

Comparative Transcriptomics Profiling of Perennial Ryegrass Infected with Wild Type or a ΔvelA Epichloë festucae Mutant Reveals Host Processes Underlying Mutualistic versus Antagonistic Interactions


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. HiSeq Results Analysis
2.3. Functional Annotation
2.4. General Bioinformatics Analyses
3. Results
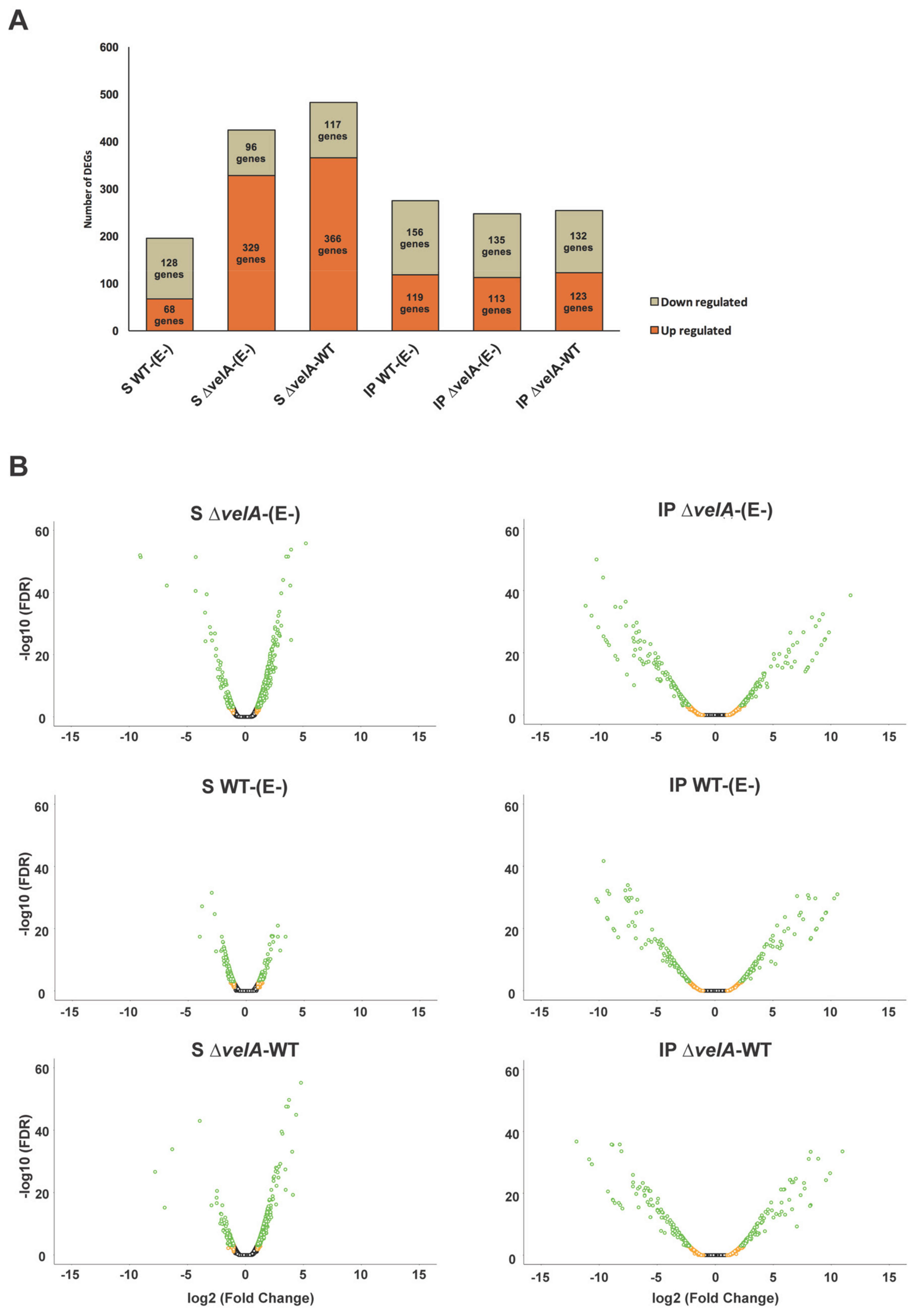
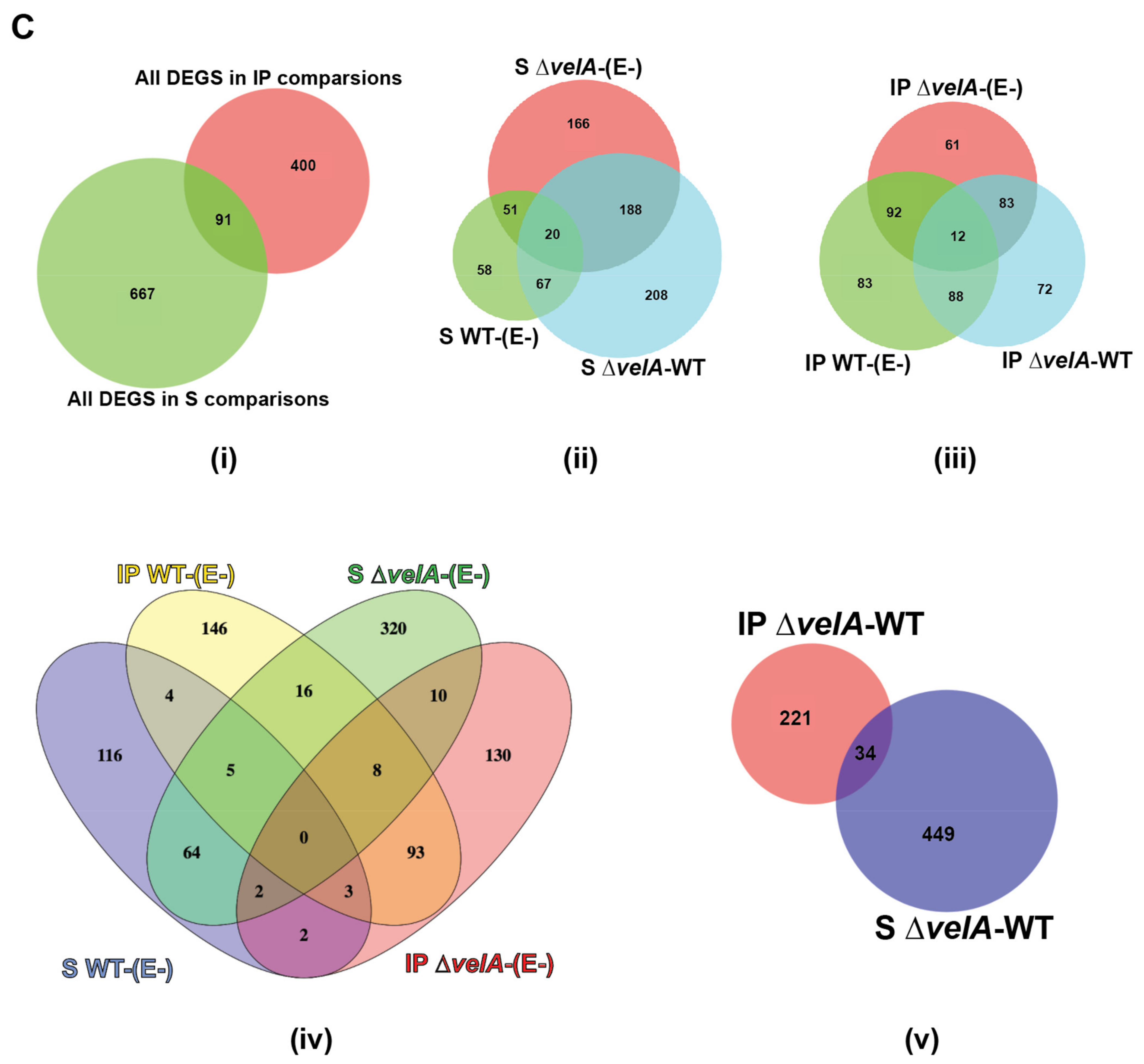
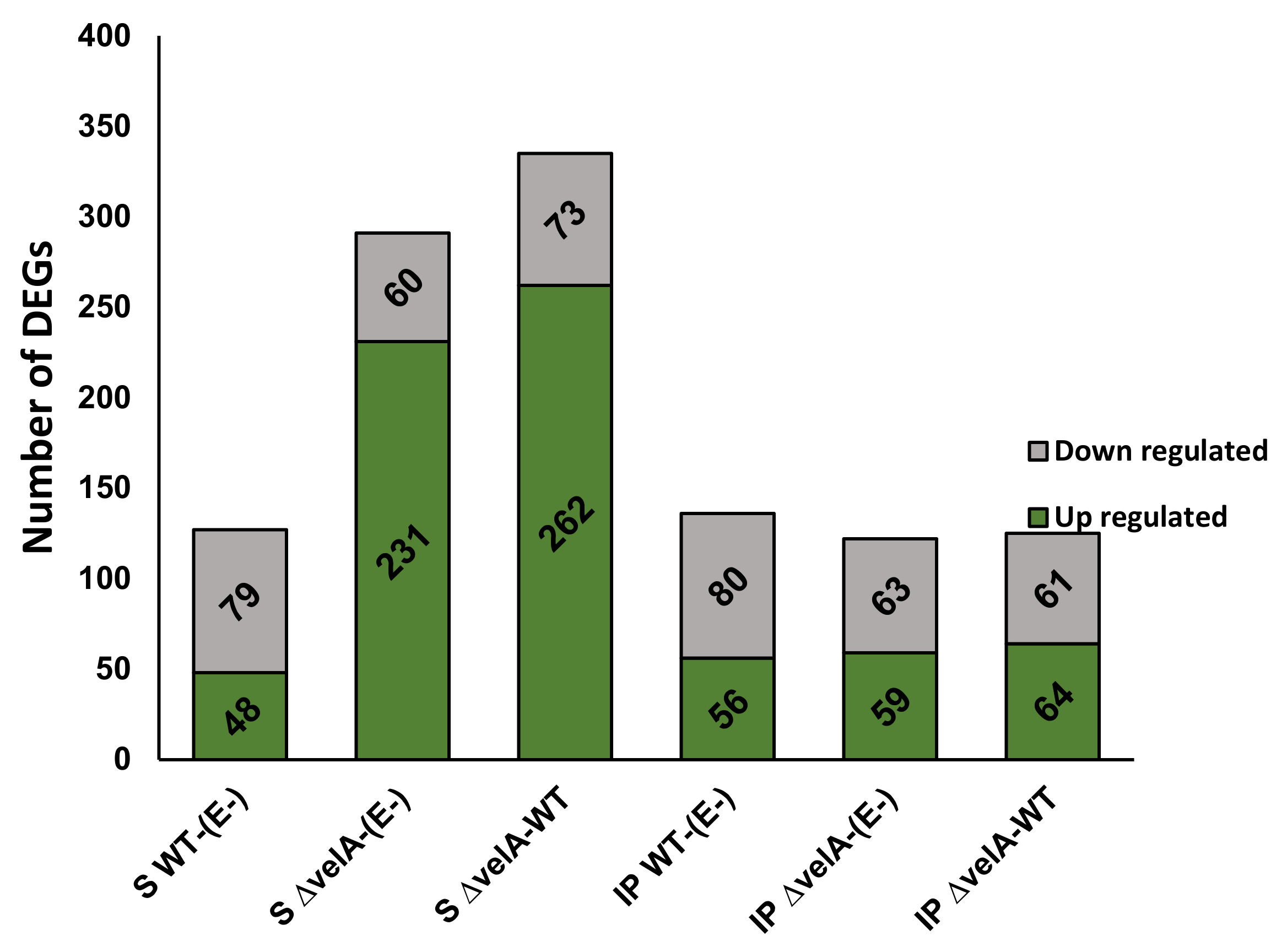
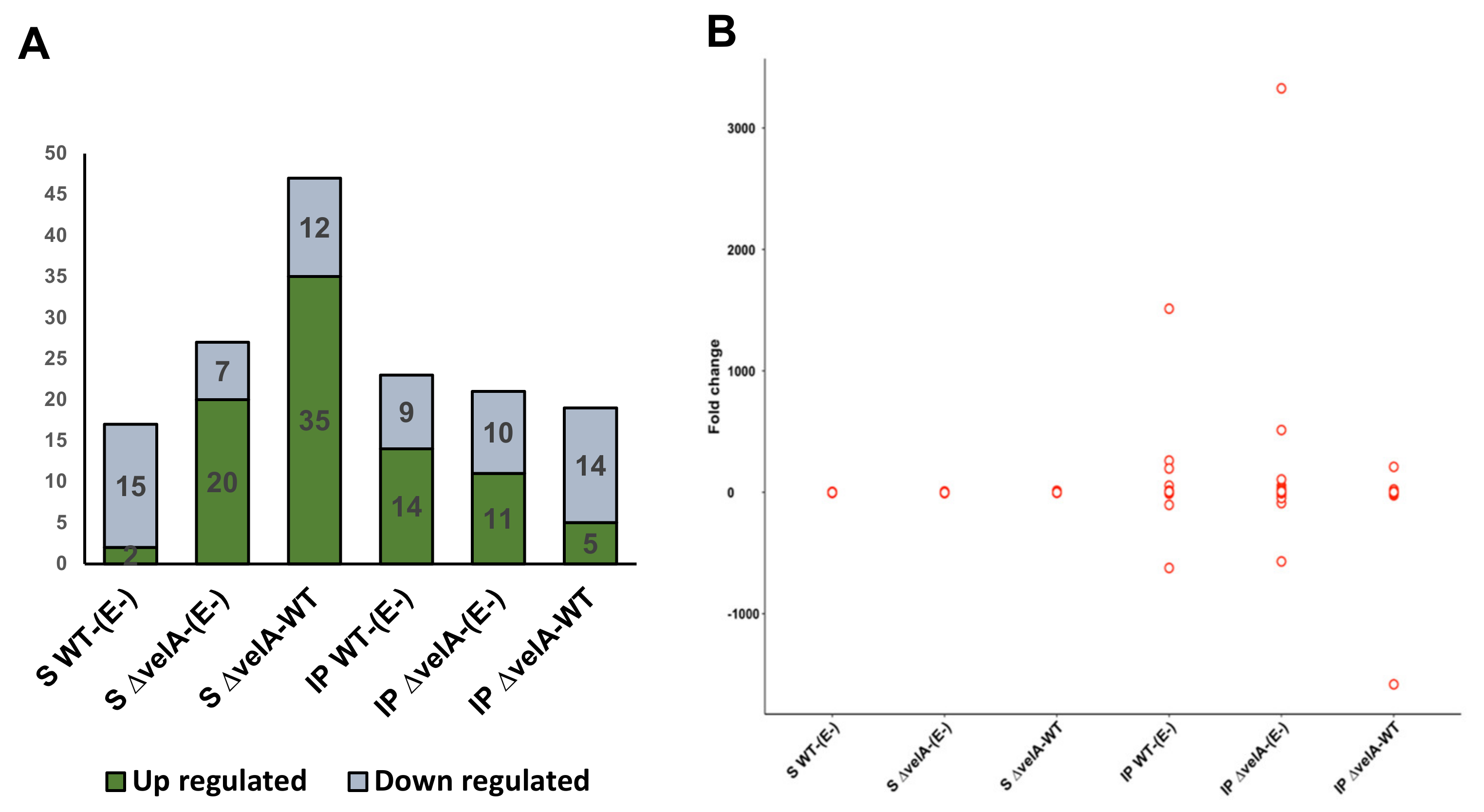
3.1. General Description of RNA-Sequencing Results
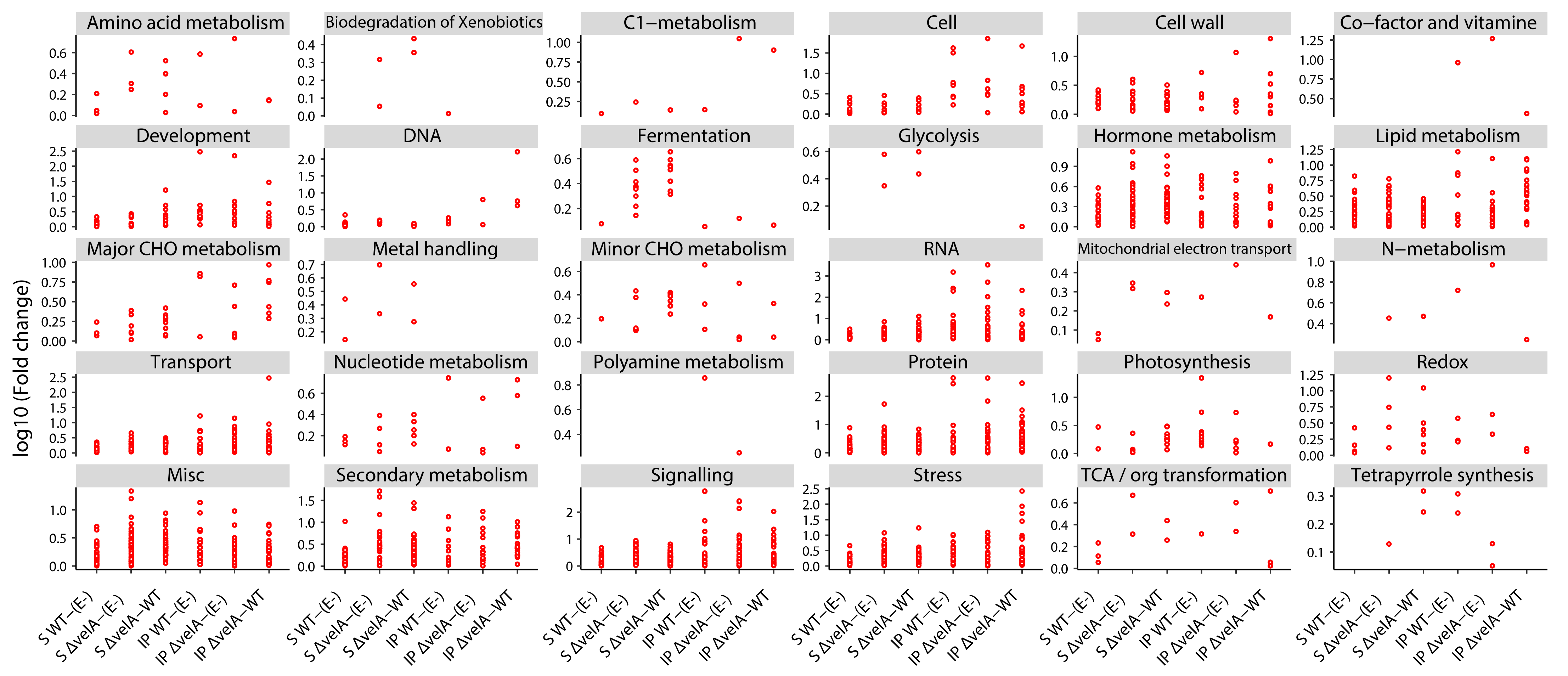
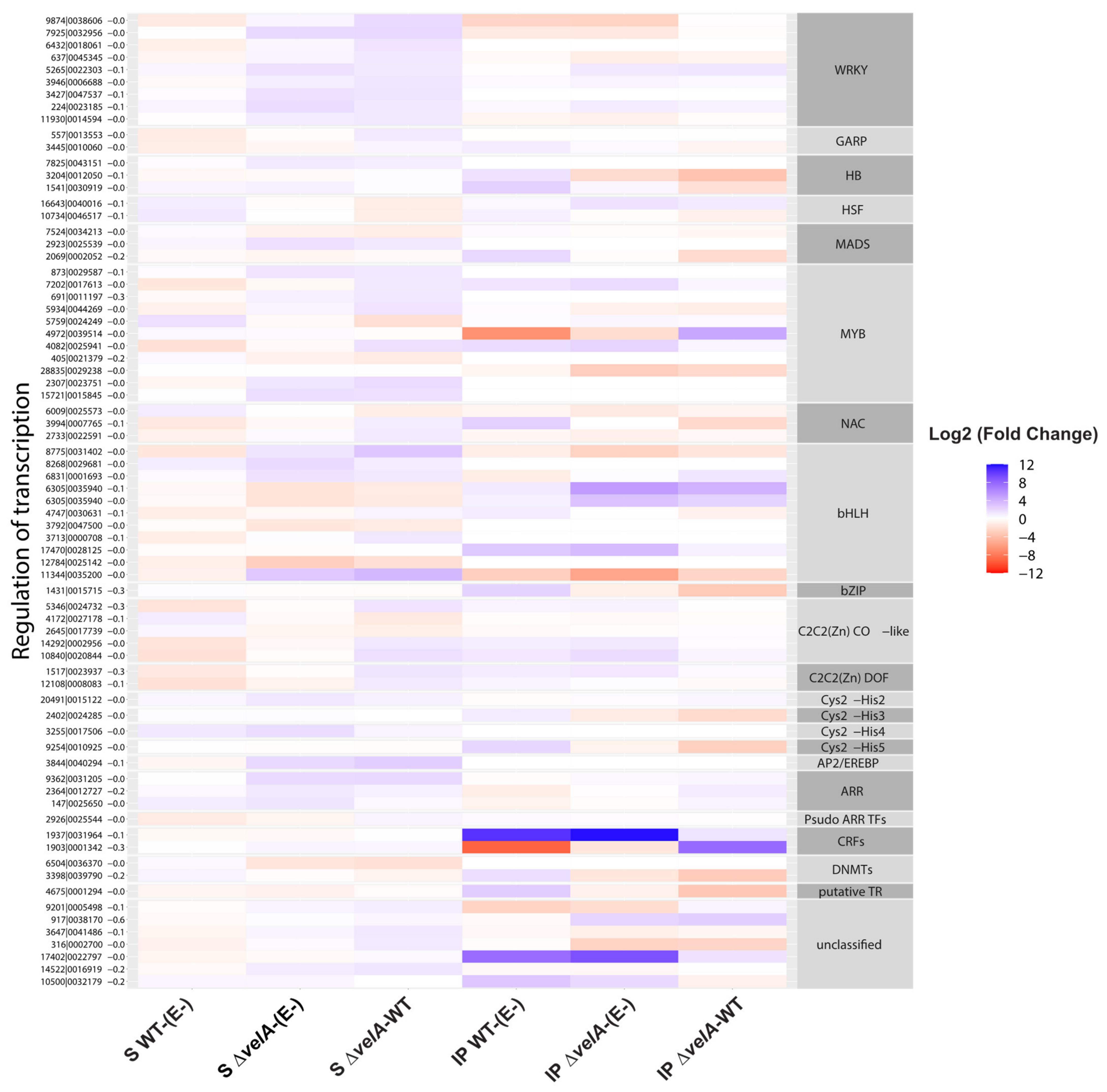
3.2. Functional Annotations of Differentially Expressed Ryegrass Genes
3.3. Mutant Endophytes Change Primary Metabolism in Their Host Plants
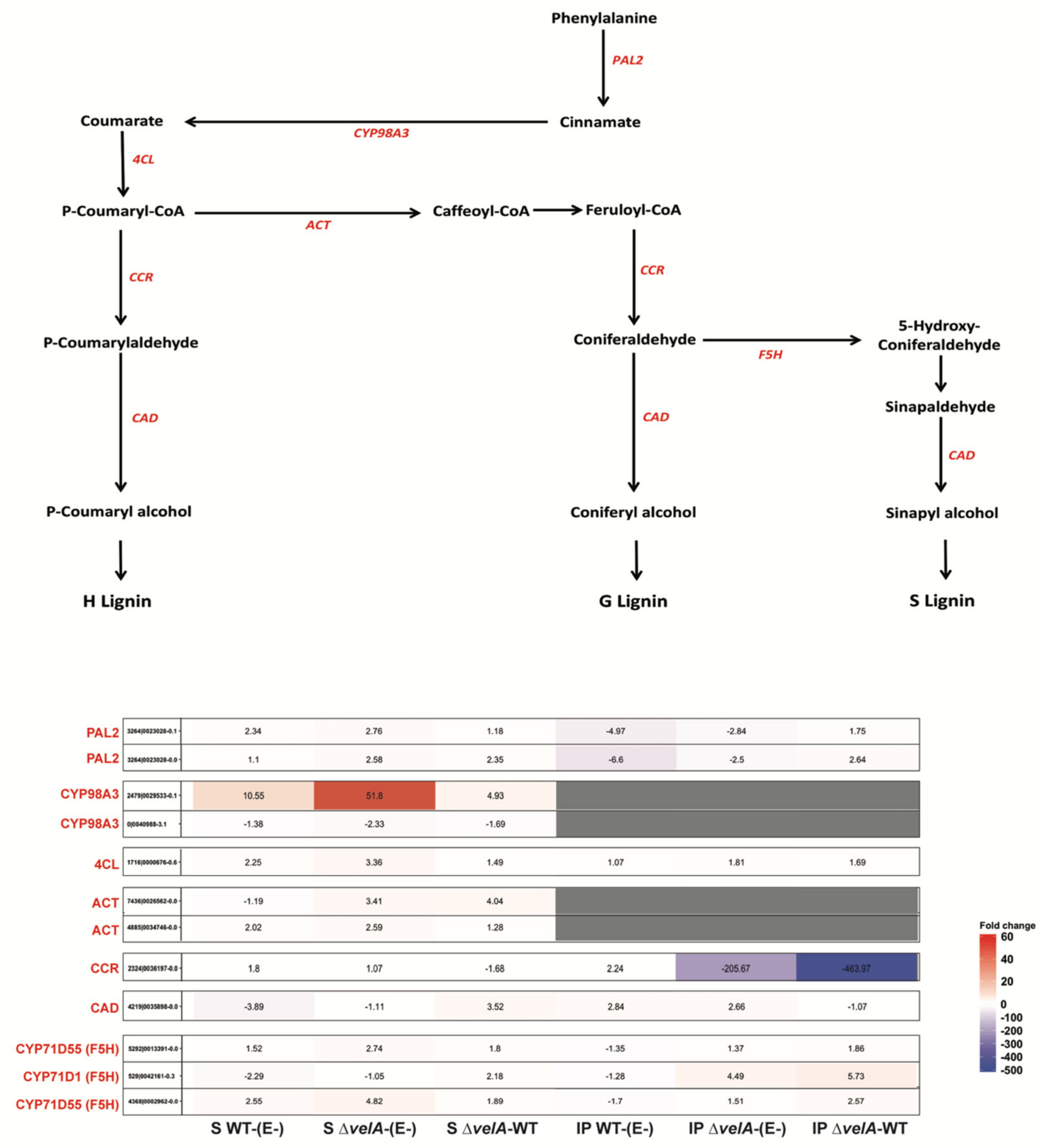
3.4. Mutant Endophytes Change Secondary Metabolism in Their Host Plants
3.5. Infecting Ryegrass with ΔvelA E. Festucae Mutant Alters the Expression of Genes Responsible for Biotic and Abiotic Stresses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leuchtmann, A.; Schardl, C.L.; Siegel, M.R. Sexual Compatibility and Taxonomy of a New Species of Epichloë Symbiotic with Fine Fescue Grasses. Mycologia 1994, 86, 802–812. [Google Scholar] [CrossRef]
- Christensen, M.J.; Bennett, R.J.; Schmid, J. Growth of Epichloë/Neotyphodium and p-endophytes in leaves of Lolium and Festuca grasses. Mycol. Res. 2002, 106, 93–106. [Google Scholar] [CrossRef]
- Schardl, C.L. The Epichloae, Symbionts of the Grass Subfamily Poöideae. Ann. Mol. Bot. Gard. 2010, 97, 646–665. [Google Scholar] [CrossRef]
- Schardl, C.L. Epichloë festucae and Related Mutualistic Symbionts of Grasses. Fungal Genet. Biol. 2001, 33, 69–82. [Google Scholar] [CrossRef]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-Symbiotic Fungi as Chemical Engineers: Multi-Genome Analysis of the Clavicipitaceae Reveals Dynamics of Alkaloid Loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [PubMed]
