Ionically Paired Layer-by-Layer Hydrogels: Water and Polyelectrolyte Uptake Controlled by Deposition Time




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
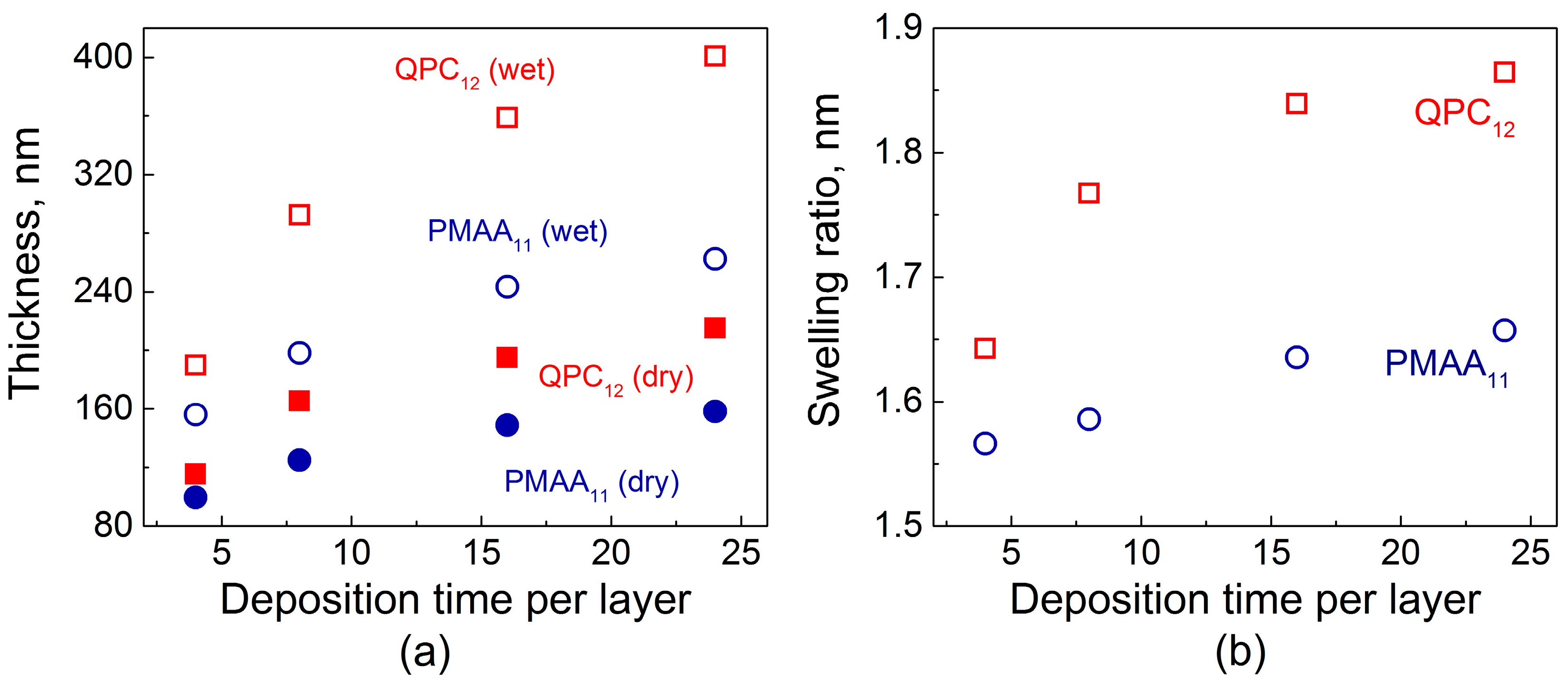
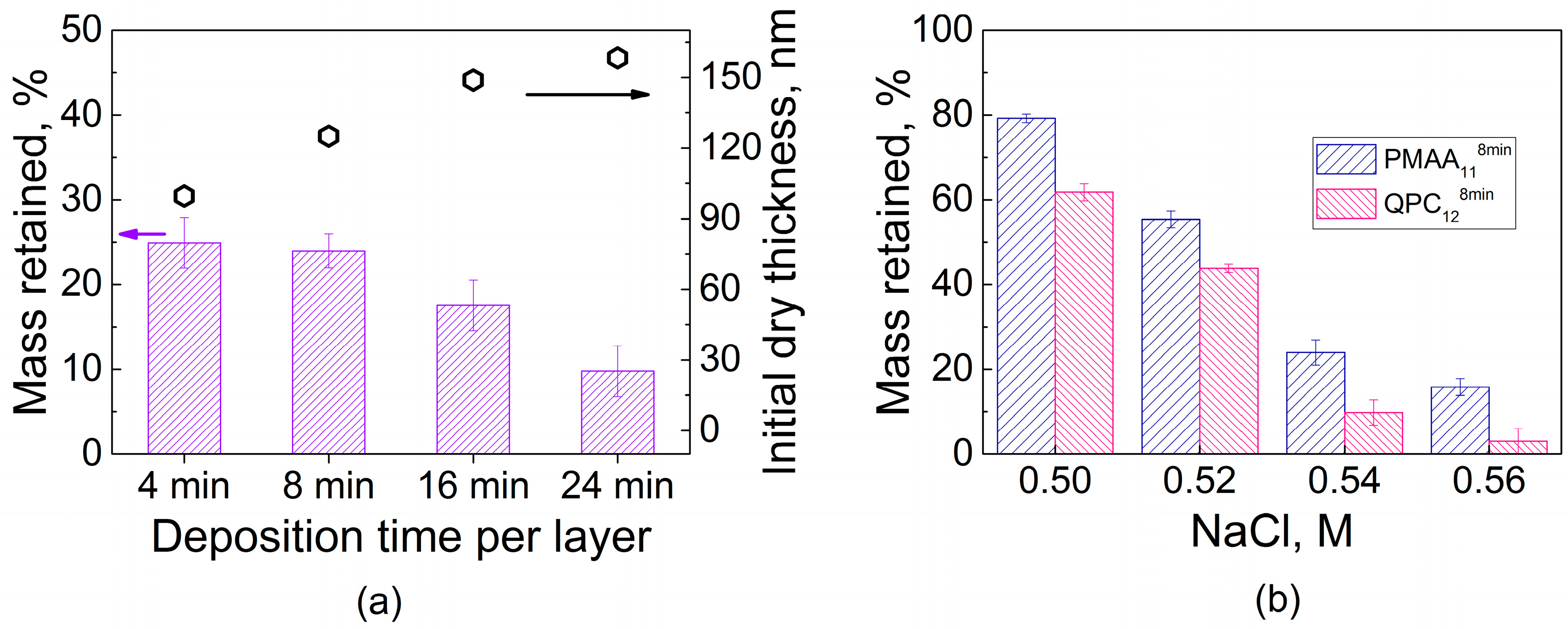
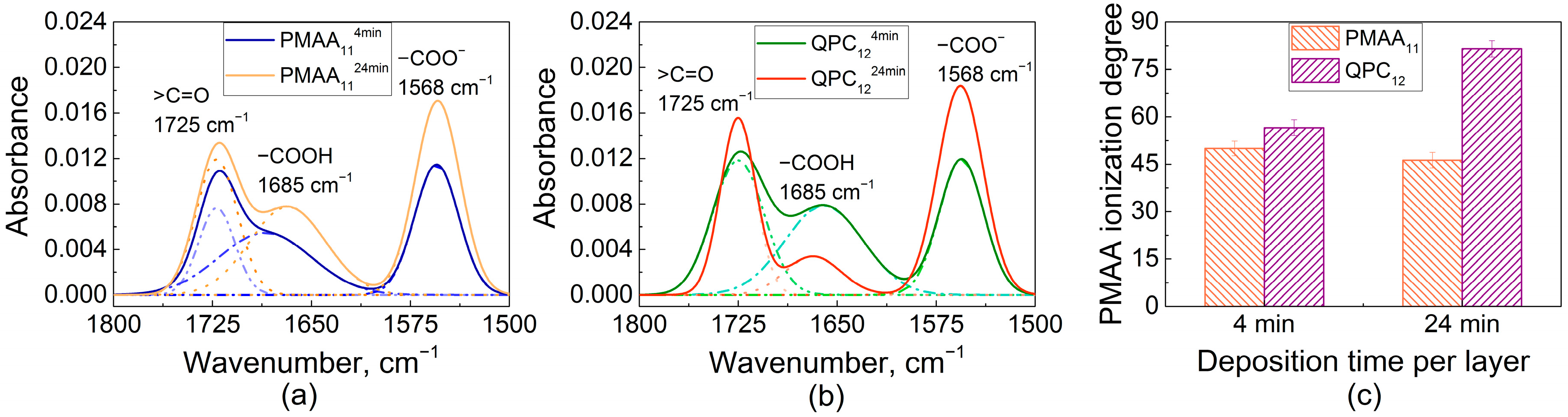
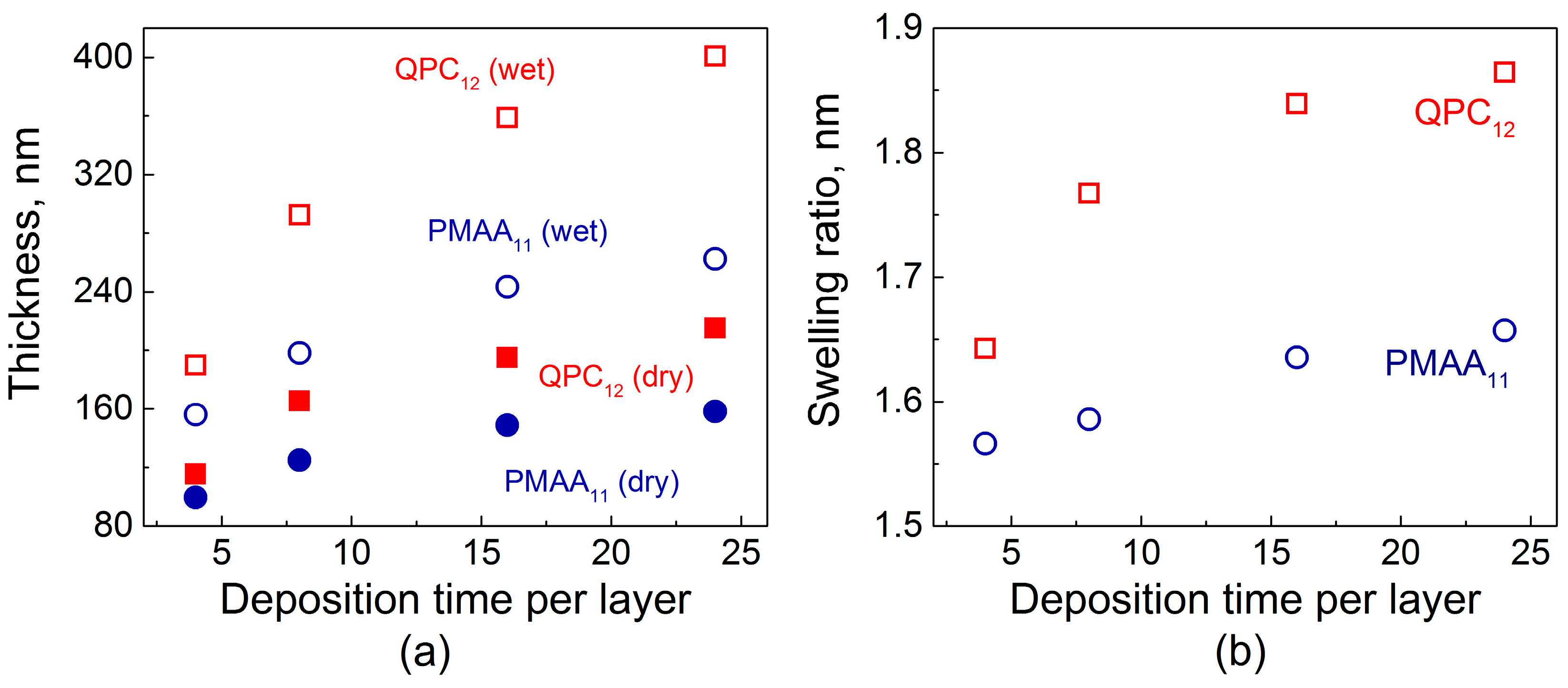
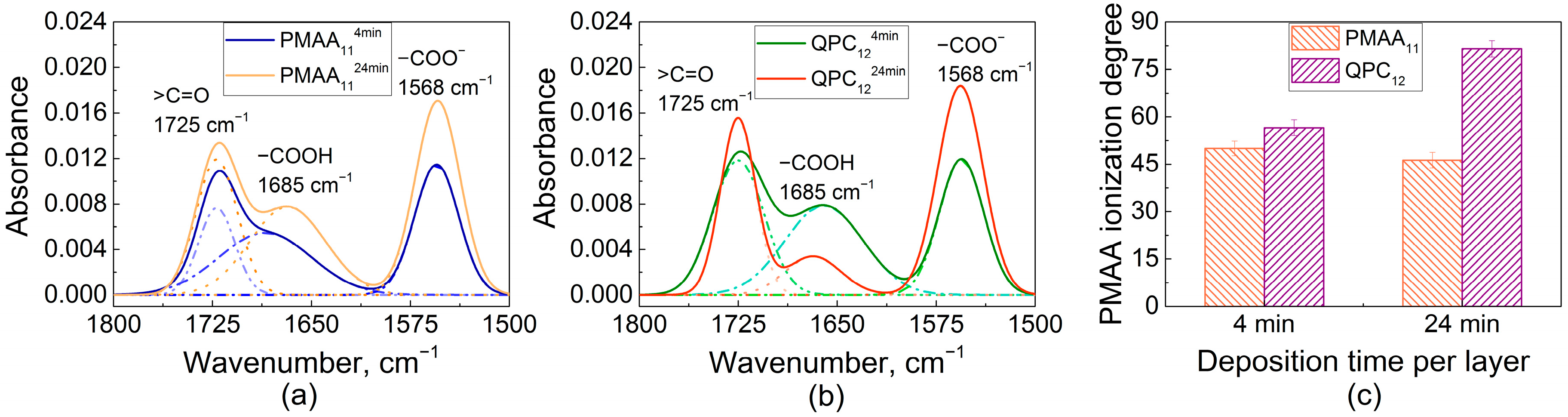
2.1. Film Swelling, Stability in Salt, and Poly(Methacrylic Acid) (PMAA) Ionization as a Function of Layer Deposition Time
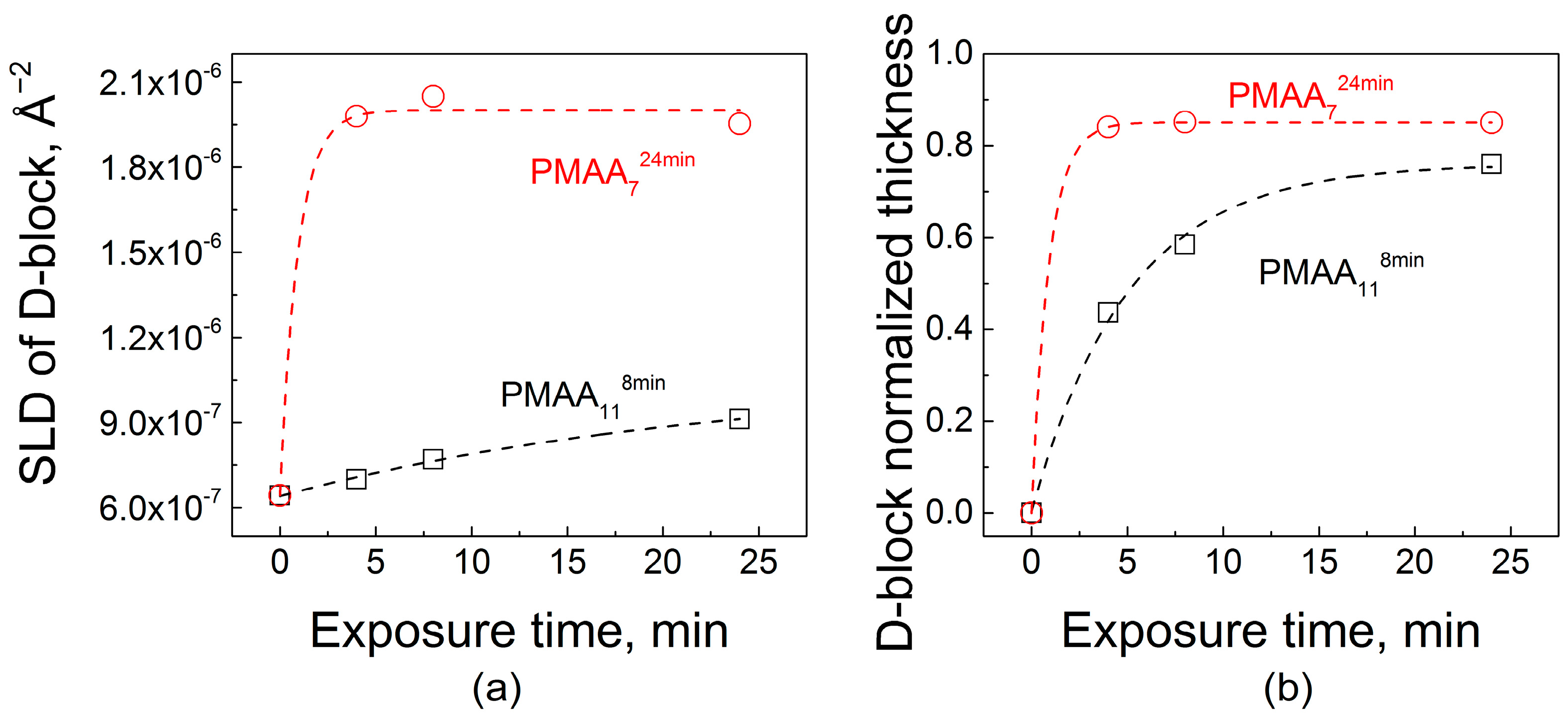
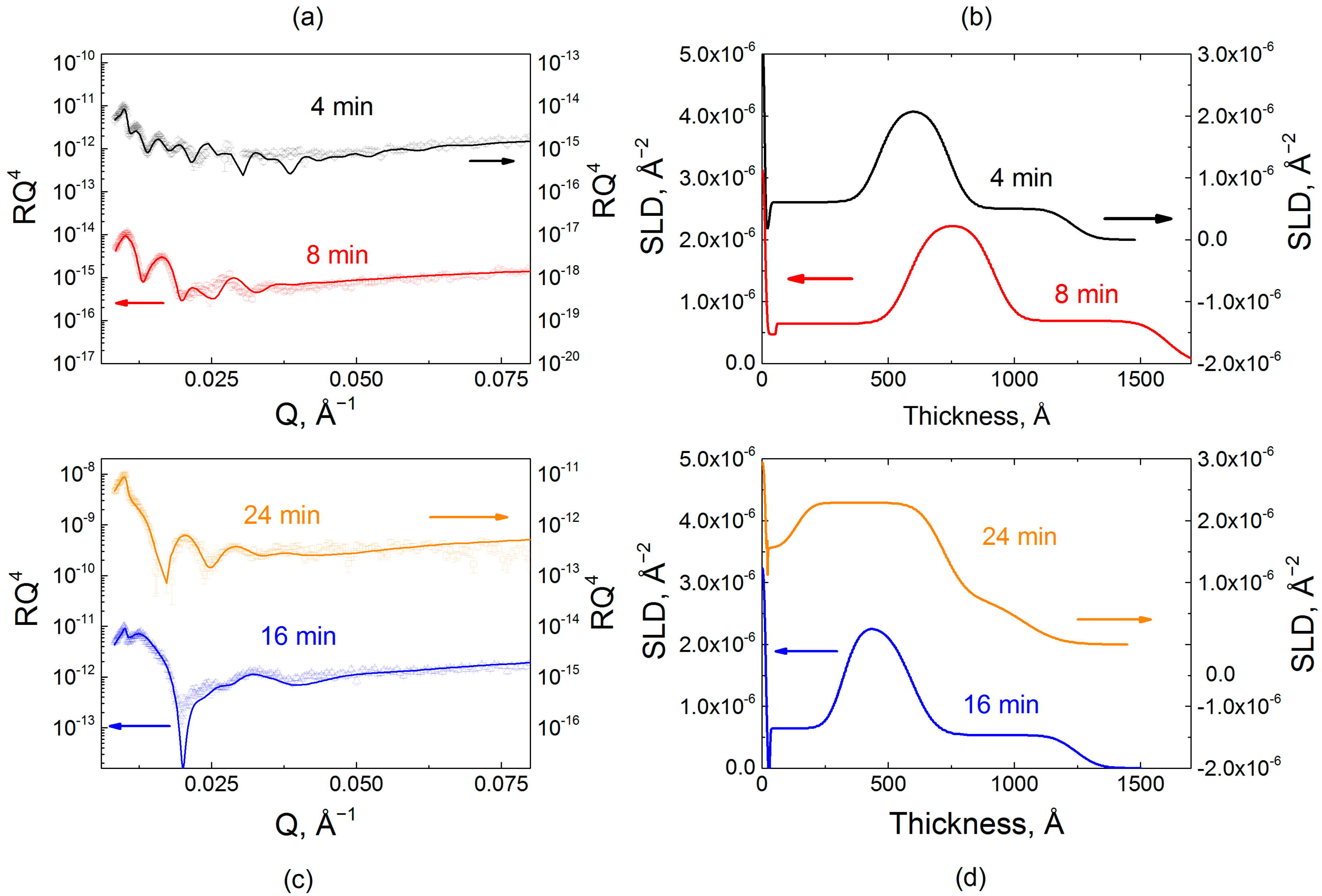
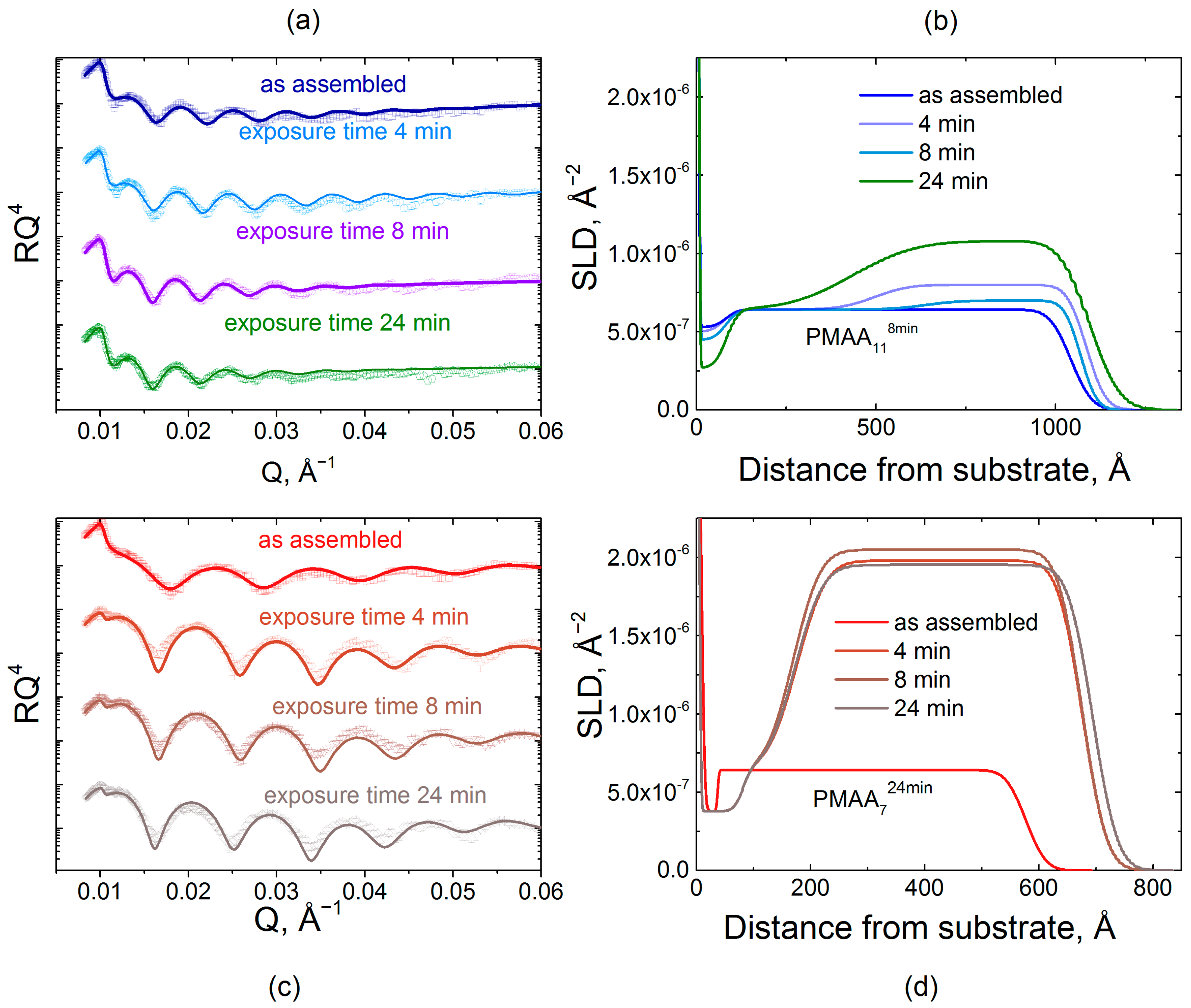
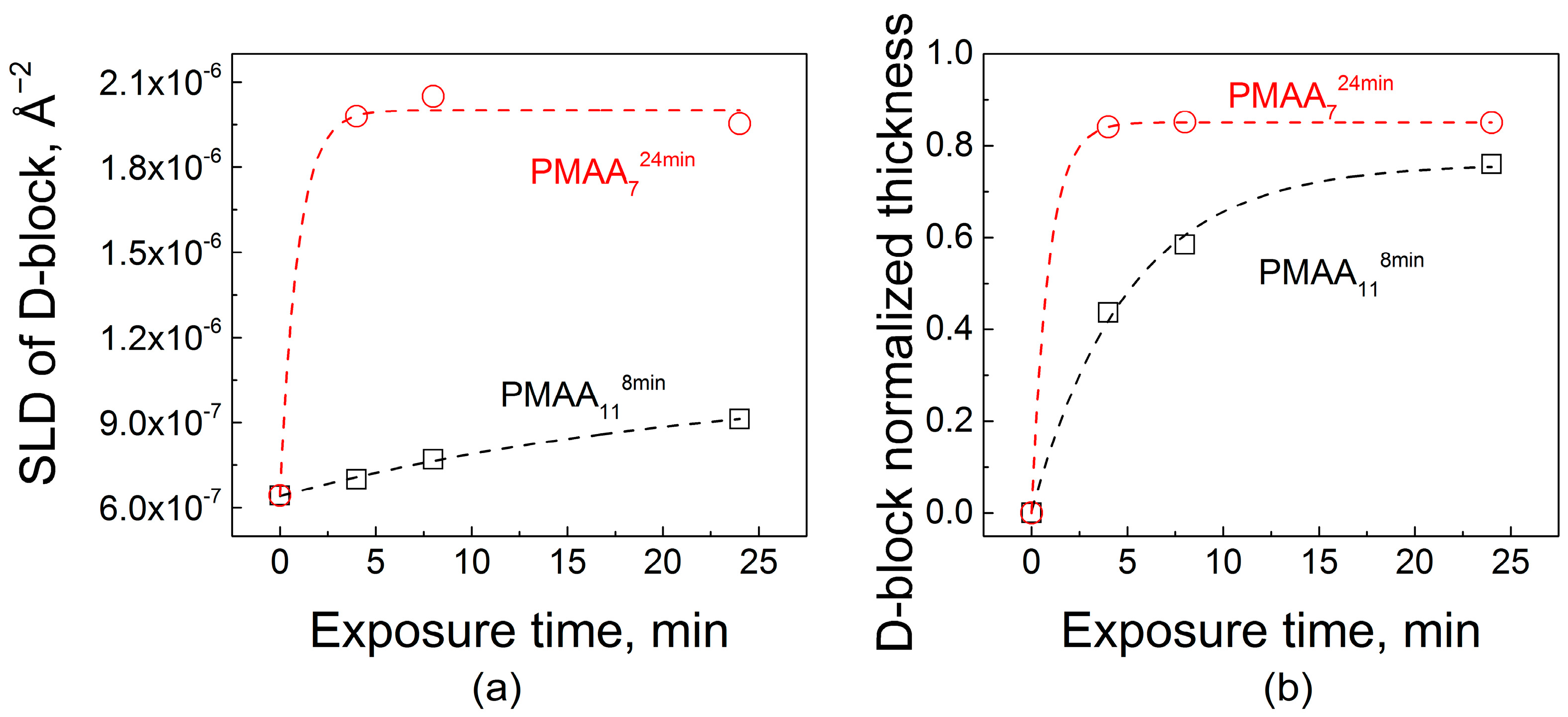
2.2. Effect of Film Internal Structure on Polyelectrolyte Uptake: Neutron Reflectometry Studies
3. Conclusions
4. Materials and Methods
4.1. Polycation Synthesis and Characterization
4.2. Multilayer Buildup and Polyelectrolyte Uptake Experiments
4.3. FTIR Analysis
4.4. Spectroscopic Ellipsometry
4.5. Neutron Reflectometry
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Decher, G. Layer-by-Layer Assembly (Putting Molecules to Work), in Multilayer Thin Films; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 1–21. [Google Scholar]
- Kharlampieva, E.; Kozlovskaya, V.; Sukhishvili, S.A. Layer-by-Layer Hydrogen-Bonded Polymer Films: From Fundamentals to Applications. Adv. Mater. 2009, 21, 3053–3065. [Google Scholar] [CrossRef]
- Borges, J.; Mano, J.F. Molecular Interactions Driving the Layer-by-Layer Assembly of Multilayers. Chem. Rev. 2014, 114, 8883–8942. