
Inborn Errors of Immunity Causing Pediatric Susceptibility to Fungal Diseases



Abstract
1. Introduction
2. Methods
2.1. Candidiasis
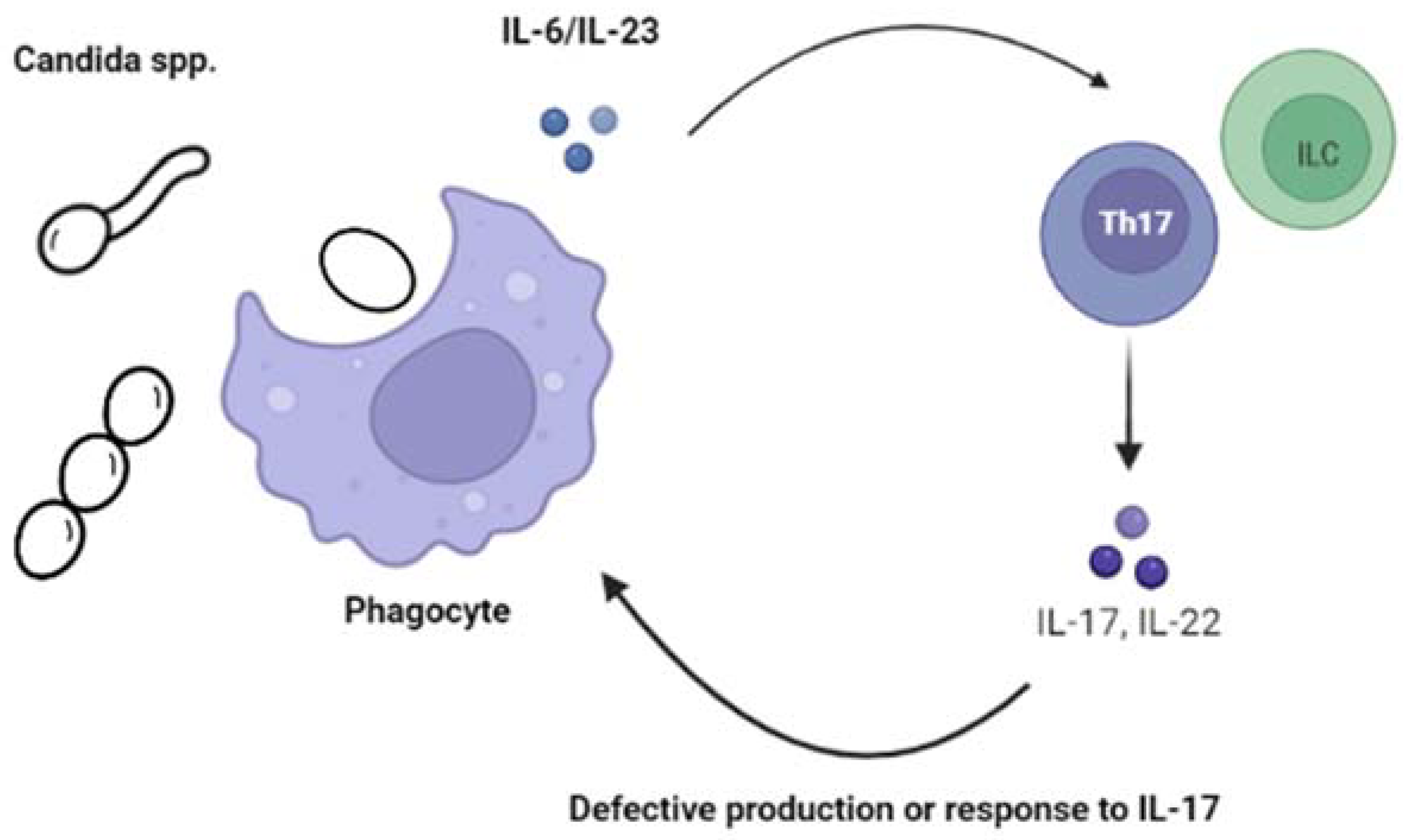
2.1.1. Chronic Mucocutaneous Candidiasis (CMC)
2.1.2. Invasive Candidiasis (IC)
2.2. Aspergillosis
Pulmonary Aspergillosis
2.3. Extrapulmonary Aspergillosis (EPA)
2.3.1. Thermally-Dimorphic Endemic Mycoses
2.3.2. Deep Dermatophytosis
2.3.3. Mucormycosis
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef] [PubMed]
- Bousfiha, A.; Moundir, A.; Tangye, S.G.; Picard, C.; Jeddane, L.; Al-Herz, W.; Rundles, C.C.; Franco, J.L.; Holland, S.M.; Klein, C.; et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J. Clin. Immunol. 2022, 42, 1508–1520. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef]
- McCarty, T.P.; White, C.M.; Pappas, P.G. Candidemia and Invasive Candidiasis. Infect. Dis. Clin. N. Am. 2021, 35, 389–413. [Google Scholar] [CrossRef]
- Turner, S.A.; Butler, G. The Candida pathogenic species complex. Cold Spring Harb. Perspect. Med. 2014, 4, a019778. [Google Scholar] [CrossRef]
- Warris, A.; Pana, Z.D.; Oletto, A.; Lundin, R.; Castagnola, E.; Lehrnbecher, T.; Groll, A.H.; Roilides, E. Etiology and Outcome of Candidemia in Neonates and Children in Europe: An 11-year Multinational Retrospective Study. Pediatr. Infect. Dis. J. 2020, 39, 114–120. [Google Scholar] [CrossRef]
- Palazzi, D.L.; Arrieta, A.; Castagnola, E.; Halasa, N.; Hubbard, S.; Brozovich, A.A.; Fisher, B.T.; Steinbach, W.J. Candida speciation, antifungal treatment and adverse events in pediatric invasive candidiasis: Results from 441 infections in a prospective, multi-national study. Pediatr. Infect. Dis. J. 2014, 33, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.P.; Steinbach, W.J. Candida Species. In Principles and Practice of Pediatric Infectious Diseases, 6th ed.; Long, S.S., Prober, C.G., Fischer, M., Kimberlin, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 243. [Google Scholar]