- Rahnama, M.; Johnson, R.D.; Voisey, C.R.; Simpson, W.R.; Fleetwood, D.J. The Global Regulatory Protein VelA Is Required for Symbiosis Between the Endophytic Fungus Epichloë festucae and Lolium perenne. Mol. Plant Microbe Interact. 2018, 31, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.; Green, K.; Berry, D. The fine balance between mutualism and antagonism in the Epichloë festucae–grass symbiotic interaction. Curr. Opin. Plant Biol. 2018, 44, 32–38. [Google Scholar] [CrossRef]
- Rahnama, M.; Maclean, P.; Fleetwood, D.J.; Johnson, R.D. The LaeA orthologue in Epichloë festucae is required for symbiotic interaction with Lolium perenne. Fungal Genet. Biol. 2019, 129, 74–85. [Google Scholar] [CrossRef]
- Rahnama, M.; Maclean, P.; Fleetwood, D.J.; Johnson, R.D. VelA and LaeA are Key Regulators of Epichloë festucae Transcriptomic Response during Symbiosis with Perennial Ryegrass. Microorganisms 2020, 8, 33. [Google Scholar] [CrossRef]
- Johnson, L.J.; Johnson, R.D.; Schardl, C.L.; Panaccione, D.G. Identification of differentially expressed genes in the mutualistic association of tall fescue with Neotyphodium coenophialum. Physiol. Mol. Plant Pathol. 2003, 63, 305–317. [Google Scholar] [CrossRef]
- Khan, A.; Bassett, S.; Voisey, C.; Gaborit, C.; Johnson, L.; Christensen, M.; McCulloch, A.; Bryan, G.; Johnson, R. Gene expression profiling of the endophytic fungus Neotyphodium lolii in association with its host plant perennial ryegrass. Australas. Plant Pathol. 2010, 39, 467–476. [Google Scholar] [CrossRef]
- Dupont, P.; Eaton, C.J.; Wargent, J.J.; Fechtner, S.; Solomon, P.; Schmid, J.; Day, R.C.; Scott, B.; Cox, M.P. Fungal endophyte infection of ryegrass reprograms host metabolism and alters development. New Phytol. 2015, 208, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.; Day, R.; Zhang, N.; Dupont, P.; Cox, M.P.; Schardl, C.L.; Minards, N.; Truglio, M.; Moore, N.; Harris, D.R.; et al. Host tissue environment directs activities of an Epichloë endophyte, while it induces systemic hormone and defense responses in its native perennial ryegrass host. Mol. Plant Microbe Interact. 2016, 30, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, R.D.; Nagabhyru, P.; Graham, M.A.; Boykin, D.; Schardl, C.L. Transcriptome response of Lolium arundinaceum to its fungal endophyte Epichloë coenophiala. New Phytol. 2017, 213, 324–337. [Google Scholar] [CrossRef]