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Johnston, A.P.R.; Such, G.K.; Zelikin, A.N.; Caruso, F. Next generation, sequentially assembled ultrathin films: Beyond electrostatics. Chem. Soc. Rev. 2007, 36, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Liljestrom, V.; Ora, A.; Hassinen, J.; Rekola, H.T.; Nonappa; Heilala, M.; Hynninen, V.; Joensuu, J.J.; Ras, R.H.A.; Torma, P.; et al. Cooperative colloidal self-assembly of metal-protein superlattice wires. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Sun, J.X.; Zhang, X.J.; Shen, D.; Yang, S.G. Hydrogen-Bonded Polymer Complex Thin Film of Poly(2-oxazoline) and Poly(acrylic acid). Polymers 2017, 9, 363. [Google Scholar] [CrossRef]
- Shukla, A.; Almeida, B. Advances in cellular and tissue engineering using layer-by-layer assembly. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Ferreira, A.M.; Callaghan, J.T.; Miller, C.A.; Atkinson, J.; Freeman, C.; Hatton, P.V. Multilayer Nanoscale Encapsulation of Biofunctional Peptides to Enhance Bone Tissue Regeneration In Vivo. Adv. Healthc. Mater. 2017, 6, 1601182. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Ghione, C.; Ferreira, A.M.; Crawford, A.; Hatton, P.V. Alginate-based hydrogels functionalised at the nanoscale using layer-by-layer assembly for potential cartilage repair. Biomater. Sci. 2017, 5, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, H.; Meng, Y. Layer-by-layer structured polysaccharides-based multilayers on cellulose acetate membrane: Towards better hemocompatibility, antibacterial and antioxidant activities. Appl. Surf. Sci. 2017, 401 (Suppl. C), 25–39. [Google Scholar] [CrossRef]
