

Hydrogel Network Architecture Design Space: Impact on Mechanical and Viscoelastic Properties

 ,
,  and
and
Abstract

1. Introduction
2. Crosslinker Architecture Design Space
2.1. Chemical Composition of Crosslinkers
2.1.1. Synthetic Crosslinkers
2.1.2. Peptide and Protein Crosslinkers
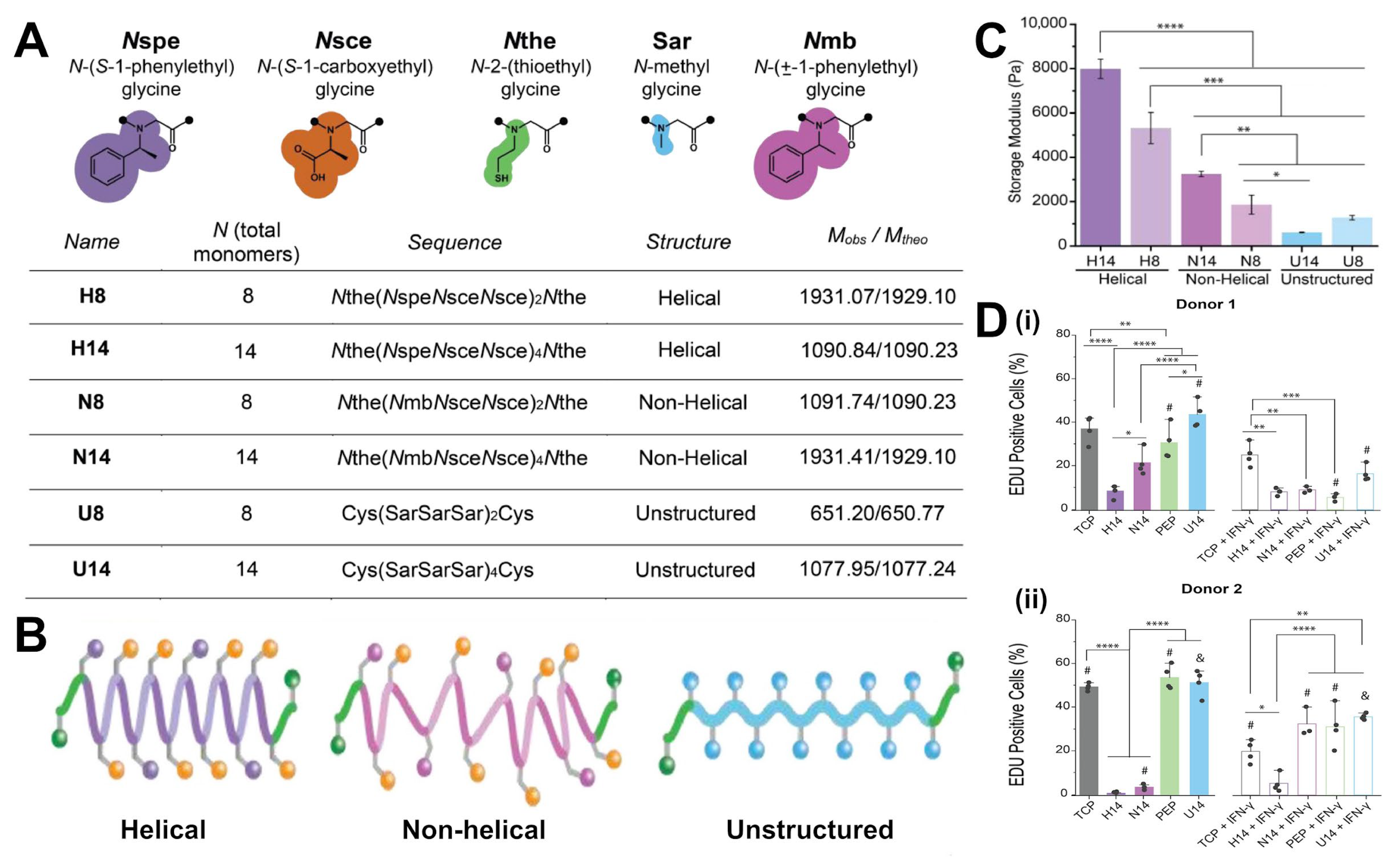
2.1.3. Peptoid and Peptidomimetic Crosslinkers
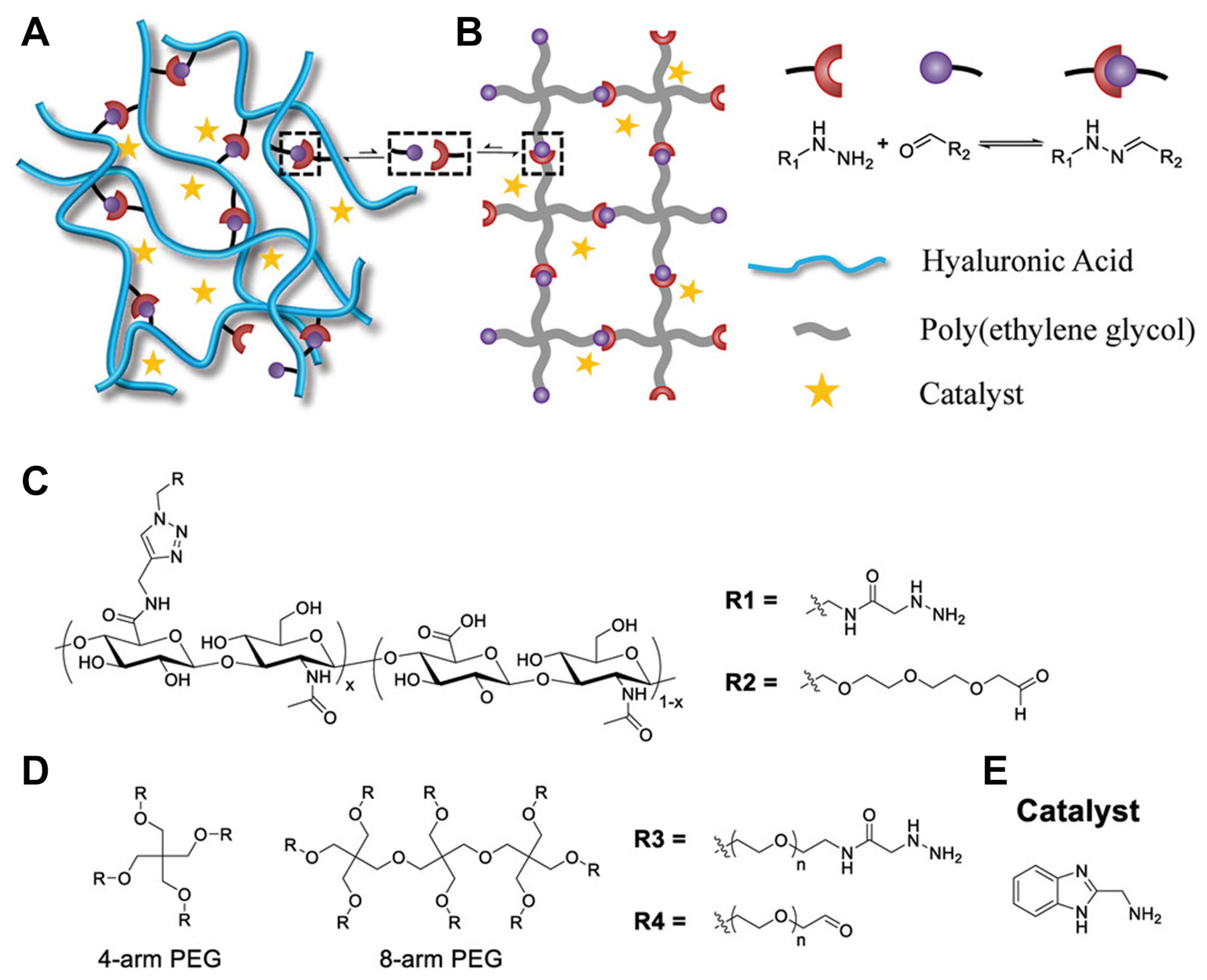
2.1.4. Polysaccharide-Based Crosslinkers
2.1.5. Hybrid and Composite Crosslinker Systems
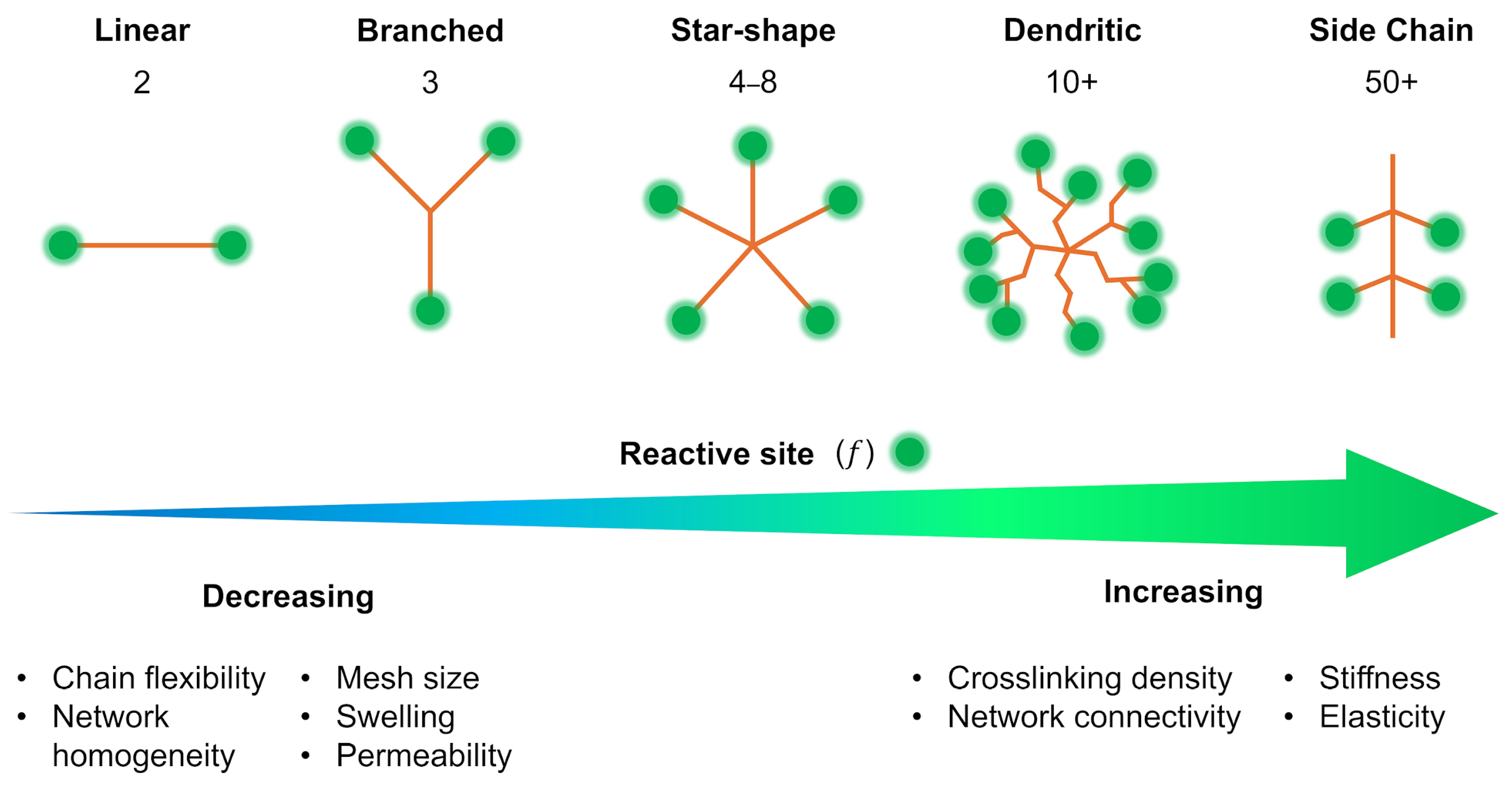
2.2. Structural Typology of Crosslinkers
2.2.1. Linear Crosslinkers (LX)
2.2.2. Star (SX) and Branched Crosslinkers
2.2.3. Side-Chain Crosslinking (SCX)
2.2.4. Dendritic Crosslinkers
3. Crosslinking Chemistry, Kinetics, and Dynamics
3.1. Covalent Crosslinking Mechanisms
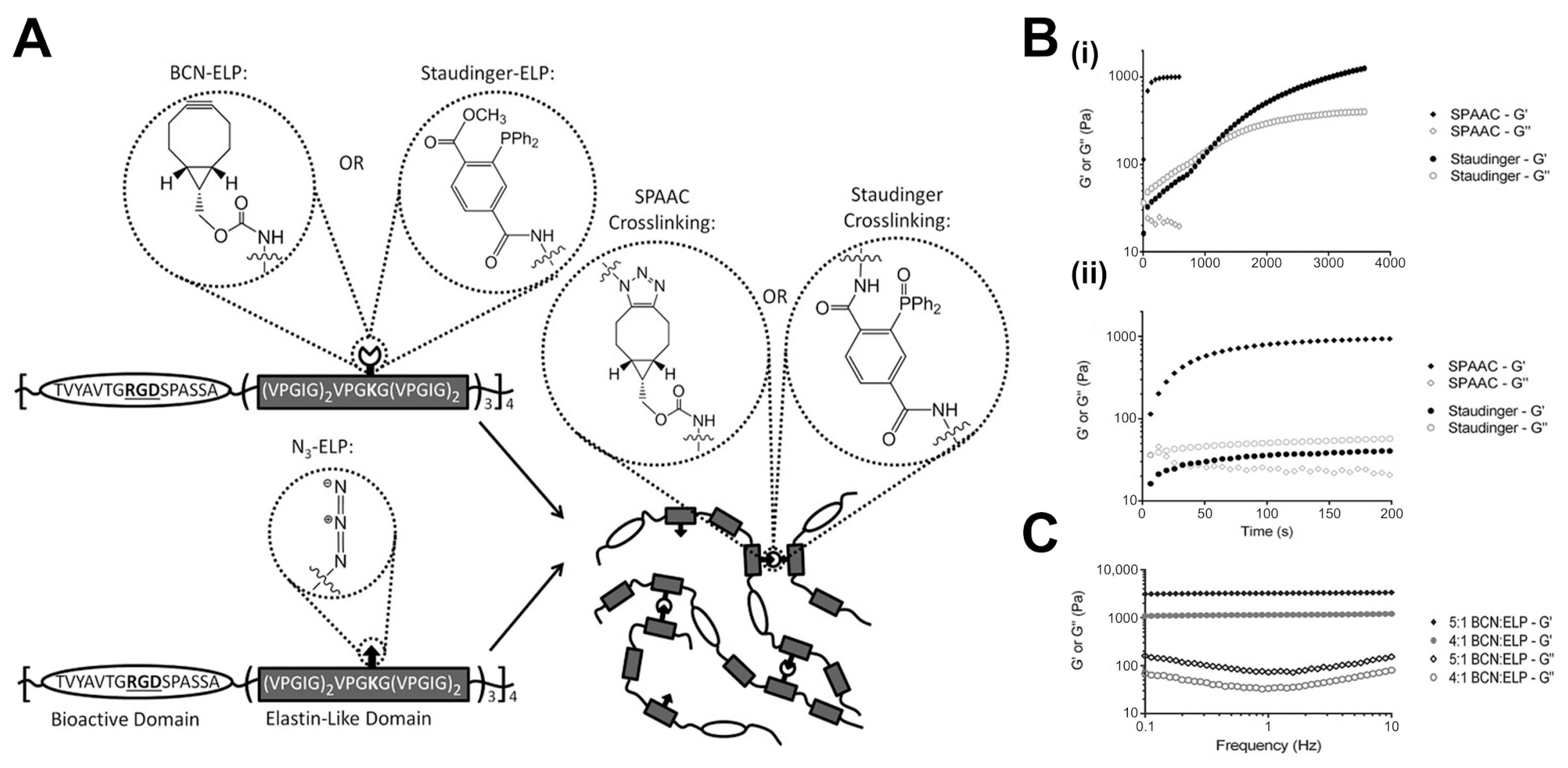
3.1.1. Photopolymerization and Click Chemistry Approaches
3.1.2. Thiol–Ene Reaction Systems
3.1.3. Michael Addition and Other Addition Reactions
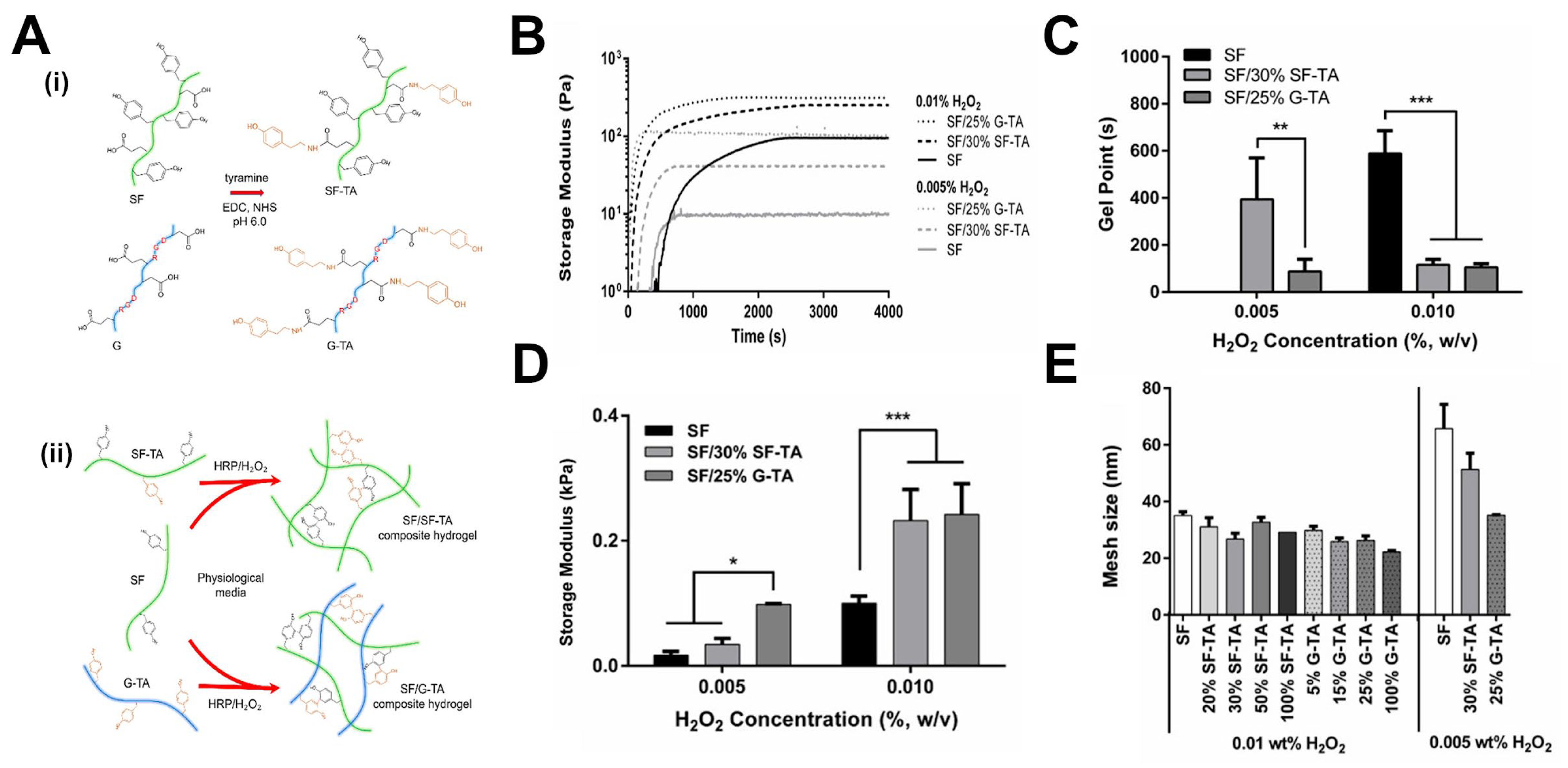
3.1.4. Effect of Crosslinking Kinetics on Resulting Network Structure and Hydrogel Properties
3.2. Dynamic Covalent Crosslinking
3.2.1. Principles of Reversible Covalent Bonds in Hydrogels
3.2.2. Stress Relaxation Behavior in Dynamic Covalent Crosslinked Hydrogels
3.2.3. Relationship Between Bond Exchange Kinetics and Viscoelasticity
3.3. Non-Covalent Crosslinking Interactions
3.3.1. Hydrogen Bonding Networks as Crosslinking Motifs
3.3.2. Host–Guest Interactions and Their Influence on Network Dynamics
3.3.3. Metal–Ligand Coordination in Crosslinked Networks
3.3.4. Combining Covalent and Non-Covalent Interactions for Complex Mechanical Responses
4. Structure–Property Relationships
4.1. Network Connectivity Effects
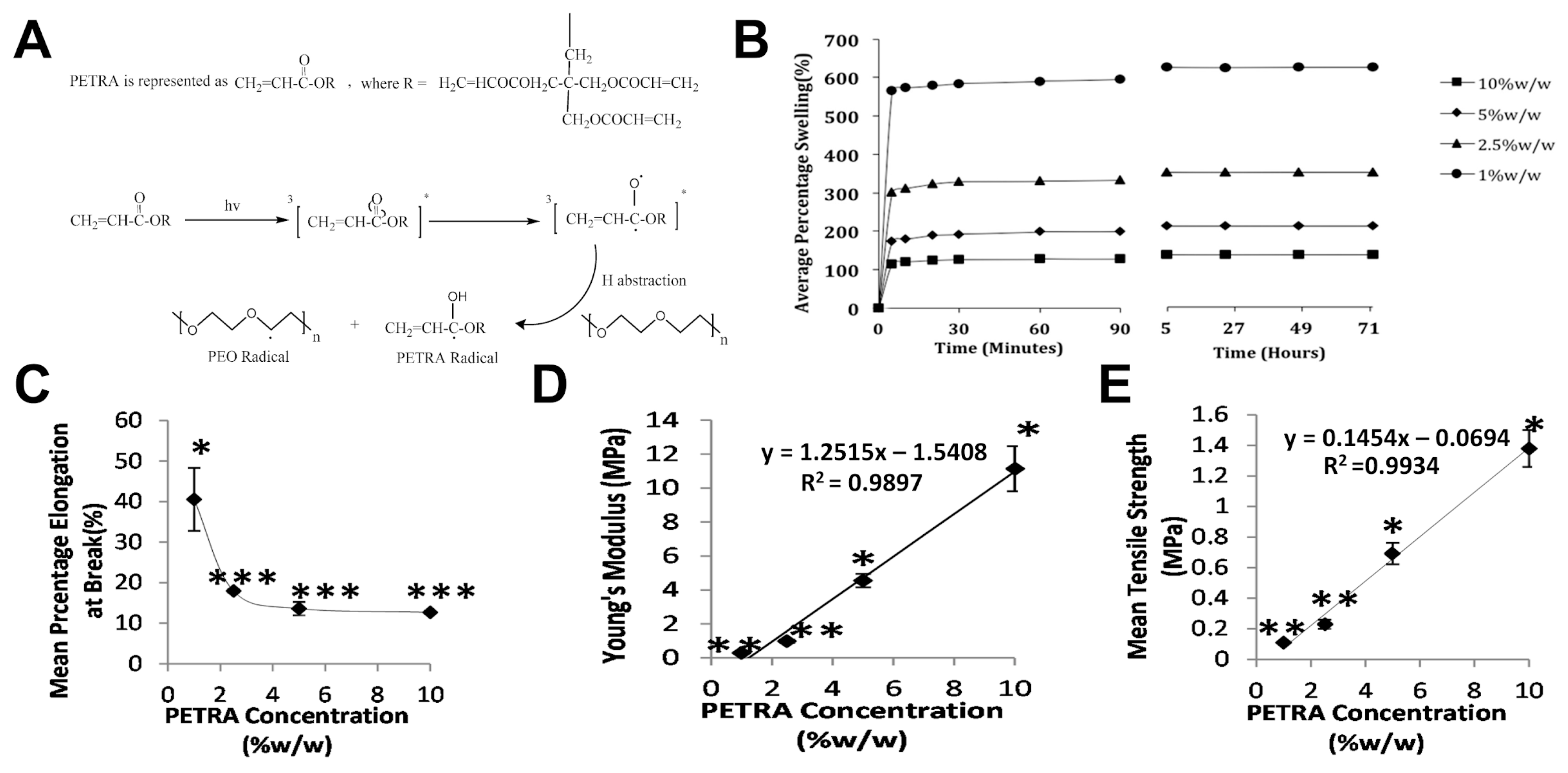
4.1.1. Crosslink Density Directly Impacts Hydrogel Mechanics
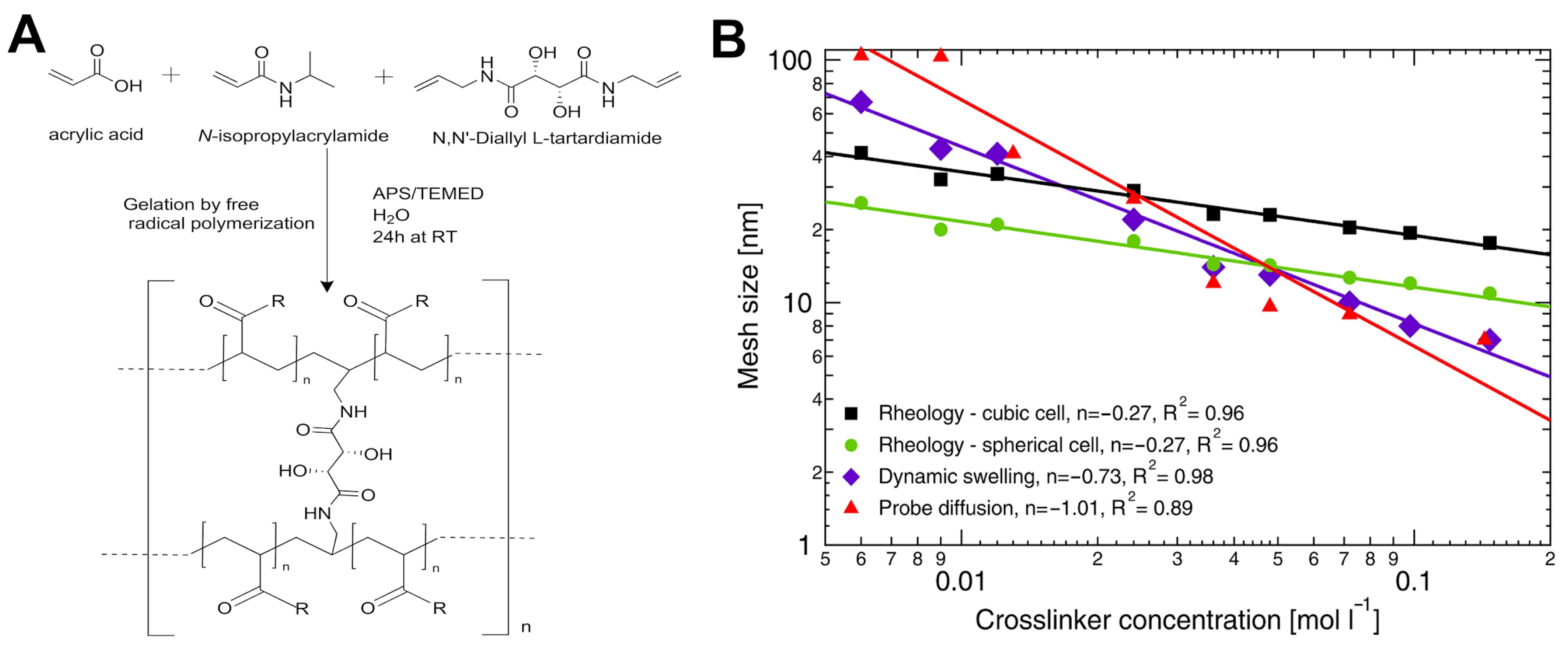
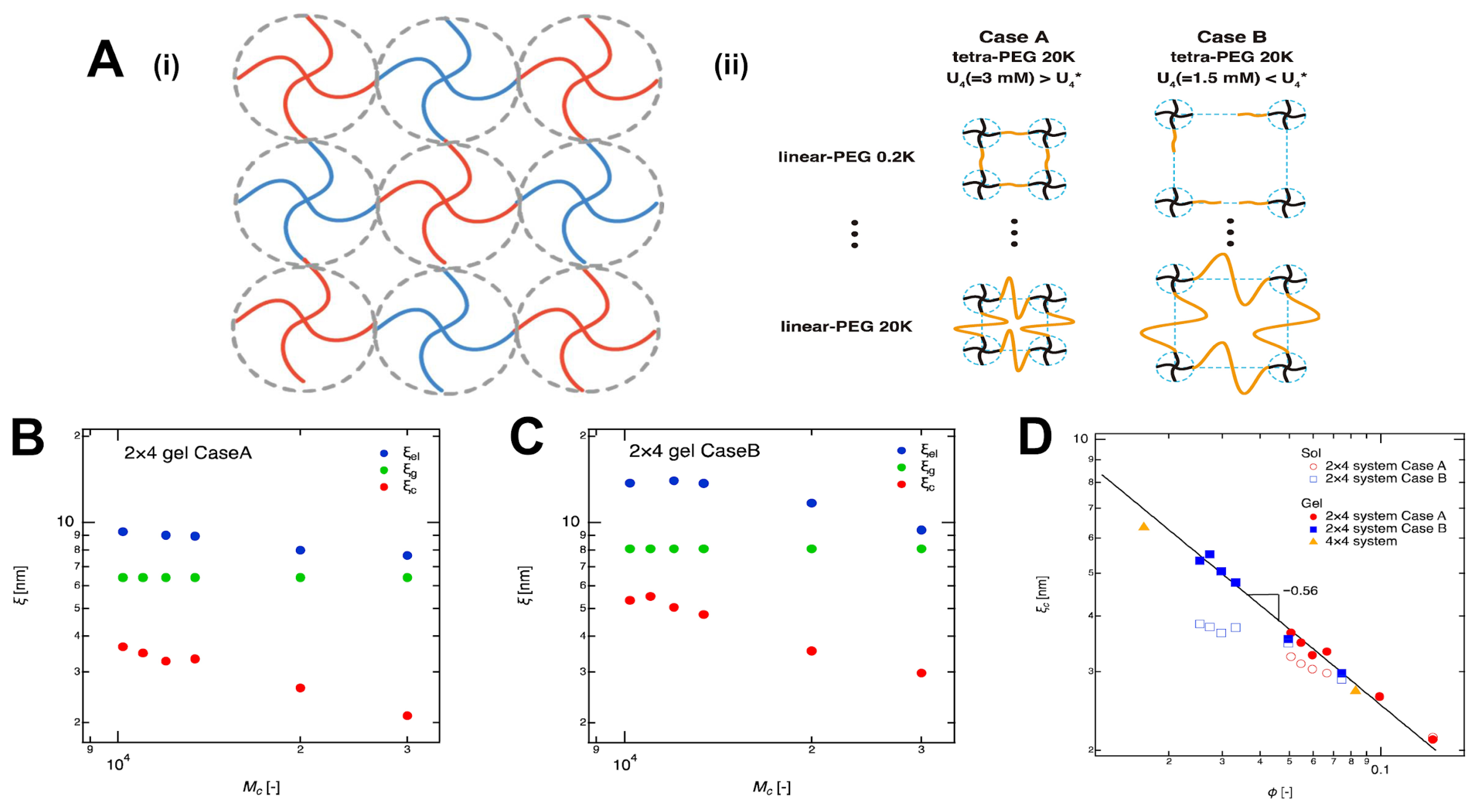
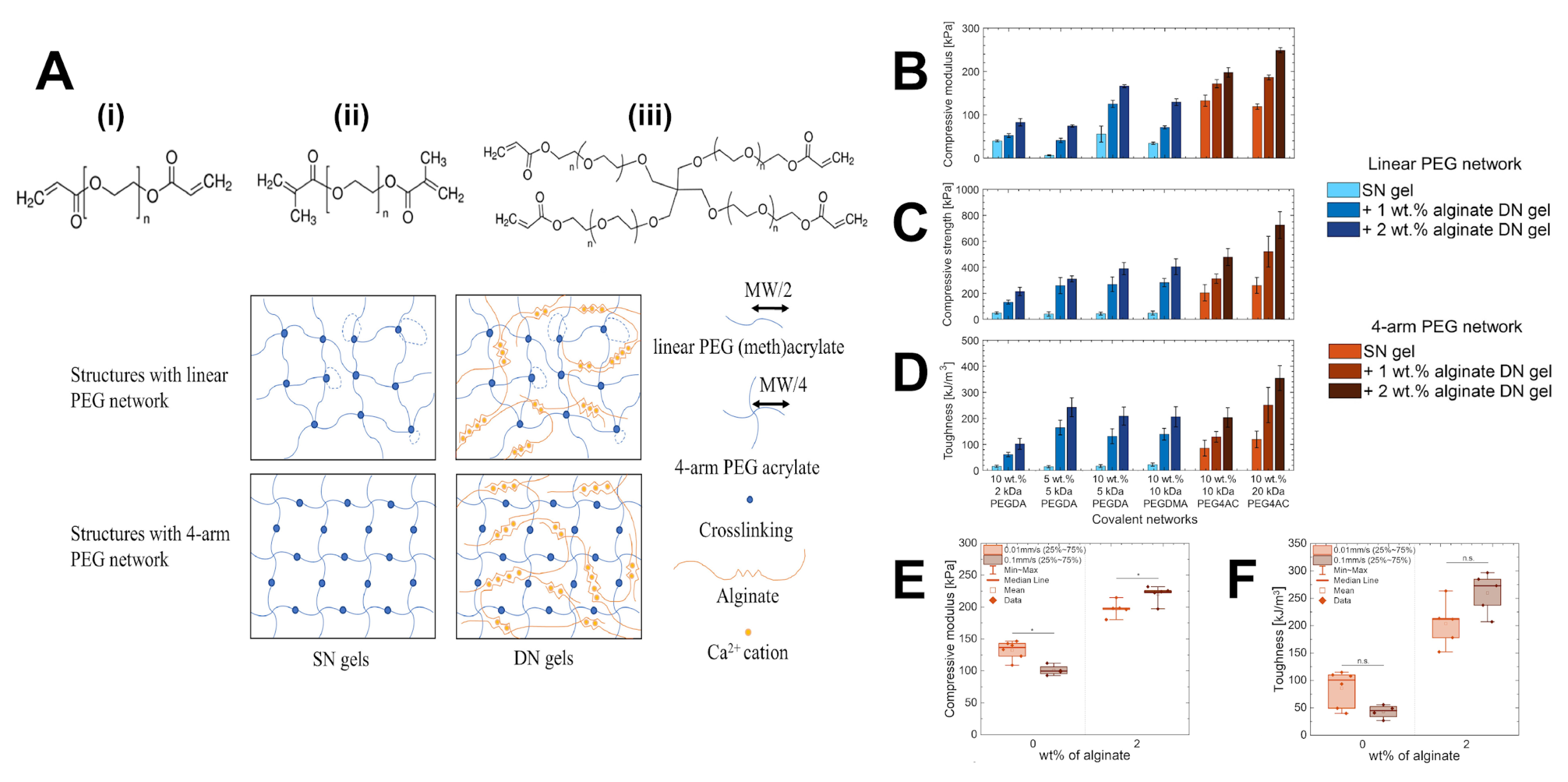
4.1.2. Crosslinker Architecture Governs Mesh Size Variation in Hydrogel Networks
4.1.3. Effect of Moisture Content on Hydrogel Mechanical Properties
4.2. Viscoelastic Behavior
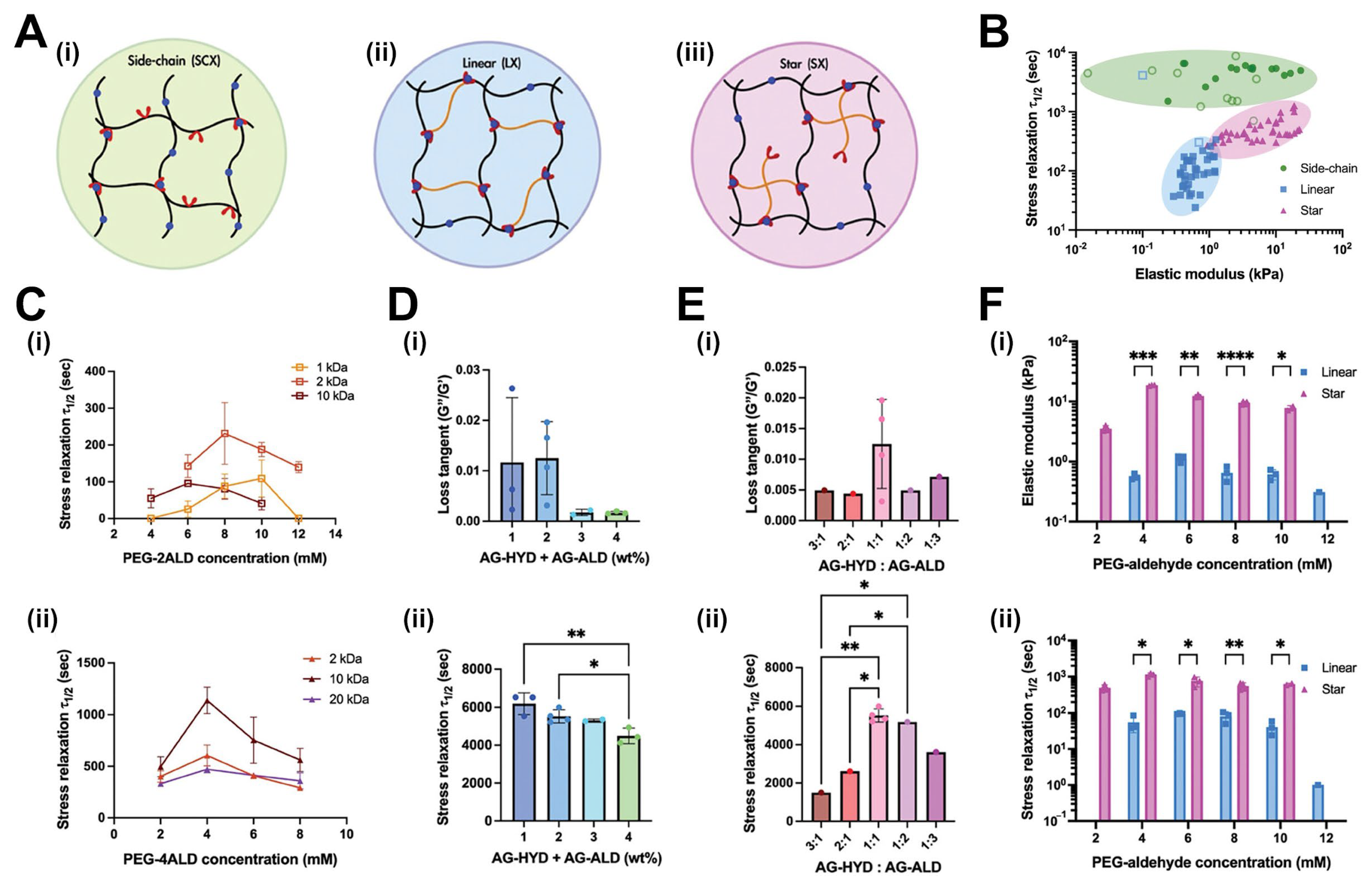
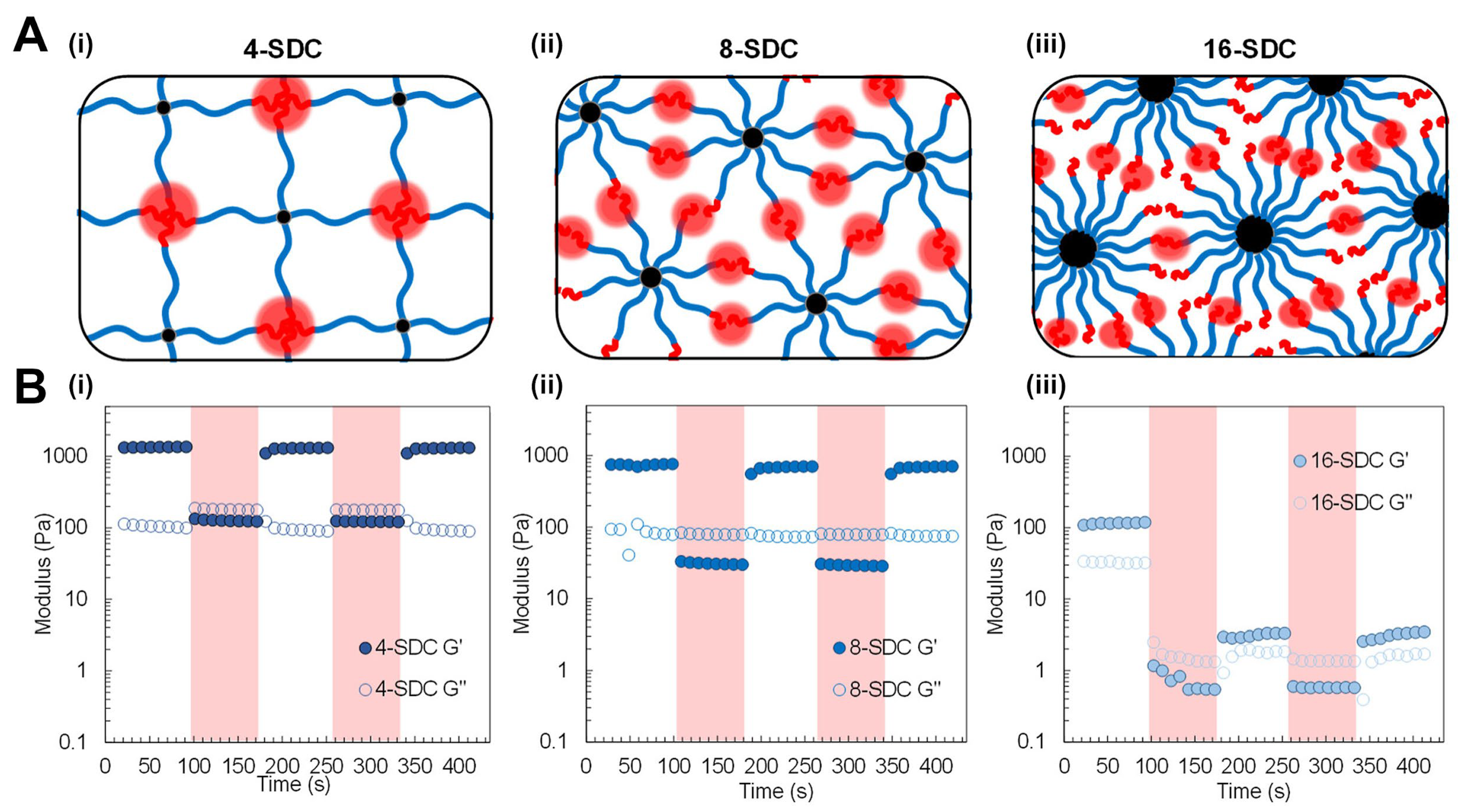
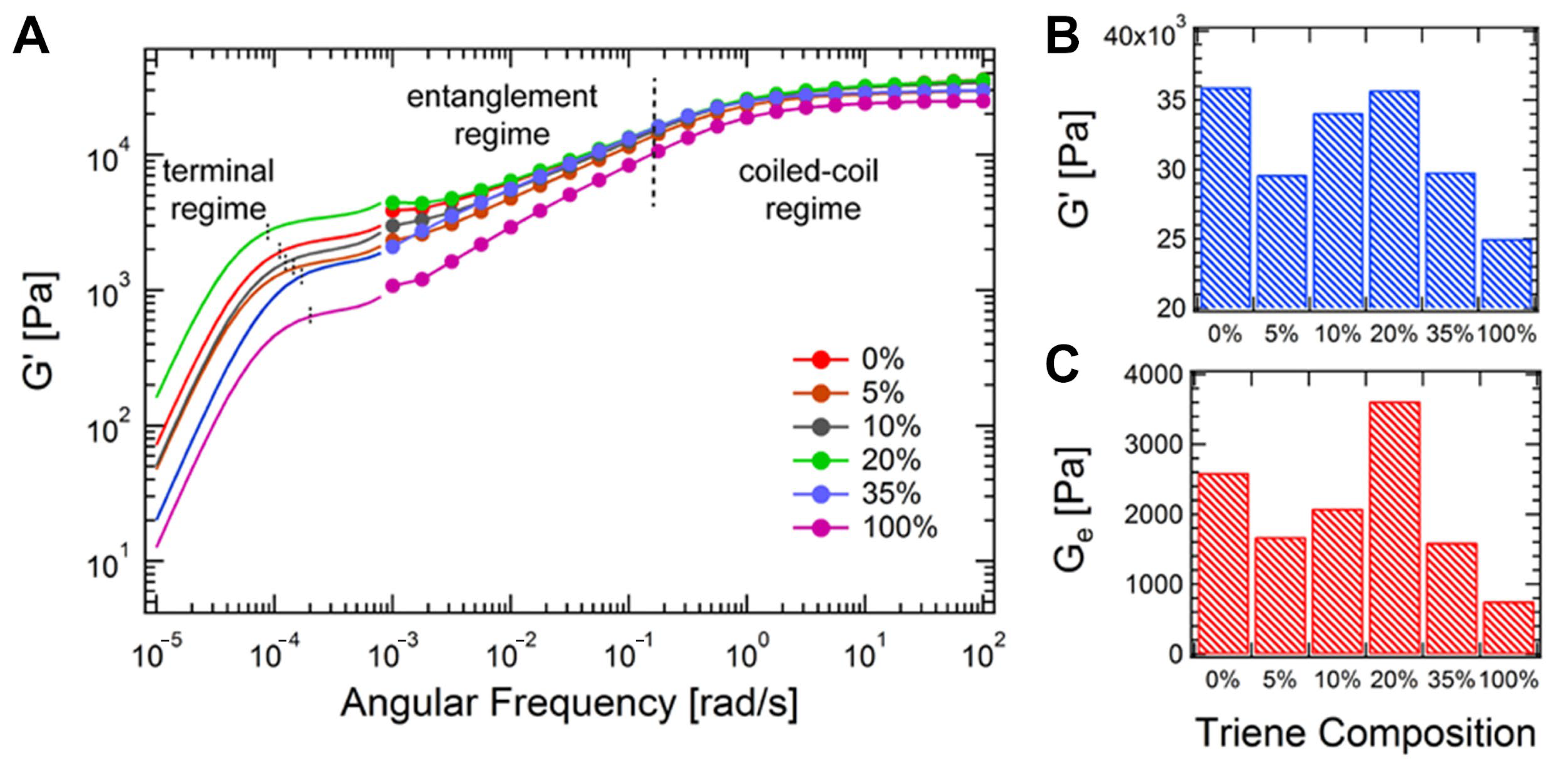
4.2.1. The Influence of Crosslink Architecture on Stress Relaxation Dynamics
4.2.2. Half-Stress Relaxation Time (t½) as a Function of Crosslinker Structure
4.2.3. Control of Creep Behavior Through Crosslinker Design
4.3. Engineering Mechanics in Hydrogels
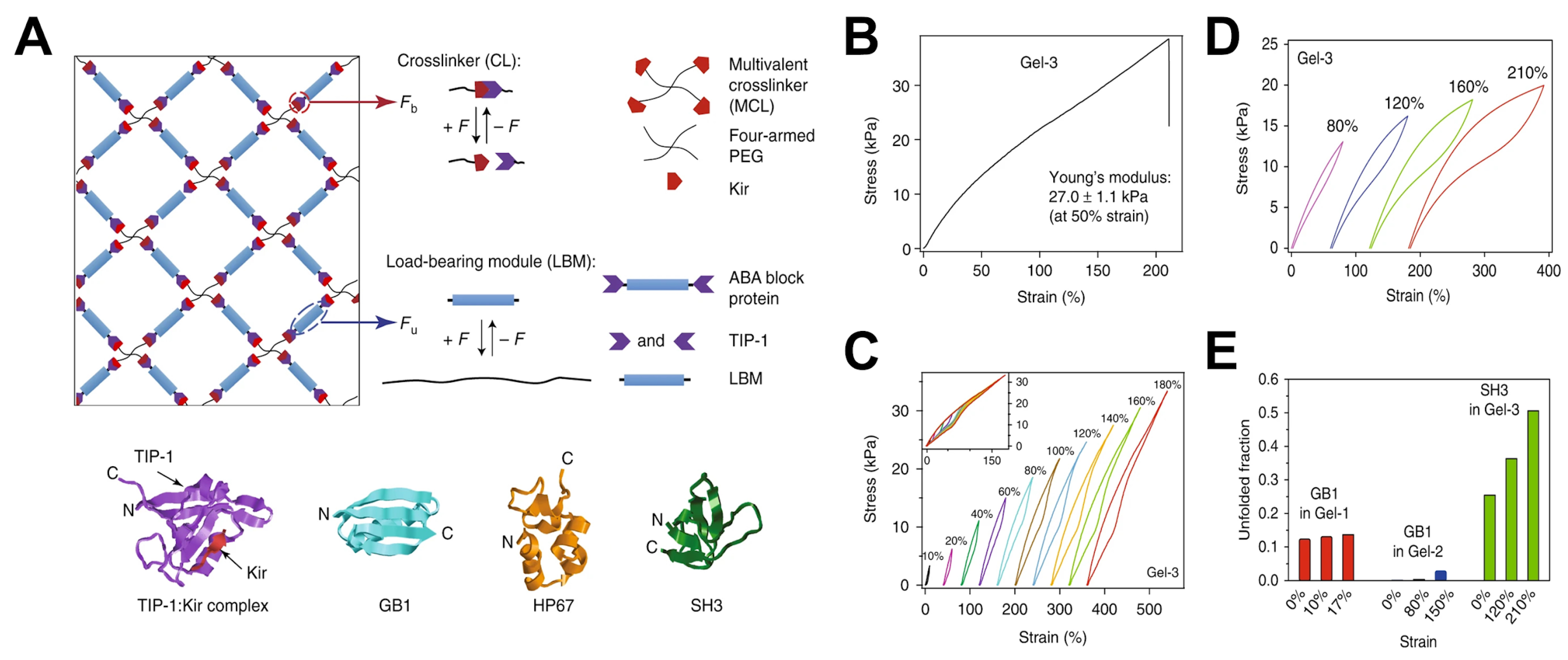
4.3.1. Biomimetic Design via Reversibly Unfolding Crosslinkers
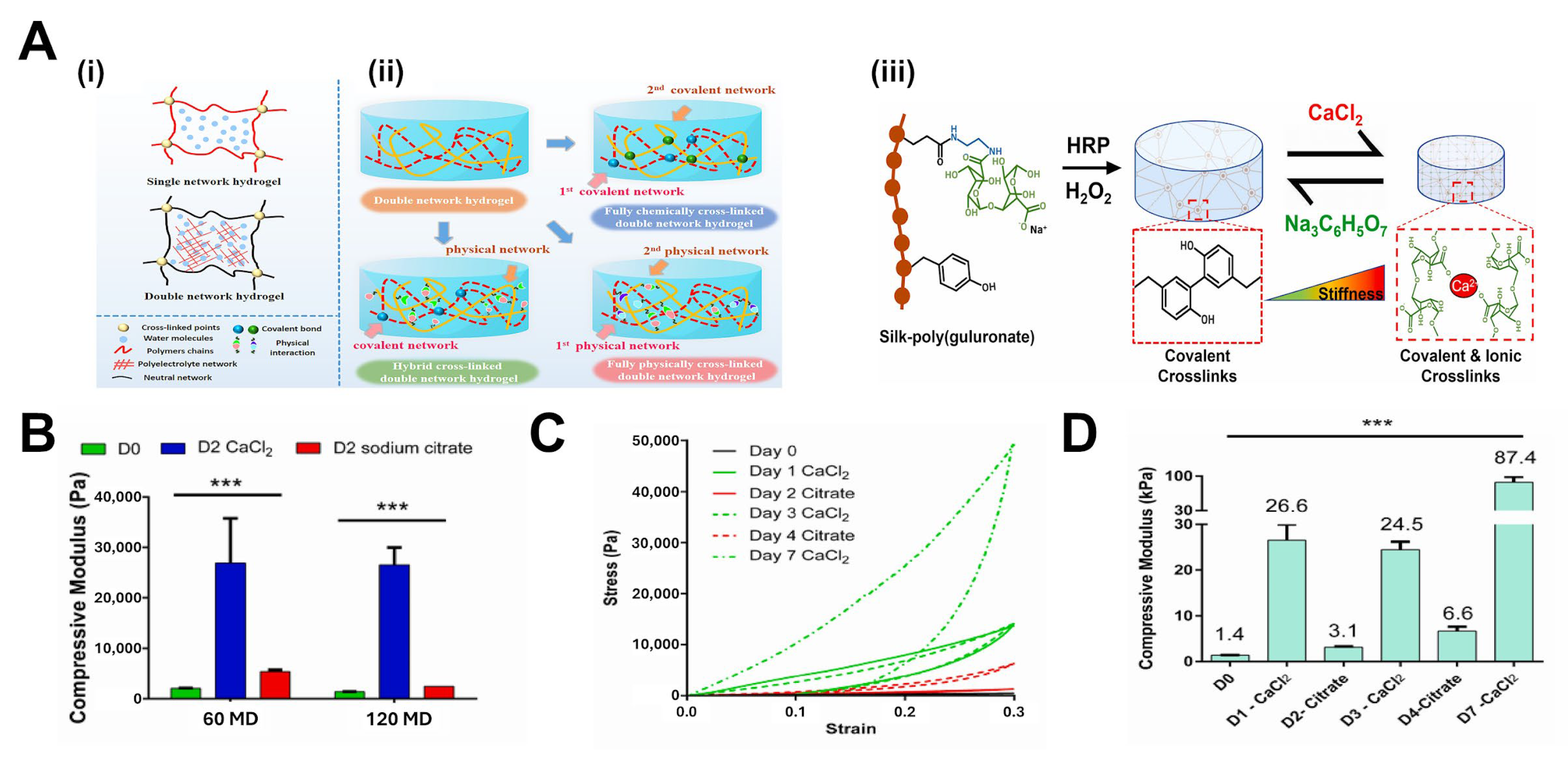
4.3.2. Double-Network Approaches with Complementary Crosslinker Architectures
4.3.3. Enhancing Mechanical Performance Through Hierarchical Crosslinking
5. Biological and Biomedical Applications
5.1. Cell-Material Interactions
5.2. Therapeutic Delivery Systems
5.3. Regenerative Medicine
6. Future Perspective and Challenges
6.1. Emerging Crosslinker Technologies
6.2. Challenges in Crosslinker Design
6.3. Future Research Directions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-APDEMS | 3-aminopropyl (diethoxy)methylsilane |
| AAc | Acrylic acid |
| ADIP | 2,2′-azobis-[2-(1-3-dimethyl-4,5-dihydro-1H-imidazol-3-ium-2-yl)] propane triflate |
| AG-ALD | Alginate-aldehyde |
| AG-HYD | Alginate-hydrazone |
| APTES | 3-aminopropyl)triethoxysilane |
| Arg-1 | Arginase-1 |
| BCN | Bicyclononyne |
| Ccl | Crosslinker concentration |
| CaCl2 | Calcium Chloride |
| CANs | Covalent adaptable networks |
| CB | Cucurbituril |
| CB[8] | Cucurbit[8]uril |
| CD-Ad | Cyclodextrin–adamantane |
| CD44 | Cluster of differentiation 44 |
| CuBGs | Copper-doped bioglass |
| DAT | N,N′-diallyltartramide |
| DMAA | N,N-dimethylacrylamide |
| DN | Double network |
| ECM | Extracellular matrix |
| EDTA | Ethylenediaminetetraacetic acid |
| EGDMA | Ethylene glycol dimethacrylate |
| ELP | Elastin-like protein |
| f | Average functionality of the crosslinkers |
| G | Bulk shear modulus |
| G-TA | Gelatin-tyramine |
| G0 | Initial shear modulus |
| GAG | Glycosaminoglycan |
| GB1 | Immunoglobulin-binding domain B1 of protein G |
| Ge | Entanglement modulus |
| GelMA | Gelatin-methacrylate |
| G′ | Storage modulus |
| G″ | Loss modulus |
| G″/G′ | Loss tangent |
| G∞ | Rubbery plateau modulus |
| H2O2 | Hydrogen peroxide |
| HA | Hyaluronic acid |
| HA-MA | Methacrylated hyaluronic acid |
| HA-SH | Thiolated hyaluronic acid |
| HA-SSPy | Pyridyl disulfide-functionalized hyaluronic acid |
| hMSCs | Human mesenchymal stem cells |
| HP67 | Folding protein 67 domain |
| HRP | Horseradish peroxidase |
| IDO | Indoleamine 2,3-dioxygenase |
| IL10 | Interleukin-10 |
| IPN | Interpenetrating network |
| k | Rate at which crosslinks can rearrange |
| keq | Equilibrium constant |
| koff | Kinetics of bond breakage |
| kon | Kinetics of bond re-formation |
| Kir | Inwardly rectifying potassium channel |
| LAP | Lithium acylphosphinate |
| LBMs | Load-bearing modules |
| LX | Linear crosslinkers |
| M2 | M2 macrophage phenotype |
| MA-GNPs | Methacrylated gelatin nanoparticles |
| MAPK | Mitogen-activated protein kinase |
| MB | N,N′-methylenebisacrylamide |
| MBAA | N,N′-methylenebis(acrylamide) |
| Mc | Molecular weight between crosslinkers |
| MMP | Matrix metalloproteinase |
| MPC | 2-methacryloyloxyethyl phosphorylcholine |
| MSCs | Mesenchymal stem cells |
| MSNs | Mesoporous silica nanoparticles |
| n | Scaling exponent |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NMR | Nuclear magnetic resonance |
| NPCs | Neural progenitor cells |
| Critical extent of reaction at the gel point | |
| PAMAM | Poly(amidoamine) |
| PAMPS | Poly(2-acrylamido-2-methylpropanesulfonic acid) |
| PEG | Polyethylene glycol |
| PEG-2ALD | Linear polyethylene glycol-dialdehyde |
| PEG-4ALD | 4-arm polyethylene glycol-aldehyde |
| PEG-VS | Polyethylene glycol-vinyl sulfone |
| PEG4AC | 4-arm polyethylene glycol |
| PEGDA | Polyethylene glycol diacrylate |
| PEGDMA | Poly(ethylene glycol) dimethacrylate |
| PETRA | Pentaerythritol tetra-acrylate |
| PGSE | Pulsed gradient spin–echo |
| pH | Potential of hydrogen |
| pHEMA | Poly(2-hydroxymethyl methacrylate) |
| pNIPAM | Poly(N-isopropylacrylamide-co-acrylic acid) |
| PVA | Poly(vinyl alcohol) |
| RGD | Arginine-Glycine-Aspartic acid |
| SCX | Side-chain crosslinkers |
| SD | Standard deviation |
| SDC | Star diblock copolypeptides |
| SF | Silk fibroin |
| SF-G | Silk-gelatin |
| SF-PG | Silk-poly(guluronate) |
| SF-TA | Silk fibroin-tyramine |
| SH3 | Src Homology 3 domain |
| SIF | Intestinal fluid |
| SilMA | Methacryloyl-modified silk fibroin |