- Dinkins, R.D.; Nagabhyru, P.; Young, C.A.; West, C.P.; Schardl, C.L. Transcriptome analysis and differential expression in tall fescue harboring different endophyte strains in response to water deficit. Plant Genome 2019, 12, 180071. [Google Scholar] [CrossRef]
- Rahnama, M.; Maclean, P.; Fleetwood, D.J.; Johnson, R.D. Comparative transcriptomics analysis of compatible wild type and incompatible ΔlaeA mutant strains of Epichloë festucae in association with perennial ryegrass. Data Brief 2019, 24, 103843. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR—Flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using MapMan to visualize and compare Omics data in plants: A case study in the crop species, Maize. Plant Cell Environ. 2009, 32, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Hester, J.; Chang, W. Devtools: Tools to make developing r packages easier. R Package Version 2016, 1, 9000. [Google Scholar]
- Team, R. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2018. [Google Scholar]
- Rohde, W.; Becker, D.; Salamini, F. Structural analysis of the waxy locus from Hordeum vulgare. Nucleic Acids Res. 1988, 16, 7185. [Google Scholar] [CrossRef]
- Bahaji, A.; Baroja-Fernández, E.; Ricarte-Bermejo, A.; Sánchez-López, Á.M.; Muñoz, F.J.; Romero, J.M.; Ruiz, M.T.; Baslam, M.; Almagro, G.; Sesma, M.T. Characterization of multiple SPS knockout mutants reveals redundant functions of the four Arabidopsis sucrose phosphate synthase isoforms in plant viability, and strongly indicates that enhanced respiration and accelerated starch turnover can alleviate the blockage of sucrose biosynthesis. Plant Sci. 2015, 238, 135–147. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant cell wall-degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef]