- Pavlukhina, S.; Sukhishvili, S. Polymer assemblies for controlled delivery of bioactive molecules from surfaces. Adv. Drug Deliv. Rev. 2011, 63, 822–836. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Han, F.; Zhao, P.; Lin, C.; Wen, X.; Ye, X. Layer-by-layer self-assembled multilayers on PEEK implants improve osseointegration in an osteoporosis rabbit model. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, B.; Kumaraswamy, G.; Caruso, F. Investigation of the Influence of Polyelectrolyte Charge Density on the Growth of Multilayer Thin Films Prepared by the Layer-by-Layer Technique. Macromolecules 2002, 35, 889–897. [Google Scholar] [CrossRef]
- Ramsden, J.J.; Lvov, Y.M.; Decher, G. Determination of optical constants of molecular films assembled via alternate polyion adsorption. Thin Solid Films 1995, 254, 246–251. [Google Scholar] [CrossRef]
- Lavalle, P.; Gergely, C.; Cuisinier, F.J.G.; Decher, G.; Schaaf, P.; Voegel, J.C.; Picart, C. Comparison of the structure of polyelectrolyte multilayer films exhibiting a linear and an exponential growth regime: An in situ atomic force microscopy study. Macromolecules 2002, 35, 4458–4465. [Google Scholar] [CrossRef]
- Kwon, S.R.; Elinski, M.B.; Batteas, J.D.; Lutkenhaus, J.L. Robust and Flexible Aramid Nanofiber/Graphene Layer-by-Layer Electrodes. ACS Appl. Mater. Interfaces 2017, 9, 17125–17135. [Google Scholar] [CrossRef] [PubMed]
- Elbert, D.L.; Herbert, C.B.; Hubbell, J.A. Thin Polymer Layers Formed by Polyelectrolyte Multilayer Techniques on Biological Surfaces. Langmuir 1999, 15, 5355–5362. [Google Scholar] [CrossRef]
- Ruths, J.; Essler, F.; Decher, G.; Riegler, H. Polyelectrolytes I: Polyanion/Polycation Multilayers at the Air/Monolayer/Water Interface as Elements for Quantitative Polymer Adsorption Studies and Preparation of Hetero-superlattices on Solid Surfaces. Langmuir 2000, 16, 8871–8878. [Google Scholar] [CrossRef]