| SPAAC | Strain-promoted azide-alkyne cycloaddition |
| SX | Star crosslinkers |
| t1/2 | Half-stress relaxation time |
| tt | Terminal relaxation time |
| TA | Tyramine |
| TIP | Tax-interacting protein |
| TIP-1 | Tax-interacting protein 1 |
| TNF-α | Tumor necrosis factor alpha |
| Tr | Relaxation time |
| UPy | Ureido-pyrimidinone |
| UPyEA | Ureido-pyrimidinone ethyl acrylate |
| UV | Ultraviolet |
| VICs | Valve interstitial cells |
| α-SMA | α-smooth muscle actin |
| γ | Strain |
| ξ | Mesh size |
| ξc | Correlation blob |
| ξel | Elastic blob |
| ξg | Geometric blob |
| σ(t) | Resulting stress |
| τ | Relaxation time |
| φ | Polymer volume fraction |
| ω | Frecuency |
References
- Ozcelik, B. 7—Degradable hydrogel systems for biomedical applications. In Biosynthetic Polymers for Medical Applications; Poole-Warren, L., Martens, P., Green, R., Eds.; Woodhead Publishing: Cambridge, UK, 2016; pp. 173–188. [Google Scholar]
- Dolmat, M.; Kozlovskaya, V.; Ankner, J.F.; Kharlampieva, E. Controlling Mechanical Properties of Poly(methacrylic acid) Multilayer Hydrogels via Hydrogel Internal Architecture. Macromolecules 2023, 56, 8054–8068. [Google Scholar] [CrossRef]
- Alavarse, A.C.; Frachini, E.C.G.; da Silva, R.L.C.G.; Lima, V.H.; Shavandi, A.; Petri, D.F.S. Crosslinkers for Polysaccharides and Proteins: Synthesis Conditions, Mechanisms, and Crosslinking Efficiency, a Review. Int. J. Biol. Macromol. 2022, 202, 558–596. [Google Scholar] [CrossRef] [PubMed]
- van Bemmelen, J.M. Das Hydrogel und das krystallinische Hydrat des Kupferoxyds. Z. Für. Anorg. Chem. 1894, 5, 466–483. [Google Scholar] [CrossRef]
- Taaca, K.L.M.; Prieto, E.I.; Vasquez, M.R. Current Trends in Biomedical Hydrogels: From Traditional Crosslinking to Plasma-Assisted Synthesis. Polymers 2022, 14, 2560. [Google Scholar] [CrossRef] [PubMed]
- Grindlay, J.H.; Clagett, O.T. A plastic sponge prosthesis for use after pneumonectomy; preliminary report of an experimental study. Proc. Staff. Meet. Mayo Clin. 1949, 24, 538. [Google Scholar] [PubMed]
- Wichterle, O.; LÍM, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Flory, P.J.; Rehner, J., Jr. Statistical Mechanics of Cross-Linked Polymer Networks II. Swelling. J. Chem. Phys. 1943, 11, 521–526. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers. I. Gelation1. J. Am. Chem. Soc. 1941, 63, 3083–3090. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched-Chain Polymers. J. Chem. Phys. 1943, 11, 45–55. [Google Scholar] [CrossRef]
- Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. [Google Scholar] [CrossRef]
- Lu, P.; Ruan, D.; Huang, M.; Tian, M.; Zhu, K.; Gan, Z.; Xiao, Z. Harnessing the potential of hydrogels for advanced therapeutic applications: Current achievements and future directions. Signal Transduct. Target. Ther. 2024, 9, 166. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Lou, J.; Stowers, R.; Nam, S.; Xia, Y.; Chaudhuri, O. Stress relaxing hyaluronic acid-collagen hydrogels promote cell spreading, fiber remodeling, and focal adhesion formation in 3D cell culture. Biomaterials 2018, 154, 213–222. [Google Scholar] [CrossRef]
- Lee, Y.; Song, W.J.; Sun, J.Y. Hydrogel soft robotics. Mater. Today Phys. 2020, 15, 100258. [Google Scholar] [CrossRef]
- Na, H.; Kang, Y.-W.; Park, C.S.; Jung, S.; Kim, H.-Y.; Sun, J.-Y. Hydrogel-based strong and fast actuators by electroosmotic turgor pressure. Science 2022, 376, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Agate, S.; Salem, K.S.; Lucia, L.; Pal, L. Hydrogel-Based Sensor Networks: Compositions, Properties, and Applications—A Review. ACS Appl. Bio Mater. 2021, 4, 140–162. [Google Scholar] [CrossRef] [PubMed]
- Hameed, H.; Faheem, S.; Paiva-Santos, A.C.; Sarwar, H.S.; Jamshaid, M. A Comprehensive Review of Hydrogel-Based Drug Delivery Systems: Classification, Properties, Recent Trends, and Applications. AAPS PharmSciTech 2024, 25, 64. [Google Scholar] [CrossRef]
- Luo, T.; Tan, B.; Zhu, L.; Wang, Y.; Liao, J. A Review on the Design of Hydrogels With Different Stiffness and Their Effects on Tissue Repair. Front. Bioeng. Biotechnol. 2022, 10, 817391. [Google Scholar] [CrossRef]
- Xue, B.; Tang, D.; Wu, X.; Xu, Z.; Gu, J.; Han, Y.; Zhu, Z.; Qin, M.; Zou, X.; Wang, W.; et al. Engineering hydrogels with homogeneous mechanical properties for controlling stem cell lineage specification. Proc. Natl. Acad. Sci. USA 2021, 118, e2110961118. [Google Scholar] [CrossRef]
- Kong, V.A.; Staunton, T.A.; Laaser, J.E. Effect of Cross-Link Homogeneity on the High-Strain Behavior of Elastic Polymer Networks. Macromolecules 2024, 57, 4670–4679. [Google Scholar] [CrossRef]
- Courbot, O.; Elosegui-Artola, A. The role of extracellular matrix viscoelasticity in development and disease. NPJ Biol. Phys. Mech. 2025, 2, 10. [Google Scholar] [CrossRef]
- Bril, M.; Fredrich, S.; Kurniawan, N.A. Stimuli-responsive materials: A smart way to study dynamic cell responses. Smart Mater. Med. 2022, 3, 257–273. [Google Scholar] [CrossRef]
- Sun, B. The Mechanics of Fibrillar Collagen Extracellular Matrix. Cell Rep. Phys. Sci. 2021, 2, 100515. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cosgrave, M.; Kaur, K.; Simpson, C.; Mielańczyk, Ł.; Murphy, C.; Murphy, R.D.; Heise, A. Tuning Star Polymer Architecture to Tailor Secondary Structures and Mechanical Properties of Diblock Polypeptide Hydrogels for Direct Ink Writing. Biomacromolecules 2025, 26, 670–678. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Lou, J.; Xia, Y.; Chaudhuri, O. Cross-Linker Architectures Impact Viscoelasticity in Dynamic Covalent Hydrogels. Adv. Healthc. Mater. 2024, 13, 2402059. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.T.; Hwang, Y.; Chen, A.C.; Varghese, S.; Sah, R.L. Cartilage-like mechanical properties of poly (ethylene glycol)-diacrylate hydrogels. Biomaterials 2012, 33, 6682–6690. [Google Scholar] [CrossRef] [PubMed]
- Bercea, M. Recent Advances in Poly(vinyl alcohol)-Based Hydrogels. Polymers 2024, 16, 2021. [Google Scholar] [CrossRef]
- Li, J.; Wang, K.; Wang, J.; Yuan, Y.; Wu, H. High-tough hydrogels formed via Schiff base reaction between PAMAM dendrimer and Tetra-PEG and their potential as dual-function delivery systems. Mater. Today Commun. 2022, 30, 103019. [Google Scholar] [CrossRef]
- Gould, S.T.; Darling, N.J.; Anseth, K.S. Small peptide functionalized thiol–ene hydrogels as culture substrates for understanding valvular interstitial cell activation and de novo tissue deposition. Acta Biomater. 2012, 8, 3201–3209. [Google Scholar] [CrossRef]
- Lutolf, M.P.; Lauer-Fields, J.L.; Schmoekel, H.G.; Metters, A.T.; Weber, F.E.; Fields, G.B.; Hubbell, J.A. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. USA 2003, 100, 5413–5418. [Google Scholar] [CrossRef]