- Hazen, S.P.; Scott-Craig, J.; Walton, J.D. Cellulose synthase-like genes of rice. Plant Physiol. 2002, 128, 336–340. [Google Scholar] [CrossRef]
- Yuan, J.S.; Yang, X.; Lai, J.; Lin, H.; Cheng, Z.; Nonogaki, H.; Chen, F. The endo-β-mannanase gene families in Arabidopsis, rice, and poplar. Funct. Integr. Genom. 2007, 7, 1–16. [Google Scholar] [CrossRef]
- Shin, J.; Jeong, D.; Park, M.C.; An, G. Characterization and Transcriptional Expression of the α-Expansin Gene Family in Rice. Mol. Cells 2005, 20, 210–218. [Google Scholar]
- Zipfel, C. Early molecular events in PAMP-triggered immunity. Curr. Opin. Plant Biol. 2009, 12, 414–420. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, Y.; Ludwiczuk, A.; Nagashima, F. Chemical constituents of bryophytes. Bio- and chemical diversity, biological activity, and chemosystematics. Prog. Chem. Org. Nat. Prod. 2013, 95, 563–605. [Google Scholar] [CrossRef]
- Woo, E.; Dunwell, J.M.; Goodenough, P.W.; Marvier, A.C.; Pickersgill, R.W. Germin is a manganese containing homohexamer with oxalate oxidase and superoxide dismutase activities. Nat. Struct. Biol. 2000, 7, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.R. Polyamines and plant disease. Phytochemistry 2003, 64, 97–107. [Google Scholar] [CrossRef]
- Custers, J.H.H.V.; Harrison, S.J.; Sela-Buurlage, M.B.; Van Deventer, E.; Lageweg, W.; Howe, P.W.; Van Der Meijs, P.J.; Ponstein, A.S.; Simons, B.H.; Melchers, L.S. Isolation and characterisation of a class of carbohydrate oxidases from higher plants, with a role in active defence. Plant J. 2004, 39, 147–160. [Google Scholar] [CrossRef]
- Hiraga, S.; Sasaki, K.; Ito, H.; Ohashi, Y.; Matsui, H. A large family of class III plant peroxidases. Plant Cell Physiol. 2001, 42, 462–468. [Google Scholar] [CrossRef]
- Rahnama, M.; Fleetwood, D.J.; Johnson, R.D. Histological methods to detect early-stage plant defense responses during artificial inoculation of lolium perenne with Epichloë festucae. Bio-Protocol 2021, 11, e4013. [Google Scholar] [CrossRef]