- Picart, C.; Lavalle, P.; Hubert, P.; Cuisinier, F.J.G.; Decher, G.; Schaaf, P.; Voegel, J.C. Buildup Mechanism for Poly(l-lysine)/Hyaluronic Acid Films onto a Solid Surface. Langmuir 2001, 17, 7414–7424. [Google Scholar] [CrossRef]
- Nestler, P.; Paßvogel, M.; Helm, C.A. Influence of Polymer Molecular Weight on the Parabolic and Linear Growth Regime of PDADMAC/PSS Multilayers. Macromolecules 2013, 46, 5622–5629. [Google Scholar] [CrossRef]
- Xu, L.; Selin, V.; Zhuk, A.; Ankner, J.F.; Sukhishvili, S.A. Molecular Weight Dependence of Polymer Chain Mobility within Multilayer Films. ACS Macro Lett. 2013, 2, 865–868. [Google Scholar] [CrossRef]
- Guzman, E.; Ritacco, H.; Rubio, J.E.F.; Rubio, R.G.; Ortega, F. Salt-induced changes in the growth of polyelectrolyte layers of poly(diallyl-dimethylammonium chloride) and poly(4-styrene sulfonate of sodium). Soft Matter 2009, 5, 2130–2142. [Google Scholar] [CrossRef]
- Dubas, S.T.; Schlenoff, J.B. Factors controlling the growth of polyelectrolyte multilayers. Macromolecules 1999, 32, 8153–8160. [Google Scholar] [CrossRef]
- Butergerds, D.; Kateloe, C.; Cramer, C.; Schonhoff, M. Influence of the Degree of Ionization on the Growth Mechanism of Poly(Diallyldimethylammonium)/Poly(Acrylic Acid) Multilayers. J. Polym. Sci. B Polym. Phys. 2017, 55, 425–434. [Google Scholar] [CrossRef]
- Alonso, T.; Irigoyen, J.; Iturri, J.J.; larena, I.L.; Moya, S.E. Study of the multilayer assembly and complex formation of poly(diallyldimethylammonium chloride) (PDADMAC) and poly(acrylic acid) (PAA) as a function of pH. Soft Matter 2013, 9, 1920–1928. [Google Scholar] [CrossRef]