- Alina, T.B.; Nash, V.A.; Spiller, K.L. Effects of Biotin-Avidin Interactions on Hydrogel Swelling. Front. Chem. 2020, 8, 593422. [Google Scholar] [CrossRef]
- Park, W.M. Coiled-Coils: The Molecular Zippers that Self-Assemble Protein Nanostructures. Int. J. Mol. Sci. 2020, 21, 3584. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Y.; Ferracci, G.; Zheng, J.; Cho, N.-J.; Lee, B.H. Gelatin methacryloyl and its hydrogels with an exceptional degree of controllability and batch-to-batch consistency. Sci. Rep. 2019, 9, 6863. [Google Scholar] [CrossRef]
- Austin, M.J.; Schunk, H.; Watkins, C.; Ling, N.; Chauvin, J.; Morton, L.; Rosales, A.M. Fluorescent Peptomer Substrates for Differential Degradation by Metalloproteases. Biomacromolecules 2022, 23, 4909–4923. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.D.; Hillsley, A.; Austin, M.J.; Rosales, A.M. Tuning hydrogel properties with sequence-defined, non-natural peptoid crosslinkers. J. Mater. Chem. B 2020, 8, 6925–6933. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.D.; Castilla-Casadiego, D.A.; Palmer, A.C.; Rosales, A.M. Crosslinker structure modulates bulk mechanical properties and dictates hMSC behavior on hyaluronic acid hydrogels. Acta Biomater. 2023, 155, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Castilla-Casadiego, D.A.; Morton, L.D.; Loh, D.H.; Pineda-Hernandez, A.; Chavda, A.P.; Garcia, F.; Rosales, A.M. Peptoid-Cross-Linked Hydrogel Stiffness Modulates Human Mesenchymal Stromal Cell Immunoregulatory Potential in the Presence of Interferon-Gamma. Macromol. Biosci. 2024, 24, 2400111. [Google Scholar] [CrossRef]
- Cooper, C.B.; Bao, Z. Using Periodic Dynamic Polymers to Form Supramolecular Nanostructures. Acc. Mater. Res. 2022, 3, 948–959. [Google Scholar] [CrossRef]
- Xu, X.; Jha, A.K.; Harrington, D.A.; Farach-Carson, M.C.; Jia, X. Hyaluronic acid-based hydrogels: From a natural polysaccharide to complex networks. Soft Matter 2012, 8, 3280–3294. [Google Scholar] [CrossRef]
- Tomić, S.L.; Babić Radić, M.M.; Vuković, J.S.; Filipović, V.V.; Nikodinovic-Runic, J.; Vukomanović, M. Alginate-Based Hydrogels and Scaffolds for Biomedical Applications. Mar. Drugs 2023, 21, 177. [Google Scholar] [CrossRef]
- Krishna, V.S.; Subashini, V.; Hariharan, A.; Chidambaram, D.; Raaju, A.; Gopichandran, N.; Nanthanalaxmi, M.P.; Lekhavadhani, S.; Shanmugavadivu, A.; Selvamurugan, N. Role of Crosslinkers in Advancing Chitosan-Based Biocomposite Scaffolds for Bone Tissue Engineering: A Comprehensive Review. Int. J. Biol. Macromol. 2024, 283, 137625. [Google Scholar] [CrossRef]
- Fan, B.; Torres García, D.; Salehi, M.; Webber, M.J.; van Kasteren, S.I.; Eelkema, R. Dynamic Covalent Dextran Hydrogels as Injectable, Self-Adjuvating Peptide Vaccine Depots. ACS Chem. Biol. 2023, 18, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Charbonier, F.; Indana, D.; Chaudhuri, O. Tuning Viscoelasticity in Alginate Hydrogels for 3D Cell Culture Studies. Curr. Protoc. 2021, 1, e124. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, S.; Ooi, H.W.; Morgan, F.L.C.; Mota, C.; Dettin, M.; Van Blitterswijk, C.; Moroni, L.; Baker, M.B. Viscoelastic Oxidized Alginates with Reversible Imine Type Crosslinks: Self-Healing, Injectable, and Bioprintable Hydrogels. Gels 2018, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Debnath, A.; Li, S.; Mondal, A.K. Polysaccharide-Based Beads and Water Purification: A Review. Int. J. Biol. Macromol. 2025, 319, 145382. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Z.; Chen, D.; Li, Y.; Zhang, Q.; Su, J. Bone Regeneration Using MMP-Cleavable Peptides-Based Hydrogels. Gels 2021, 7, 199. [Google Scholar] [CrossRef]
- Zengin, A.; Hafeez, S.; Habibovic, P.; Baker, M.; van Rijt, S. Extracellular matrix mimetic supramolecular hydrogels reinforced with covalent crosslinked mesoporous silica nanoparticles. J. Mater. Chem. B 2024, 12, 12577–12588. [Google Scholar] [CrossRef]
- Lysenkov, E.; Klepko, V.; Bulavin, L.; Lebovka, N. Physico-Chemical Properties of Laponite®/Polyethylene-oxide Based Composites. Chem. Rec. 2024, 24, e202300166. [Google Scholar] [CrossRef]
- Song, H.; Fang, Z.; Jin, B.; Pan, P.; Zhao, Q.; Xie, T. Synergetic Chemical and Physical Programming for Reversible Shape Memory Effect in a Dynamic Covalent Network with Two Crystalline Phases. ACS Macro Lett. 2019, 8, 682–686. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, S.; Jiang, W.; Zhang, Q.; Liu, N.; Wang, Z.; Li, Z.; Zhang, D. Double-Network Hydrogels for Biomaterials: Structure-Property Relationships and Drug Delivery. Eur. Polym. J. 2023, 185, 111807. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting Integrin Pathways: Mechanisms and Advances in Therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Steudter, T.; Lam, T.; Pirmahboub, H.; Stoppel, C.; Kloke, L.; Pearson, S.; del Campo, A. A Comparative Study between Thiol-Ene and Acrylate Photocrosslinkable Hyaluronic Acid Hydrogel Inks for Digital Light Processing. Macromol. Biosci. 2025, 25, 2400535. [Google Scholar] [CrossRef]
- Ahmadi, M.; Ehrmann, K.; Koch, T.; Liska, R.; Stampfl, J. From Unregulated Networks to Designed Microstructures: Introducing Heterogeneity at Different Length Scales in Photopolymers for Additive Manufacturing. Chem. Rev. 2024, 124, 3978–4020. [Google Scholar] [CrossRef] [PubMed]
- Maafa, I.M. Inhibition of Free Radical Polymerization: A Review. Polymers 2023, 15, 488. [Google Scholar] [CrossRef]
- Jiang, H.; Qin, S.; Dong, H.; Lei, Q.; Su, X.; Zhuo, R.; Zhong, Z. An injectable and fast-degradable poly(ethylene glycol) hydrogel fabricated via bioorthogonal strain-promoted azide–alkyne cycloaddition click chemistry. Soft Matter 2015, 11, 6029–6036. [Google Scholar] [CrossRef]
- Lv, L.; Cheng, W.; Wang, S.; Lin, S.; Dang, J.; Ran, Z.; Zhu, H.; Xu, W.; Huang, Z.; Xu, P.; et al. Poly(β-amino ester) Dual-Drug-Loaded Hydrogels with Antibacterial and Osteogenic Properties for Bone Repair. ACS Biomater. Sci. Eng. 2023, 9, 1976–1990. [Google Scholar] [CrossRef]
- Savoca, M.P.; Tonoli, E.; Atobatele, A.G.; Verderio, E.A.M. Biocatalysis by Transglutaminases: A Review of Biotechnological Applications. Micromachines 2018, 9, 562. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Zhao, X.-H. Structure and property changes of whey protein isolate in response to the chemical modification mediated by horseradish peroxidase, glucose oxidase and d-glucose. Food Chem. 2022, 373, 131328. [Google Scholar] [CrossRef]
- Øvrebø, Ø.; Perale, G.; Wojciechowski, J.P.; Echalier, C.; Jeffers, J.R.T.; Stevens, M.M.; Haugen, H.J.; Rossi, F. Design and clinical application of injectable hydrogels for musculoskeletal therapy. Bioeng. Transl. Med. 2022, 7, e10295. [Google Scholar] [CrossRef]