- Dixon, D.P.; Skipsey, M.; Edwards, R. Roles for glutathione transferases in plant secondary metabolism. Phytochemistry 2010, 71, 338–350. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Kim, S.A.; Yun, H.K. Glutathione S-transferase genes differently expressed by pathogen-infection in Vitis flexuosa. Plant Breed. Biotechnol. 2016, 4, 61–70. [Google Scholar] [CrossRef]
- Gadjev, I.; Stone, J.M.; Gechev, T.S. Programmed cell death in plants: New insights into redox regulation and the role of hydrogen peroxide. Int. Rev. Cell Mol. Biol. 2008, 270, 87–144. [Google Scholar] [CrossRef]
- Divi, U.K.; Krishna, P. Brassinosteroid: A biotechnological target for enhancing crop yield and stress tolerance. New Biotechnol. 2009, 26, 131–136. [Google Scholar] [CrossRef]
- Sirhindi, G.; Kumar, M.; Kumar, S.; Bhardwaj, R. Brassinosteroids: Physiology and stress management in plants. In Abiotic Stress Response in Plants; John Wiley & Sons.: Hoboken, NJ, USA, 2016; pp. 279–314. [Google Scholar] [CrossRef]
- Heil, M.; Bostock, R.M. Induced systemic resistance (ISR) against pathogens in the context of induced plant defences. Ann. Bot. 2002, 89, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Irfan, M.; Ahmad, A. Brassinosteroids: Under biotic stress. In Brassinosteroids: A Class of Plant Hormone; Springer: Berlin/Heidelberg, Germany, 2011; pp. 345–360. [Google Scholar]
- De Vleesschauwer, D.; Gheysen, G.; Höfte, M. Hormone defense networking in rice: Tales from a different world. Trends Plant Sci. 2013, 18, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Dave, A.; Graham, I.A. Oxylipin signaling: A distinct role for the jasmonic acid precursor cis-( )-12-oxo-phytodienoic acid (cis-OPDA). Front. Plant Sci. 2012, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Song, J.T.; Koo, Y.J.; Seo, H.S.; Kim, M.C.; Do Choi, Y.; Kim, J.H. Overexpression of AtSGT1, an Arabidopsis salicylic acid glucosyltransferase, leads to increased susceptibility to Pseudomonas syringae. Phytochemistry 2008, 69, 1128–1134. [Google Scholar] [CrossRef]