- Lavalle, P.; Picart, C.; Mutterer, J.; Gergely, C.; Reiss, H.; Voegel, J.-C.; Senger, B.; Schaaf, P. Modeling the Buildup of Polyelectrolyte Multilayer Films Having Exponential Growth. J. Phys. Chem. B 2004, 108, 635–648. [Google Scholar] [CrossRef]
- Picart, C.; Mutterer, J.; Richert, L.; Luo, Y.; Prestwich, G.D.; Schaaf, P.; Voegel, J.-C.; Lavalle, P. Molecular basis for the explanation of the exponential growth of polyelectrolyte multilayers. Proc. Natl. Acad. Sci. USA 2002, 99, 12531–12535. [Google Scholar] [CrossRef] [PubMed]
- Haynie, D.T.; Cho, E.; Waduge, P. In and out diffusion hypothesis of exponential multilayer film buildup revisited. Langmuir 2011, 27, 5700–5704. [Google Scholar] [CrossRef] [PubMed]
- Porcel, C.; Lavalle, P.; Decher, G.; Senger, B.; Voegel, J.C.; Schaaf, P. Influence of the Polyelectrolyte Molecular Weight on Exponentially Growing Multilayer Films in the Linear Regime. Langmuir 2007, 23, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Porcel, C.; Lavalle, P.; Ball, V.; Decher, G.; Senger, B.; Voegel, J.-C.; Schaaf, P. From Exponential to Linear Growth in Polyelectrolyte Multilayers. Langmuir 2006, 22, 4376–4383. [Google Scholar] [CrossRef] [PubMed]
- Fares, H.M.; Schlenoff, J.B. Diffusion of Sites versus Polymers in Polyelectrolyte Complexes and Multilayers. J. Am. Chem. Soc. 2017, 139, 14656–14667. [Google Scholar] [CrossRef] [PubMed]
- Selin, V.; Ankner, J.F.; Sukhishvili, S.A. Nonlinear Layer-by-Layer Films: Effects of Chain Diffusivity on Film Structure and Swelling. Macromolecules 2017, 50, 6192–6201. [Google Scholar] [CrossRef]