- Lou, J.; Friedowitz, S.; Will, K.; Qin, J.; Xia, Y. Predictably Engineering the Viscoelastic Behavior of Dynamic Hydrogels via Correlation with Molecular Parameters. Adv. Mater. 2021, 33, 2104460. [Google Scholar] [CrossRef] [PubMed]
- Sajjadi, M.; Jamali, R.; Kiyani, T.; Mohamadnia, Z.; Moradi, A.-R. Characterization of Schiff base self-healing hydrogels by dynamic speckle pattern analysis. Sci. Rep. 2024, 14, 27950. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovic, M.; Rikkers, M.; Mihajlovic, M.; Viola, M.; Schuiringa, G.; Ilochonwu, B.C.; Masereeuw, R.; Vonk, L.; Malda, J.; Ito, K.; et al. Viscoelastic Chondroitin Sulfate and Hyaluronic Acid Double-Network Hydrogels with Reversible Cross-Links. Biomacromolecules 2022, 23, 1350–1365. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhang, X.; Wang, Q.; Yong, Q.; Xu, W.; Xu, M.; Xu, C.; Wang, X. Dynamic reversible disulfide bonds hydrogel of thiolated galactoglucomannan/cellulose nanofibril with self-healing property for protein release. Ind. Crops Prod. 2023, 206, 117615. [Google Scholar] [CrossRef]
- Marozas, I.A.; Anseth, K.S.; Cooper-White, J.J. Adaptable boronate ester hydrogels with tunable viscoelastic spectra to probe timescale dependent mechanotransduction. Biomaterials 2019, 223, 119430. [Google Scholar] [CrossRef]
- Kramer, R.K.; Belgacem, M.N.; Carvalho, A.J.F.; Gandini, A. Thermally reversible nanocellulose hydrogels synthesized via the furan/maleimide Diels-Alder click reaction in water. Int. J. Biol. Macromol. 2019, 141, 493–498. [Google Scholar] [CrossRef]
- Lozinsky, V.I. Cryotropic gelation of poly(vinyl alcohol) solutions. Russ. Chem. Rev. 1998, 67, 573–586. [Google Scholar] [CrossRef]
- Schnell, I.; Langer, B.; Söntjens, S.H.M.; Sijbesma, R.P.; van Genderen, M.H.P.; Wolfgang Spiess, H. Quadruple hydrogen bonds of ureido-pyrimidinone moieties investigated in the solid state by 1H double-quantum MAS NMR spectroscopy. Phys. Chem. Chem. Phys. 2002, 4, 3750–3758. [Google Scholar] [CrossRef]
- Tu, C.-W.; Tsai, F.-C.; Chen, J.-K.; Wang, H.-P.; Lee, R.-H.; Zhang, J.; Chen, T.; Wang, C.-C.; Huang, C.-F. Preparations of Tough and Conductive PAMPS/PAA Double Network Hydrogels Containing Cellulose Nanofibers and Polypyrroles. Polymers 2020, 12, 2835. [Google Scholar] [CrossRef]
- Hou, N.; Wang, R.; Geng, R.; Wang, F.; Jiao, T.; Zhang, L.; Zhou, J.; Bai, Z.; Peng, Q. Facile preparation of self-assembled hydrogels constructed from poly-cyclodextrin and poly-adamantane as highly selective adsorbents for wastewater treatment. Soft Matter 2019, 15, 6097–6106. [Google Scholar] [CrossRef]
- Madl, A.C.; Madl, C.M.; Myung, D. Injectable Cucurbit[8]uril-Based Supramolecular Gelatin Hydrogels for Cell Encapsulation. ACS Macro Lett. 2020, 9, 619–626. [Google Scholar] [CrossRef]
- Zeng, Y.-B.; Zhang, D.-M.; Li, H.; Sun, H. Binding of Ni2+ to a histidine- and glutamine-rich protein, Hpn-like. JBIC J. Biol. Inorg. Chem. 2008, 13, 1121–1131. [Google Scholar] [CrossRef]
- Fullenkamp, D.E.; Barrett, D.G.; Miller, D.R.; Kurutz, J.W.; Messersmith, P.B. pH-dependent cross-linking of catechols through oxidation via Fe3+ and potential implications for mussel adhesion. RSC Adv. 2014, 4, 25127–25134. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Narkar, A.; Wilharm, R. Effect of metal ion type on the movement of hydrogel actuator based on catechol-metal ion coordination chemistry. Sens. Actuators B Chem. 2016, 227, 248–254. [Google Scholar] [CrossRef]
- Lee, B.P.; Lin, M.-H.; Narkar, A.; Konst, S.; Wilharm, R. Modulating the movement of hydrogel actuator based on catechol–iron ion coordination chemistry. Sens. Actuators B Chem. 2015, 206, 456–462. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, H.; Zhu, L.; Zheng, J. Fundamentals of double network hydrogels. J. Mater. Chem. B 2015, 3, 3654–3676. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Li, X.; Xiong, Y. Application of “Click” Chemistry in Biomedical Hydrogels. ACS Omega 2022, 7, 36918–36928. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. In situ forming poly(ethylene glycol)-based hydrogels via thiol-maleimide Michael-type addition. J. Biomed. Mater. Res. Part A 2011, 98A, 201–211. [Google Scholar] [CrossRef]
- Song, W.; Ko, J.; Choi, Y.H.; Hwang, N.S. Recent advancements in enzyme-mediated crosslinkable hydrogels: In vivo-mimicking strategies. APL Bioeng. 2021, 5, 021502. [Google Scholar] [CrossRef]
- Wang, Z.J.; Zhu, C.N.; Hong, W.; Wu, Z.L.; Zheng, Q. Cooperative deformations of periodically patterned hydrogels. Sci. Adv. 2017, 3, e1700348. [Google Scholar] [CrossRef]
- Ooi, H.W.; Mota, C.; ten Cate, A.T.; Calore, A.; Moroni, L.; Baker, M.B. Thiol–Ene Alginate Hydrogels as Versatile Bioinks for Bioprinting. Biomacromolecules 2018, 19, 3390–3400. [Google Scholar] [CrossRef]
- Ortiz-Cárdenas, J.E.; Zatorski, J.M.; Arneja, A.; Montalbine, A.N.; Munson, J.M.; Luckey, C.J.; Pompano, R.R. Towards spatially-organized organs-on-chip: Photopatterning cell-laden thiol-ene and methacryloyl hydrogels in a microfluidic device. Organs-on-a-Chip 2022, 4, 100018. [Google Scholar] [CrossRef]
- Liu, H.; Sui, X.; Xu, H.; Zhang, L.; Zhong, Y.; Mao, Z. Self-Healing Polysaccharide Hydrogel Based on Dynamic Covalent Enamine Bonds. Macromol. Mater. Eng. 2016, 301, 725–732. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.-p.; Lippens, E.; Duda, G.N.; et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 2016, 15, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Drozdov, A.D.; deClaville Christiansen, J. Tuning the viscoelastic response of hydrogel scaffolds with covalent and dynamic bonds. J. Mech. Behav. Biomed. Mater. 2022, 130, 105179. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Richardson, B.M.; Anseth, K.S. Dynamic covalent hydrogels as biomaterials to mimic the viscoelasticity of soft tissues. Prog. Mater. Sci. 2021, 120, 100738. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Dynamic Diselenide Bonds: Exchange Reaction Induced by Visible Light without Catalysis. Angew. Chem. Int. Ed. 2014, 53, 6781–6785. [Google Scholar] [CrossRef]
- Zheng, J.; Png, Z.M.; Ng, S.H.; Tham, G.X.; Ye, E.; Goh, S.S.; Loh, X.J.; Li, Z. Vitrimers: Current research trends and their emerging applications. Mater. Today 2021, 51, 586–625. [Google Scholar] [CrossRef]
- Hoti, G.; Caldera, F.; Cecone, C.; Rubin Pedrazzo, A.; Anceschi, A.; Appleton, S.L.; Khazaei Monfared, Y.; Trotta, F. Effect of the Cross-Linking Density on the Swelling and Rheological Behavior of Ester-Bridged β-Cyclodextrin Nanosponges. Materials 2021, 14, 478. [Google Scholar] [CrossRef]
- Li, L.; Eyckmans, J.; Chen, C.S. Designer biomaterials for mechanobiology. Nat. Mater. 2017, 16, 1164–1168. [Google Scholar] [CrossRef]
