Sergeants-and-Soldiers Principle in Organogels. Chem. Eur. J. 2008, 14, 6040–6043. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Kandanelli, R.; Linnanto, J.; Bose, K.; Maitra, U. Supramolecular Chirality in Organogels: A Detailed Spectroscopic and Rheological Investigation of Gels (and Xerogels) Derived from Alkyl Pyrenyl Urethanes. Langmuir 2010, 26, 16141–16149. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Llansola, F.; Hermida-Merino, D.; Nieto-Ortega, B.; Ramirez, F.J.; Navarette, J.T.L.; Casado, J.; Hamley, I.W.; Escuder, B.; Hayes, W.; Miravet, J.F. Self-Assembly Studies of a Chiral Bisurea-Based Superhydrogelator. Chem. Eur. J. 2012, 18, 14725–14731. [Google Scholar] [CrossRef] [PubMed]
- Lascialfari, L.; Berti, D.; Brandi, A.; Cicchi, S.; Mannini, M.; Pescitelli, G.; Procacci, P. Chiral/ring Closed vs. Achiral/open Chain Triazine-based Organogelators: Induction and Amplification of Supramolecular Chirality in Organic Gels. Soft Matter 2014, 10, 3762–3770. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Paul, M.; Sakurai, T.; Matsuda, W.; Seki, S.; Ghosh, S. Supramolecular Chirality Issues in Unorthodox Naphthalene Diimide Gelators. Chem. Eur. J. 2018, 24, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Smith, D.K.; Feiters, M.C.; Geurts, H.P.M. Two-Component Dendritic Gels: Easily Tunable Materials. J. Am. Chem. Soc. 2003, 125, 9010–9011. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Smith, D.K.; Feiters, M.C.; Geurts, H.P.M. Two-Component Dendritic Gel: Effect of Spacer Chain Length on the Supramolecular Chiral Assembly. Langmuir 2004, 20, 7070–7077. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Smith, D.K. Self-Assembly of Two-Component Peptidic Dendrimers: Dendritic Effects on Gel-Phase Materials. Org. Biomol. Chem. 2004, 2, 2965–2971. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Hirst, A.R.; Smith, D.K.; Castelletto, V.; Hamley, I.W. A Direct Comparison of One- and Two-Component Dendritic Self-Assembled Materials: Elucidating Molecular Recognition Pathways. J. Am. Chem. Soc. 2005, 127, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Smith, D.K.; Harrington, J.P. Unique Nanoscale Morphologies Underpinning Organic Gel-Phase Materials. Chem. Eur. J. 2005, 11, 6552–6559. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.R.; Hirst, A.R.; Smith, D.K. Exploring Molecular Recognition Pathways in One- and Two-Component Gels Formed by Dendritic Lysine-Based Gelators. Soft Matter 2012, 8, 3399–3406. [Google Scholar] [CrossRef]
- Edwards, W.; Smith, D.K. Enantioselective Component Selection in Multicomponent Supramolecular Gels. J. Am. Chem. Soc. 2014, 136, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.R.; Smith, D.K.; Feiters, M.C.; Geurts, H.P.M. Two-Component Dendritic Gel: Effect of Stereochemistry on the Supramolecular Chiral Assembly. Chem. Eur. J. 2004, 10, 5901–5910. [Google Scholar] [CrossRef] [PubMed]
- Escuder, B.; LLusar, M.; Miravet, J.F. Insight on the NMR Study of Supramolecular Gels and Its Application to Monitor Molecular Recogition on Self-Assembled Fibers. J. Org. Chem. 2006, 71, 7747–7752. [Google Scholar] [CrossRef]