- Xu, L.; Pristinski, D.; Zhuk, A.; Stoddart, C.; Ankner, J.F.; Sukhishvili, S.A. Linear versus Exponential Growth of Weak Polyelectrolyte Multilayers: Correlation with Polyelectrolyte Complexes. Macromolecules 2012, 45, 3892–3901. [Google Scholar] [CrossRef]
- Xu, L.; Ankner, J.F.; Sukhishvili, S.A. Steric Effects in Ionic Pairing and Polyelectrolyte Interdiffusion within Multilayered Films: A Neutron Reflectometry Study. Macromolecules 2011, 44, 6518–6524. [Google Scholar] [CrossRef]
- Selin, V.; Ankner, J.F.; Sukhishvili, S.A. Diffusional Response of Layer-by-Layer Assembled Polyelectrolyte Chains to Salt Annealing. Macromolecules 2015, 48, 3983–3990. [Google Scholar] [CrossRef]
- Korneev, D.; Lvov, Y.; Decher, G.; Schmitt, J.; Yaradaikin, S. Neutron Reflectivity Analysis of Self-Assembled Film Superlattices with Alternate Layers of Deuterated and Hydrogenated Polysterenesulfonate and Polyallylamine. Phys. B 1995, 213, 954–956. [Google Scholar] [CrossRef]
- Lösche, M.; Schmitt, J.; Decher, G.; Bouwman, W.G.; Kjaer, K. Detailed Structure of Molecularly Thin Polyelectrolyte Multilayer Films on Solid Substrates as Revealed by Neutron Reflectometry. Macromolecules 1998, 31, 8893–8906. [Google Scholar] [CrossRef]
- Kharlampieva, E.; Kozlovskaya, V.; Ankner, J.F.; Sukhishvili, S.A. Hydrogen-bonded polymer multilayers probed by neutron reflectivity. Langmuir 2008, 24, 11346–11349. [Google Scholar] [CrossRef] [PubMed]
- Kharlampieva, E.L.; Sukhishvili, S.A. Ionization and pH stability of multilayers formed by self-assembly of weak polyelectrolytes. Langmuir 2003, 19, 1235–1243. [Google Scholar] [CrossRef]