- Wong, R.S.; Ashton, M.; Dodou, K. Effect of Crosslinking Agent Concentration on the Properties of Unmedicated Hydrogels. Pharmaceutics 2015, 7, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Koetting, M.C.; Guido, J.F.; Gupta, M.; Zhang, A.; Peppas, N.A. pH-responsive and enzymatically-responsive hydrogel microparticles for the oral delivery of therapeutic proteins: Effects of protein size, crosslinking density, and hydrogel degradation on protein delivery. J. Control. Release 2016, 221, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, A.; Atala, A. Tissue Engineering: Toward a New Era of Medicine. Annu. Rev. Med. 2017, 68, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Madl, C.M.; Katz, L.M.; Heilshorn, S.C. Bio-Orthogonally Crosslinked, Engineered Protein Hydrogels with Tunable Mechanics and Biochemistry for Cell Encapsulation. Adv. Funct. Mater. 2016, 26, 3612–3620. [Google Scholar] [CrossRef]
- Hasturk, O.; Jordan, K.E.; Choi, J.; Kaplan, D.L. Enzymatically crosslinked silk and silk-gelatin hydrogels with tunable gelation kinetics, mechanical properties and bioactivity for cell culture and encapsulation. Biomaterials 2020, 232, 119720. [Google Scholar] [CrossRef]
- Richbourg, N.R.; Peppas, N.A. The swollen polymer network hypothesis: Quantitative models of hydrogel swelling, stiffness, and solute transport. Prog. Polym. Sci. 2020, 105, 101243. [Google Scholar] [CrossRef]
- Peppas, N.A.; Merrill, E.W. Poly(vinyl alcohol) hydrogels: Reinforcement of radiation-crosslinked networks by crystallization. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 441–457. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Wisniewska, M.A.; Seland, J.G.; Wang, W. Determining the scaling of gel mesh size with changing crosslinker concentration using dynamic swelling, rheometry, and PGSE NMR spectroscopy. J. Appl. Polym. Sci. 2018, 135, 46695. [Google Scholar] [CrossRef]
- Sabater i Serra, R.; León-Boigues, L.; Sánchez-Laosa, A.; Gómez-Estrada, L.; Gómez Ribelles, J.L.; Salmeron-Sanchez, M.; Gallego Ferrer, G. Role of chemical crosslinking in material-driven assembly of fibronectin (nano)networks: 2D surfaces and 3D scaffolds. Colloids Surf. B: Biointerfaces 2016, 148, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B.G. Hydrogel Mesh Size and Its Impact on Predictions of Mathematical Models of the Solute Diffusion Coefficient. Macromolecules 2022, 55, 8399–8408. [Google Scholar] [CrossRef]
- Rehmann, M.S.; Skeens, K.M.; Kharkar, P.M.; Ford, E.M.; Maverakis, E.; Lee, K.H.; Kloxin, A.M. Tuning and Predicting Mesh Size and Protein Release from Step Growth Hydrogels. Biomacromolecules 2017, 18, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.T.; Wysoczynski, K.; Hadley, D.J.; Silva, E.A. Computational-Based Design of Hydrogels with Predictable Mesh Properties. ACS Biomater. Sci. Eng. 2020, 6, 308–319. [Google Scholar] [CrossRef]
- Tsuji, Y.; Li, X.; Shibayama, M. Evaluation of Mesh Size in Model Polymer Networks Consisting of Tetra-Arm and Linear Poly(ethylene glycol)s. Gels 2018, 4, 50. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, Z.; Liu, Z.; Sharma, P. Concurrent Stiffening and Softening in Hydrogels under Dehydration. Sci. Adv. 2023, 9, eade3240. [Google Scholar] [CrossRef]
- Grad, E.M.; Tunn, I.; Voerman, D.; de Léon, A.S.; Hammink, R.; Blank, K.G. Influence of Network Topology on the Viscoelastic Properties of Dynamically Crosslinked Hydrogels. Front. Chem. 2020, 8, 536. [Google Scholar] [CrossRef]
- Kadam, V.; Nicolai, T.; Nicol, E.; Benyahia, L. Structure and Rheology of Self-Assembled Telechelic Associative Polymers in Aqueous Solution before and after Photo-Cross-Linking. Macromolecules 2011, 44, 8225–8232. [Google Scholar] [CrossRef]
- Song, X.; Chi, H.; Li, Z.; Li, T.; Wang, F. Star-Shaped Crosslinker for Multifunctional Shape Memory Polyurethane. Polymers 2020, 12, 740. [Google Scholar] [CrossRef]
- Ahmed, J. Rheology and Rheological Measurements. In Kirk-Othmer Encyclopedia of Chemical Technology; Howe-Grant, M., Ed.; Wiley: New York, NY, USA, 2021; pp. 1–70. [Google Scholar] [CrossRef]
- Tang, S.; Olsen, B.D. Controlling topological entanglement in engineered protein hydrogels with a variety of thiol coupling chemistries. Front. Chem. 2014, 2, 23. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, C.; Li, H.; Zhao, P.; Zhao, Y.; Li, B.; Zhou, W.; Fan, G.; Guan, D.; Zheng, Y. Visible light-responsive hydrogels for cellular dynamics and spatiotemporal viscoelastic regulation. Nat. Commun. 2025, 16, 1365. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; You, M.; Xu, J.; Zhou, J.; Jin, Y.; Li, D.; Xu, Z.; Li, J.; Chen, C. Mechanically robust, flexible, conductive, and anti-freezing hydrogels reinforced by cellulose of wood skeleton. Int. J. Biol. Macromol. 2025, 307, 142049. [Google Scholar] [CrossRef] [PubMed]
- Huerta-López, C.; Alegre-Cebollada, J. Protein Hydrogels: The Swiss Army Knife for Enhanced Mechanical and Bioactive Properties of Biomaterials. Nanomaterials 2021, 11, 1656. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, P.; Dong, C.; Jiang, H.; Bin, X.; Gao, X.; Qin, M.; Wang, W.; Bin, C.; Cao, Y. Rationally designed synthetic protein hydrogels with predictable mechanical properties. Nat. Commun. 2018, 9, 620. [Google Scholar] [CrossRef]
- Biswas, S.; Yashin, V.V.; Balazs, A.C. Harnessing biomimetic cryptic bonds to form self-reinforcing gels. Soft Matter 2020, 16, 5120–5131. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, S.-H.; Chuang, W.-T.; Dai, N.-T.; Hsu, S.-h. Biomimetic Strain-Stiffening in Chitosan Self-Healing Hydrogels. ACS Appl. Mater. Interfaces 2022, 14, 16032–16046. [Google Scholar] [CrossRef]
- Xu, X.; Jerca, V.V.; Hoogenboom, R. Bioinspired double network hydrogels: From covalent double network hydrogels via hybrid double network hydrogels to physical double network hydrogels. Mater. Horiz. 2021, 8, 1173–1188. [Google Scholar] [CrossRef]
- Xin, H. Double-Network Tough Hydrogels: A Brief Review on Achievements and Challenges. Gels 2022, 8, 247. [Google Scholar] [CrossRef]
- Yin, Y.; Gu, Q.; Liu, X.; Liu, F.; McClements, D.J. Double network hydrogels: Design, fabrication, and application in biomedicines and foods. Adv. Colloid. Interface Sci. 2023, 320, 102999. [Google Scholar] [CrossRef]
- Hasturk, O.; Sahoo, J.K.; Kaplan, D.L. Synthesis and characterization of silk-poly(guluronate) hybrid polymers for the fabrication of dual crosslinked, mechanically dynamic hydrogels. Polymer 2023, 281, 126129. [Google Scholar] [CrossRef]
- Huang, Y.; Jayathilaka, P.B.; Islam, M.S.; Tanaka, C.B.; Silberstein, M.N.; Kilian, K.A.; Kruzic, J.J. Structural aspects controlling the mechanical and biological properties of tough, double network hydrogels. Acta Biomater. 2022, 138, 301–312. [Google Scholar] [CrossRef]