- Arachevaleta, M.; Bacon, C.W.; Hoveland, C.S.; Radcliffe, D.E. Effect of the tall fescue endophyte on plant response to environmental stress. Agron. J. 1989, 81, 83–90. [Google Scholar] [CrossRef]
- Malinowski, D.P.; Belesky, D.P. Adaptations of endophyte-infected cool-season grasses to environmental stresses: Mechanisms of drought and mineral stress tolerance. Crop Sci. 2000, 40, 923–940. [Google Scholar] [CrossRef]
- Wang, J.; Hou, W.; Christensen, M.J.; Li, X.; Xia, C.; Li, C.; Nan, Z. Role of Epichloë endophytes in improving host grass resistance ability and soil properties. J. Agric. Food Chem. 2020, 68, 6944–6955. [Google Scholar] [CrossRef]
- West, C.P. Physiology and drought tolerance of endophyte-lnfected grasses. In Biotechnology of Endophytic Fungi of Grasses; CRC Press: Boca Raton, FL, USA, 2018; pp. 87–99. [Google Scholar]
- Wiewióra, B.; Żurek, G. The response of the associations of grass and epichloë endophytes to the increased content of heavy metals in the soil. Plants 2021, 10, 429. [Google Scholar] [CrossRef]
- Ambrose, K.V.; Belanger, F.C. SOLiD-SAGE of endophyte-infected red fescue reveals numerous effects on host transcriptome and an abundance of highly expressed fungal secreted proteins. PLoS ONE 2012, 7, e53214. [Google Scholar] [CrossRef]
- Nagabhyru, P.; Dinkins, R.D.; Schardl, C.L. Transcriptomics of Epichloë-grass symbioses in host vegetative and reproductive stages. Mol. Plant-Microbe Interact. 2019, 32, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Nagabhyru, P.; Dinkins, R.D.; Wood, C.L.; Bacon, C.W.; Schardl, C.L. Tall fescue endophyte effects on tolerance to water-deficit stress. BMC Plant Biol. 2013, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Goodwin, S.B. MVE1, encoding the velvet gene product homolog in Mycosphaerella graminicola, is associated with aerial mycelium formation, melanin biosynthesis, hyphal swelling, and light signaling. Appl. Environ. Microbiol. 2011, 77, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Hoff, B.; Kamerewerd, J.; Sigl, C.; Mitterbauer, R.; Zadra, I.; Kürnsteiner, H.; Kück, U. Two components of a velvet-like complex control hyphal morphogenesis, conidiophore development, and penicillin biosynthesis in Penicillium chrysogenum. Eukaryot. Cell 2010, 9, 1236–1250. [Google Scholar] [CrossRef]