- Hirst, A.R.; Coates, I.A.; Boucheteau, T.R.; Miravet, J.F.; Escuder, B.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Low-Molecular-Weight Gelators: Elucidating the Priciples of Gelation Based on Gelator Solubility and a Cooperative Self-Assembly Model. J. Am. Chem. Soc. 2008, 130, 9113–9121. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, Y.E. Structure and Dynamics of Hydrogels and Organogels: An NMR Spectroscopy Approach. Prog. Polym. Sci. 2011, 36, 1184–1253. [Google Scholar] [CrossRef]
- Nebot, V.J.; Armengol, J.; Smets, J.; Prieto, S.F.; Escuder, B.; Miravet, J.F. Molecular Hydrogels from Bolaform Amino Acid Derivatives: A Structure-Properties Study Based on the Thermodynamics of Gel Solubilization. Chem. Eur. J. 2012, 18, 4063–4072. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D. Win Some, Lose Some: Enthalpy-Entropy Compensation in Weak Intermolecular Interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Q.X. Isokinetic Relationship, Isoequilibrium Relationship and Enthalpy-Entropy Compensation. Chem. Rev. 2001, 101, 673–695. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amine | l,l,l | d,d,d | d,l,l | l,d,d |
|---|---|---|---|---|
| C4 | 51 | 51 | 71 | 71 |
| C5 | 64 | 64 | 65 | 65 |
| C6 | 70 | 70 | 70 | 70 |
| C7 | 53 | 53 | 69 | 69 |
| C8 | 45 | 45 | 54 | 54 |
| Dendron | Amine | T100% a/°C | Tgel b/°C | [Insol]@Tgel c/mM | MGC d/mM |
|---|---|---|---|---|---|
| L,L,L | C8 | 49 | 45 | 2.8 | 4.0 |
| L,L,L + D,D,D | C8 | 67 | 65 | 0.5 | 2.8 |
| L,D,D | C5 | 66 | 65 | 0.4 | 6.0 |
| L,D,D + D,L,L | C5 | 77 | 75 | 0.7 | 1.0 |
| Peptide Dendron | Amine. | ΔHdiss a/kJmol−1 | ΔSdiss a/Jmol−1 K−1 |
|---|---|---|---|
| L,L,L | C8 | 67.6 | 172 |
| L,L,L + D,D,D | C8 | 41.7 | 84.6 |
| L,D,D | C5 | 64.0 | 152 |
| L,D,D + D,L,L | C5 | 70.0 | 163 |
| Peptide Dendron | T100% a/°C | Tgel b/°C | [Insol]@Tgel c/mM | MGC d/mM |
|---|---|---|---|---|
| L,L,L | 72 | 69 | 0.5 | 0.6 |
| L,L,L + D,D,D | 75 | 74 | 1.2 | 1.4 |
| D,D,D | 55 | 53 | 1.3 | 1.6 |
| Peptide Dendron | ΔHdiss/kJmol−1 | ΔSdiss/Jmol−1 K−1 |
|---|---|---|
| L,L,L | 56.2 | 125 |
| L,L,L + D,D,D | 56.8 | 125 |
| D,D,D | 61.5 | 149 |
| Amines | Thermal Stability | Thermal Stability of Peptide Mixtures |
|---|---|---|
| Achiral Amines | L,L,L = D,D,D | Racemic Mixture Best |
| Chiral ‘R’ Amines | L,L,L > D,D,D | L,L,L ~ Racemic Mix >> D,D,D |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, W.; Smith, D.K. Chiral Assembly Preferences and Directing Effects in Supramolecular Two-Component Organogels. Gels 2018, 4, 31. https://doi.org/10.3390/gels4020031
Edwards W, Smith DK. Chiral Assembly Preferences and Directing Effects in Supramolecular Two-Component Organogels. Gels. 2018; 4(2):31. https://doi.org/10.3390/gels4020031
Chicago/Turabian StyleEdwards, William, and David K. Smith. 2018. "Chiral Assembly Preferences and Directing Effects in Supramolecular Two-Component Organogels" Gels 4, no. 2: 31. https://doi.org/10.3390/gels4020031
APA StyleEdwards, W., & Smith, D. K. (2018). Chiral Assembly Preferences and Directing Effects in Supramolecular Two-Component Organogels. Gels, 4(2), 31. https://doi.org/10.3390/gels4020031