- Zhang, Y.; Heher, P.; Hilborn, J.; Redl, H.; Ossipov, D.A. Hyaluronic acid-fibrin interpenetrating double network hydrogel prepared in situ by orthogonal disulfide cross-linking reaction for biomedical applications. Acta Biomater. 2016, 38, 23–32. [Google Scholar] [CrossRef]
- Xue, S.; Wu, Y.; Liu, G.; Guo, M.; Liu, Y.; Zhang, T.; Wang, Z. Hierarchically reversible crosslinking polymeric hydrogels with highly efficient self-healing, robust mechanical properties, and double-driven shape memory behavior. J. Mater. Chem. A 2021, 9, 5730–5739. [Google Scholar] [CrossRef]
- Pardo, A.; Gomez-Florit, M.; Davidson, M.D.; Öztürk-Öncel, M.Ö.; Domingues, R.M.A.; Burdick, J.A.; Gomes, M.E. Hierarchical Design of Tissue-Mimetic Fibrillar Hydrogel Scaffolds. Adv. Healthc. Mater. 2024, 13, 2303167. [Google Scholar] [CrossRef] [PubMed]
- Yesilyurt, V.; Ayoob, A.M.; Appel, E.A.; Borenstein, J.T.; Langer, R.; Anderson, D.G. Mixed Reversible Covalent Crosslink Kinetics Enable Precise, Hierarchical Mechanical Tuning of Hydrogel Networks. Adv. Mater. 2017, 29, 1605947. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Yang, J.; Zheng, Q.; Wu, N.; Lv, Z.; Jiang, Z. Ultra-stretchable hydrogels with hierarchical hydrogen bonds. Sci. Rep. 2020, 10, 11727. [Google Scholar] [CrossRef]
- Hafeez, S.; Islam, A.; Durrani, A.K.; Butt, M.T.Z.; Rehmat, S.; Khurshid, A.; Khan, S.M. Fabrication of pectin-based stimuli responsive hydrogel for the controlled release of ceftriaxone. Chem. Pap. 2023, 77, 1809–1819. [Google Scholar] [CrossRef]
- Rasool, A.; Ata, S.; Islam, A.; Khan, R.U. Fabrication of novel carrageenan based stimuli responsive injectable hydrogels for controlled release of cephradine. RSC Adv. 2019, 9, 12282–12290. [Google Scholar] [CrossRef]
- Qiu, T.; Hanna, E.; Dabbous, M.; Borislav, B.; Toumi, M. Regenerative medicine regulatory policies: A systematic review and international comparison. Health Policy 2020, 124, 701–713. [Google Scholar] [CrossRef]
- Duan, B.; Hockaday, L.A.; Kapetanovic, E.; Kang, K.H.; Butcher, J.T. Stiffness and adhesivity control aortic valve interstitial cell behavior within hyaluronic acid based hydrogels. Acta Biomater. 2013, 9, 7640–7650. [Google Scholar] [CrossRef]
- Jiang, L.-B.; Ding, S.-L.; Ding, W.; Su, D.-H.; Zhang, F.-X.; Zhang, T.-W.; Yin, X.-F.; Xiao, L.; Li, Y.-L.; Yuan, F.-L.; et al. Injectable sericin based nanocomposite hydrogel for multi-modal imaging-guided immunomodulatory bone regeneration. Chem. Eng. J. 2021, 418, 129323. [Google Scholar] [CrossRef]
- Tunn, I.; Harrington, M.J.; Blank, K.G. Bioinspired Histidine–Zn2+ Coordination for Tuning the Mechanical Properties of Self-Healing Coiled Coil Cross-Linked Hydrogels. Biomimetics 2019, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Jian, G.; Li, D.; Ying, Q.; Chen, X.; Zhai, Q.; Wang, S.; Mei, L.; Cannon, R.D.; Ji, P.; Liu, W.; et al. Dual Photo-Enhanced Interpenetrating Network Hydrogel with Biophysical and Biochemical Signals for Infected Bone Defect Healing. Adv. Healthc. Mater. 2023, 12, 2300469. [Google Scholar] [CrossRef] [PubMed]
- Neffe, A.T. Cell–Material Interactions 2022. Int. J. Mol. Sci. 2023, 24, 6057. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef]
- Sanz-Herrera, J.A.; Reina-Romo, E. Cell-Biomaterial Mechanical Interaction in the Framework of Tissue Engineering: Insights, Computational Modeling and Perspectives. Int. J. Mol. Sci. 2011, 12, 8217–8244. [Google Scholar] [CrossRef]
- Pineda-Hernandez, A.; Castilla-Casadiego, D.A.; Morton, L.D.; Giordano-Nguyen, S.A.; Halwachs, K.N.; Rosales, A.M. Tunable hydrogel networks by varying secondary structures of hydrophilic peptoids provide viable 3D cell culture platforms for hMSCs. Biomater. Sci. 2025, 13, 3380–3394. [Google Scholar] [CrossRef]
- Buwalda, S.J.; Vermonden, T.; Hennink, W.E. Hydrogels for Therapeutic Delivery: Current Developments and Future Directions. Biomacromolecules 2017, 18, 316–330. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Yu, H.; Gan, Y.; Wang, Y.; Mei, L.; et al. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef]
- Mashabela, L.T.; Maboa, M.M.; Miya, N.F.; Ajayi, T.O.; Chasara, R.S.; Milne, M.; Mokhele, S.; Demana, P.H.; Witika, B.A.; Siwe-Noundou, X.; et al. A Comprehensive Review of Cross-Linked Gels as Vehicles for Drug Delivery to Treat Central Nervous System Disorders. Gels 2022, 8, 563. [Google Scholar] [CrossRef]
- Hasturk, O.; Smiley, J.A.; Arnett, M.; Sahoo, J.K.; Staii, C.; Kaplan, D.L. Cytoprotection of Human Progenitor and Stem Cells through Encapsulation in Alginate Templated, Dual Crosslinked Silk and Silk–Gelatin Composite Hydrogel Microbeads. Adv. Healthc. Mater. 2022, 11, 2200293. [Google Scholar] [CrossRef]
- Shahin-Shamsabadi, A.; Cappuccitti, J. Muscle-specific acellular ECM fibers made with anchored cell sheet engineering support regeneration in rat models of volumetric muscle loss. Acta Biomater. 2025, 200, 416–431. [Google Scholar] [CrossRef]
- Osman, M.; Sharmin, Z.; Suchy, S.; Gao, F.; Kaminski, A.; Mitchell, J.C.; Sigar, I.M.; Carrilho, M.R. Bioinspired smart dentin ECM-chitosan hydrogels for dentin-pulp complex regeneration. J. Dent. 2025, 159, 105811. [Google Scholar] [CrossRef] [PubMed]
- Chocarro-Wrona, C.; Pleguezuelos-Beltrán, P.; López de Andrés, J.; Antich, C.; de Vicente, J.; Jiménez, G.; Arias-Santiago, S.; Gálvez-Martín, P.; López-Ruiz, E.; Marchal, J.A. A bioactive three-layered skin substitute based on ECM components effectively promotes skin wound healing and regeneration. Mater. Today Bio 2025, 31, 101592. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, S.; Chen, Y.; Dong, S.; Zhang, C.; Liao, L.; Zhang, W. Hydrogels mimicking the viscoelasticity of extracellular matrix for regenerative medicine: Design, application, and molecular mechanism. Chem. Eng. J. 2024, 498, 155206. [Google Scholar] [CrossRef]
- Si, H.; Xing, T.; Ding, Y.; Zhang, H.; Yin, R.; Zhang, W. 3D Bioprinting of the Sustained Drug Release Wound Dressing with Double-Crosslinked Hyaluronic-Acid-Based Hydrogels. Polymers 2019, 11, 1584. [Google Scholar] [CrossRef]
- Krakowski, P.; Rejniak, A.; Sobczyk, J.; Karpiński, R. Cartilage Integrity: A Review of Mechanical and Frictional Properties and Repair Approaches in Osteoarthritis. Healthcare 2024, 12, 1648. [Google Scholar] [CrossRef]
- Castilla-Casadiego, D.A.; Loh, D.H.; Pineda-Hernandez, A.; Rosales, A.M. Stimuli-Responsive Substrates to Control the Immunomodulatory Potential of Stromal Cells. Biomacromolecules 2024, 25, 6319–6337. [Google Scholar] [CrossRef]