- Dubas, S.T.; Schlenoff, J.B. Swelling and Smoothing of Polyelectrolyte Multilayers by Salt. Langmuir 2001, 17, 7725–7727. [Google Scholar] [CrossRef]
- Xu, L.; Kozlovskaya, V.; Kharlampieva, E.; Ankner, J.F.; Sukhishvili, S.A. Anisotropic Diffusion of Polyelectrolyte Chains within Multilayer Films. ACS Macro Lett. 2012, 1, 127–130. [Google Scholar] [CrossRef] [PubMed]
- McAloney, R.A.; Dudnik, V.; Goh, M.C. Kinetics of Salt-Induced Annealing of a Polyelectrolyte Multilayer Film Morphology. Langmuir 2003, 19, 3947–3952. [Google Scholar] [CrossRef]
- Dubas, S.T.; Schlenoff, J.B. Polyelectrolyte Multilayers Containing a Weak Polyacid: Construction and Deconstruction. Macromolecules 2001, 34, 3736–3740. [Google Scholar] [CrossRef]
- Zhuk, A.; Selin, V.; Zhuk, I.; Belov, B.; Ankner, J.F.; Sukhishvili, S.A. Chain conformation and dynamics in spin-assisted weak polyelectrolyte multilayers. Langmuir 2015, 31, 3889–3896. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Nkwantabisah, S.; Gammana, M.; Tripp, C.P. Dynamics of Layer-by-Layer Growth of a Polyelectrolyte Multilayer Studied in Situ Using Attenuated Total Reflectance Infrared Spectroscopy. Langmuir 2014, 30, 11696–11703. [Google Scholar] [CrossRef] [PubMed]
- Sukhishvili, S.A.; Granick, S. Layered, Erasable Polymer Multilayers Formed by Hydrogen-Bonded Sequential Self-Assembly. Macromolecules 2002, 35, 301–310. [Google Scholar] [CrossRef]
- Choi, J.; Rubner, M.F. Influence of the degree of ionization on weak polyelectrolyte multilayer assembly. Macromolecules 2005, 38, 116–124. [Google Scholar] [CrossRef]
- Burke, S.E.; Barrett, C.J. Acid−Base Equilibria of Weak Polyelectrolytes in Multilayer Thin Films. Langmuir 2003, 19, 3297–3303. [Google Scholar] [CrossRef]