- Karimi Aghcheh, R.; Nemeth, Z.; Atanasova, L.; Fekete, E.; Paholcsek, M.; Sándor, E.; Aquino, B.; Druzhinina, I.S.; Karaffa, L.; Kubicek, C.P. The VELVET A orthologue VEL1 of Trichoderma reesei regulates fungal development and is essential for cellulase gene expression. PLoS ONE 2014, 9, e112799. [Google Scholar] [CrossRef]
- Li, S.; Myung, K.; Guse, D.; Donkin, B.; Proctor, R.H.; Grayburn, W.S.; Calvo, A.M. FvVE1 regulates filamentous growth, the ratio of microconidia to macroconidia and cell wall formation in Fusarium verticillioides. Mol. Microbiol. 2006, 62, 1418–1432. [Google Scholar] [CrossRef]
- Lind, A.L.; Wisecaver, J.H.; Smith, T.D.; Feng, X.; Calvo, A.M.; Rokas, A. Examining the evolution of the regulatory circuit controlling secondary metabolism and development in the fungal genus Aspergillus. PLoS Genet. 2015, 11, e1005096. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Kenerley, C.M. Regulation of morphogenesis and biocontrol properties in Trichoderma virens by a VELVET protein, Vel1. Appl. Environ. Microbiol. 2010, 76, 2345–2352. [Google Scholar] [CrossRef]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Nagabhyru, P.; Schardl, C.L.; Dinkins, R.D. Differential gene expression in tall fescue tissues in response to water deficit. Plant Genome 2022, 15, e20199. [Google Scholar] [CrossRef]
- Thalmann, M.; Santelia, D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017, 214, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Plant cell wall extensibility: Connecting plant cell growth with cell wall structure, mechanics, and the action of wall-modifying enzymes. J. Exp. Bot. 2016, 67, 463–476. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Change | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene ID | Bincode Name | Best Annotation | S WT-(E-) | S ΔvelA-(E-) | S ΔvelA-WT | IP WT-(E-) | IP ΔvelA-(E-) | IP ΔvelA-WT |