- El-Husseiny, H.M.; Mady, E.A.; Hamabe, L.; Abugomaa, A.; Shimada, K.; Yoshida, T.; Tanaka, T.; Yokoi, A.; Elbadawy, M.; Tanaka, R. Smart/stimuli-responsive hydrogels: Cutting-edge platforms for tissue engineering and other biomedical applications. Mater. Today Bio 2022, 13, 100186. [Google Scholar] [CrossRef]
- Bordbar-Khiabani, A.; Gasik, M. Smart Hydrogels for Advanced Drug Delivery Systems. Int. J. Mol. Sci. 2022, 23, 3665. [Google Scholar] [CrossRef]
- Acharya, R.; Dutta, S.D.; Mallik, H.; Patil, T.V.; Ganguly, K.; Randhawa, A.; Kim, H.; Lee, J.; Park, H.; Mo, C.; et al. Physical stimuli-responsive DNA hydrogels: Design, fabrication strategies, and biomedical applications. J. Nanobiotechnol. 2025, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xiao, X.; Zhang, Y.; Liao, L. High strength dual-crosslinked hydrogels with photo-switchable color changing behavior. Eur. Polym. J. 2019, 116, 545–553. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, L.; Wang, M.; Zang, D.; Long, H.; Weng, L.; Guo, N.; Gao, J.; Liu, Y.; Xu, B.B. A wearable ionic hydrogel strain sensor with double cross-linked network for human–machine interface. Adv. Compos. Hybrid. Mater. 2024, 8, 17. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Liu, J.; Zhang, H.; Fu, D. Advances in the application of natural/synthetic hybrid hydrogels in tissue engineering and delivery systems: A comprehensive review. Int. J. Pharm. 2025, 672, 125323. [Google Scholar] [CrossRef]
- Masuda, T.; Saegusa, Y.; Tsuji, T.; Takai, M. Development of deformable and adhesive biocompatible polymer hydrogels by a simple one-pot method using ADIP as a cationic radical initiator. Polym. J. 2025, 57, 819. [Google Scholar] [CrossRef]
- Alonso, J.M.; Andrade del Olmo, J.; Perez Gonzalez, R.; Saez-Martinez, V. Injectable Hydrogels: From Laboratory to Industrialization. Polymers 2021, 13, 650. [Google Scholar] [CrossRef]
- Choi, S.M.; Shin, E.J.; Zo, S.M.; Kummara, M.R.; Kim, C.M.; Kumar, A.; Bae, H.J.; Sood, A.; Han, S.S. Development of Scalable Elastic Gelatin Hydrogel Films Crosslinked with Waterborne Polyurethane for Enhanced Mechanical Properties and Strain Recovery. Gels 2025, 11, 49. [Google Scholar] [CrossRef]
- Xu, F.; Dawson, C.; Lamb, M.; Mueller, E.; Stefanek, E.; Akbari, M.; Hoare, T. Hydrogels for Tissue Engineering: Addressing Key Design Needs Toward Clinical Translation. Front. Bioeng. Biotechnol. 2022, 10, 849831. [Google Scholar] [CrossRef]
- Xue, B.; Xu, Z.; Li, L.; Guo, K.; Mi, J.; Wu, H.; Li, Y.; Xie, C.; Jin, J.; Xu, J.; et al. Hydrogels with programmed spatiotemporal mechanical cues for stem cell-assisted bone regeneration. Nat. Commun. 2025, 16, 3633. [Google Scholar] [CrossRef]
- Shin, J.; Kang, R.; Hyun, K.; Li, Z.; Kumar, H.; Kim, K.; Park, S.S.; Kim, K. Machine Learning-Enhanced Optimization for High-Throughput Precision in Cellular Droplet Bioprinting. Adv. Sci. 2025, 12, 2412831. [Google Scholar] [CrossRef]
- Zhang, C.W.; Chen, C.; Duan, S.; Yan, Y.; He, P.; He, X. Hydrogel-based soft bioelectronics for personalized healthcare. Med-X 2024, 2, 20. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crosslinker Type | Hydrogel Systems | Range of Mechanical and Viscoelastic Properties | Key Features | Advantages | Limitations | Reference |
|---|---|---|---|---|---|---|
| Synthetic | PEGDA, PEG-(multiarm), PVA, MBAA, PAMAM dendrimers | PVA/PEG-Tensile strength: 2.93–4.41 MPa, Elongation at break: ~450–573%, Compressive tangent modulus: 0–4.6 MPa at strain from 10–60% | Biocompatible, tunable mechanics, chemically versatile, often inert | Predictable structure, facile functionalization, customizable network mechanics | Lacks bioactivity, requires bioactive cues | [20,27,28,29,30] |
| Peptide/Protein | MMP-cleavable peptides, avidin-biotin, coiled-coils, GelMA | GelMA—Compressive modulus: 20–300 kPa, Young’s modulus: 27.1–114.4 kPa, Stress–strain: 0–15 kPa at strain from 0–20% | Biological origin, enzyme-degradable, responsive, cell interactive | Cell-responsive degradation/remodeling, bioactive | Less mechanically robust, susceptible to proteolysis | [31,32,33,34,35] |
| Peptoid/ Peptidomimetic | Helical peptoids, D-peptides, foldamers | Helical peptoid—Compressive modulus: ~2–7 kPa, Storage modulus: ~0.5–2.3 kPa | Sequence-defined, protease-resistant, some structural control | High stability, tunable stiffness, long-term integrity | Synthetic complexity, cost | [36,37,38,39,40] |
| Polysaccharide-Based | Alginate, HA, Chitosan, Dextran | Alginate-GelMa—Storage modulus: ~5–50 Pa, Loss modulus: ~10–350 Pa, Compressive modulus: ~25–280 kPa | Natural, hydrophilic, often ionically/covalently modified | Biocompatibility, bioactivity, mild gelation | Mechanical weakness, reversible (ionic) bonding | [3,41,42,43,44,45,46,47] |
| Hybrid/Composite | PEG-peptide, silica nanoparticles, nanoclays | Self-assembling peptide—Storage modulus: 14.43–452,400 Pa, Loss modulus: 2.569–80,680 Pa, Yield stress: 0.32–7012 Pa | Combines synthetic and biological elements | Synergistic properties, biofunctionality + robustness | Complex design, potential heterogeneity | [48,49,50] |
| Linear | PEGDA, diamines, MBAA | PEG-2ALD—Storage modulus: Young’s modulus: 3–11 kPa, >1000 Pa, Loss modulus: ~10–100 Pa, Stress relaxation half time: 1–100 s | Difunctional, forms chain-end linkages | Simple, homogeneous networks possible | Difficulty inducing gelation | [20,28,30,51] |
| Star/Branched | Multi-arm PEG, streptavidin | 4-arm PEG—Storage modulus: ~0–15,000 Pa, Loss modulus: ~1–5000 Pa | Multivalent, increased connectivity | Higher stiffness, more robust networks | May lead to loss of homogeneity | [20,27,33] |
| Side-Chain Crosslinking | GelMA, HAMA | AG-HYD—Young’s modulus: 1–17 kPa, Storage modulus: ~1000 Pa, Loss modulus: <1 Pa, Stress relaxation half time: 4000–6000 s | Crosslinking along backbone | Stiff networks, higher crosslink density | Loop formation, brittle behavior | [24,27,35] |
| Dendritic | PAMAM dendrimers, hyperbranched polyesters, polyglycerols | PAMAM—Storage modulus: ~0.2–31 kPa, Adhesive strength: 25–29 kPa | Tree-like architecture with high terminal group density, very high functionality | Extremely high crosslink density, rapid gelation, tunable surfaces for biofunctionality or stimuli-responsiveness | Synthetic complexity, potential loss of homogeneity, cost, intra-particle looping | [30] |
| Crosslinking Mechanism and Sub-Type | Bond Nature | Typical Gelation Kinetics/ Network Uniformity | Signature Mechanical or Dynamic Behaviors | Key Advantages and Representative Uses | Main Limitations/ Design Caveats | Reference |
|---|---|---|---|---|---|---|