- Müller, M.; Rieser, T.; Lunkwitz, K.; Berwald, S.; Meier-Haack, J.; Jehnichen, D. An in-situ ATR-FTIR study on polyelectrolyte multilayer assemblies on solid surfaces and their susceptibility to fouling. Macromol. Rapid Commun. 1998, 19, 333–336. [Google Scholar]
- Müller, M.; Briššová, M.; Rieser, T.; Powers, A.C.; Lunkwitz, K. Deposition and properties of polyelectrolyte multilayers studied by ATR-FTIR spectroscopy. Mater. Sci. Eng. C 1999, 8–9 (Suppl. C), 163–169. [Google Scholar] [CrossRef]
- Zhang, X.; Xia, J.H.; Gaynor, S.G.; Matyjaszewski, K. Atom transfer radical polymerization using functional initiators containing carboxylic acid group. Abstr. Pap. Am. Chem. Soc. 1998, 216, U872. [Google Scholar]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selin, V.; Ankner, J.F.; Sukhishvili, S.A. Ionically Paired Layer-by-Layer Hydrogels: Water and Polyelectrolyte Uptake Controlled by Deposition Time. Gels 2018, 4, 7. https://doi.org/10.3390/gels4010007
Selin V, Ankner JF, Sukhishvili SA. Ionically Paired Layer-by-Layer Hydrogels: Water and Polyelectrolyte Uptake Controlled by Deposition Time. Gels. 2018; 4(1):7. https://doi.org/10.3390/gels4010007
Chicago/Turabian StyleSelin, Victor, John F. Ankner, and Svetlana A. Sukhishvili. 2018. "Ionically Paired Layer-by-Layer Hydrogels: Water and Polyelectrolyte Uptake Controlled by Deposition Time" Gels 4, no. 1: 7. https://doi.org/10.3390/gels4010007
APA StyleSelin, V., Ankner, J. F., & Sukhishvili, S. A. (2018). Ionically Paired Layer-by-Layer Hydrogels: Water and Polyelectrolyte Uptake Controlled by Deposition Time. Gels, 4(1), 7. https://doi.org/10.3390/gels4010007