| 13063|0011328-0.1 | ‘major CHO metabolism.synthesis.starch.starch synthase’ | starch synthase 1 | −1.12 | 1.04 | 1.16 | −7.45 | 1.25 | 9.31 |
| 1617|0046842-0.0 | ‘major CHO metabolism.degradation.starch.starch cleavage.beta amylase’ | beta-amylase 9-like | −1.83 | −1.89 | −1.03 | 7.20 | 5.13 | −1.40 |
| 1952|0042706-0.4 | ‘major CHO metabolism.degradation.starch.starch cleavage.beta amylase’ | beta-amylase 6 | −2.24 | −1.55 | 1.45 | −1.02 | −1.25 | −1.22 |
| 6792|0008466-0.0 | ‘cell wall.hemicellulose synthesis.glucuronoxylan’ | plant glycogenin-like starch initiation protein 2 | 2.44 | 4.00 | 1.64 | −1.34 | −1.23 | 1.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahnama, M.; Maclean, P.; Fleetwood, D.J.; Johnson, R.D. Comparative Transcriptomics Profiling of Perennial Ryegrass Infected with Wild Type or a ΔvelA Epichloë festucae Mutant Reveals Host Processes Underlying Mutualistic versus Antagonistic Interactions. J. Fungi 2023, 9, 190. https://doi.org/10.3390/jof9020190
Rahnama M, Maclean P, Fleetwood DJ, Johnson RD. Comparative Transcriptomics Profiling of Perennial Ryegrass Infected with Wild Type or a ΔvelA Epichloë festucae Mutant Reveals Host Processes Underlying Mutualistic versus Antagonistic Interactions. Journal of Fungi. 2023; 9(2):190. https://doi.org/10.3390/jof9020190
Chicago/Turabian StyleRahnama, Mostafa, Paul Maclean, Damien J. Fleetwood, and Richard D. Johnson. 2023. "Comparative Transcriptomics Profiling of Perennial Ryegrass Infected with Wild Type or a ΔvelA Epichloë festucae Mutant Reveals Host Processes Underlying Mutualistic versus Antagonistic Interactions" Journal of Fungi 9, no. 2: 190. https://doi.org/10.3390/jof9020190
APA StyleRahnama, M., Maclean, P., Fleetwood, D. J., & Johnson, R. D. (2023). Comparative Transcriptomics Profiling of Perennial Ryegrass Infected with Wild Type or a ΔvelA Epichloë festucae Mutant Reveals Host Processes Underlying Mutualistic versus Antagonistic Interactions. Journal of Fungi, 9(2), 190. https://doi.org/10.3390/jof9020190