| Chain-growth photopolymerization | Covalent (radical polymerization) | Seconds to minutes, can outrun diffusion | Elastic solid, initial high stiffness, microgels can act as defects | Rapid spatiotemporal control, light-patterning for 3D culture, in situ curing | O2 inhibition, residual sol fraction, can be brittle | [54,55,56] |
| Step-growth photopolymerization | Covalent (radical, step-growth) | Seconds, uniform conversion, oxygen-tolerant | More homogeneous modulus, good toughness | Fast, cytocompatible, bio-inks, microfluidics | Requires photoinitiator and light access, stoichiometry critical | [54] |
| SPAAC click | Covalent (azide + cyclooctyne) | Minutes, catalyst-free, uniform | Elastic, can be degradable | Injectable, bio-orthogonal in vivo gels | High cost, limited light control unless photo-uncage used | [57] |
| Michael-type additions | Covalent (base-catalyzed) | Tunable: seconds to hours, homogeneous if mixed well | Elastic, very cell-friendly | No radicals, no UV, easy bio-functionalization during gelation | Competing mono-thiol motifs lower crosslink density, pH-dependent | [58,59,60,61] |
| Kinetic-tuning (catalyzed hydrazone, etc.) | Same as parent bond | Catalyst decouples gel time from final modulus | Allows tunable injectability vs. modulus | Optimize delivery vs. defect density | Extra catalyst must be biocompatible or cleared | [59,62] |
| Dynamic covalent: imine/hydrazone/oxime | Reversible covalent | Tunable exchange rate (pH controlled) | Stress-relaxing, self-healing, viscoelastic | Cell-responsive matrices, injectable shear-thinning gels | Hydrolytic drift shifts equilibrium, slower at neutral pH for imines | [63,64] |
| Dynamic covalent: disulfide, Diels–Alder, boronate ester | Reversible covalent | Redox- or T-dependent, from minutes to hours | Tunable τ, shape-memory or thermo-morphing | Injectable depots, on-demand remolding | Requires stimulus for re-flow, can creep under load | [65,66,67] |
| Hydrogen-bonded networks | Non-covalent (H-bonds) | Rapid, but reversibility across wide timescales | Tough, strain–stiffening, pronounced stress relaxation | Self-healing, temperature-responsive | Weak under high humidity/heat, hysteresis on cycling | [68,69,70] |
| Host–guest inclusion | Non-covalent (inclusion complex) | Fast association, dissociation usually μs to s | Shear-thinning, quick self-recovery | 3D printing, injectable therapeutics | Affinity set by host/guest choice | [71,72] |
| Metal–ligand coordination | Non-covalent (coordination) | Very fast, pH/ionic-strength controlled | Sacrificial bond toughness, viscoelastic | Tough DN gels, wound adhesives, pH-switchable systems | Chelators or pH shifts dissolve network, metal toxicity must be managed | [73,74,75,76] |
| Hybrid/multi-network | Often covalent and non-covalent | Determined by fastest chemistry | High modulus and high toughness, multi-phase relaxation | Load-bearing soft tissues, stretchable sensors | More complex synthesis, balance of network fractions is critical | [51,77,78] |
| Properties | SCX | LX | SX |
|---|---|---|---|
| Storage modulus—G′ (Pa) | ~103 | <103 | 103 ~ 104 |
| Loss modulus—G″ (Pa) | <101 | 101 ~ 102 | ~102 |
| No. | Molar Ratio DMMA/AAc/UPyEA | Modulus (kPa) | Max Stress (kPa) | Strain at Break | Strain Energy Density (kJ m−3) |
|---|---|---|---|---|---|
| 1 | 50/50/0 | 74 ± 10 | 74 ± 5 | 3150 ± 240 | 2370 ± 140 |
| 2 | 50/50/0.2 | 283 ± 18 | 87 ± 11 | 3850 ± 230 | 3230 ± 370 |
| 3 | 50/50/0.4 | 409 ± 36 | 132 ±17 | 3900 ± 280 | 4330 ± 320 |
| 4 | 50/50/0.8 | 1252 ± 108 | 234 ± 16 | 4340 ± 320 | 7160 ± 670 |
| Biological and Biomedical Application | Hydrogel Type and Crosslinking Strategy | Key Outcomes | Reference |
|---|---|---|---|
| Immunomodulation of hMSCs | HA hydrogels crosslinked with peptoids; mechanical tuning without altering network connectivity | G′ range ~0.6–8 kPa; softer hydrogels (~0.6–3.2 kPa) increased proliferation (44–54%), IDO expression, circularity | [38] |
| hMSC adhesion and viability | SF-TA and Gel-TA enzymatically crosslinked (HRP/H2O2); crosslinking density modified | Softer hydrogels with lower density promoted spreading and viability; stiffer ones reduced adhesion and metabolic activity | [97] |
| Injectable cytoprotective delivery of hMSCs and NPCs | DN microbeads: SF-TA, Gel-TA (HRP/H2O2) + ionic alginate crosslinking | Higher viability after extrusion, ~70–80% under adverse conditions; unencapsulated cells dropped below ~50% | [129] |
| pH-responsive drug release of ceftriaxone | Pectin/PVA hydrogel crosslinked with 3-APDEMS; physical and chemical bonds | Porosity 61–79%, swelling 300–1275%, >90% drug release at 180 min | [130] |
| Sustained cephradine delivery | PVA/carrageenan hydrogel crosslinked with APTES; dual chemical and physical network | ~85% release in 7.5 h; swelling 20–200%; release tuned by crosslinker content and pH | [131] |
| Bone regeneration in infected defects | DN hydrogel: SilMA, MA-GNPs, CuBGs; UV-curable, self-healing IPN | Bone volume increased from ~35% to ~60% between 2 and 4 weeks post-implantation | [132] |
| Aortic valve ECM mimicry | HA hydrogel with variable stiffness (via HA MW and crosslinking density) + Me-Gel for RGD motifs | Softer hydrogels increase GAG; stiffer hydrogels increase α-SMA (fibroblast vs. myofibroblast phenotype) | [133] |
| Bone regeneration and immunomodulation | Injectable alginate–tyramine hydrogel with sericin and GO; HRP/H2O2 crosslinking | M2 polarization increase IL-10, Arg-1; osteogenesis enhanced; scaffold degradable and injectable | [134] |
| Wound healing and skin regeneration | 3D-bioprintable HA-based hydrogel (HA-MA + HA-SH); dual crosslinking (thiol–click + UV) | Swelling ~95%, degradation ~90% in 11 days, sustained Nafcillin release for infection control | [135] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roca-Arroyo, A.F.; Gutierrez-Rivera, J.A.; Morton, L.D.; Castilla-Casadiego, D.A. Hydrogel Network Architecture Design Space: Impact on Mechanical and Viscoelastic Properties. Gels 2025, 11, 588. https://doi.org/10.3390/gels11080588
Roca-Arroyo AF, Gutierrez-Rivera JA, Morton LD, Castilla-Casadiego DA. Hydrogel Network Architecture Design Space: Impact on Mechanical and Viscoelastic Properties. Gels. 2025; 11(8):588. https://doi.org/10.3390/gels11080588
Chicago/Turabian StyleRoca-Arroyo, Andres F., Jhonatan A. Gutierrez-Rivera, Logan D. Morton, and David A. Castilla-Casadiego. 2025. "Hydrogel Network Architecture Design Space: Impact on Mechanical and Viscoelastic Properties" Gels 11, no. 8: 588. https://doi.org/10.3390/gels11080588
APA StyleRoca-Arroyo, A. F., Gutierrez-Rivera, J. A., Morton, L. D., & Castilla-Casadiego, D. A. (2025). Hydrogel Network Architecture Design Space: Impact on Mechanical and Viscoelastic Properties. Gels, 11(8), 588. https://doi.org/10.3390/gels11080588