- Kirkpatrick, C.H. Chronic mucocutaneous candidiasis. Pediatr. Infect. Dis. J. 2001, 20, 197–206. [Google Scholar] [CrossRef]
- Okada, S.; Puel, A.; Casanova, J.L.; Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin. Transl. Immunol. 2016, 5, e114. [Google Scholar] [CrossRef]
- Puel, A. Human inborn errors of immunity underlying superficial or invasive candidiasis. Hum. Genet. 2020, 139, 1011–1022. [Google Scholar] [CrossRef]
- Millsop, J.W.; Fazel, N. Oral candidiasis. Clin. Dermatol. 2016, 34, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; Vinh, D.C. Yeast infections—Human genetics on the rise. N. Engl. J. Med. 2009, 361, 1798–1801. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C. Insights into human antifungal immunity from primary immunodeficiencies. Lancet Infect. Dis. 2011, 11, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S.; Iliev, I.D.; Hohl, T.M. Immunity against fungi. JCI Insight 2017, 2, e93156. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C. The molecular immunology of human susceptibility to fungal diseases: Lessons from single gene defects of immunity. Expert Rev. Clin. Immunol. 2019, 15, 461–486. [Google Scholar] [CrossRef]
- Break, T.J.; Oikonomou, V.; Dutzan, N.; Desai, J.V.; Swidergall, M.; Freiwald, T.; Chauss, D.; Harrison, O.J.; Alejo, J.; Williams, D.W.; et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 2021, 371, eaay5731. [Google Scholar] [CrossRef] [PubMed]
- Philippot, Q.; Casanova, J.L.; Puel, A. Candidiasis in patients with APS-1: Low IL-17, high IFN-γ, or both? Curr. Opin. Immunol. 2021, 72, 318–323. [Google Scholar] [CrossRef]
- Slatter, M.A.; Gennery, A.R. Advances in the treatment of severe combined immunodeficiency. Clin. Immunol. 2022, 242, 109084. [Google Scholar] [CrossRef]
- Sponzilli, I.; Notarangelo, L.D. Severe combined immunodeficiency (SCID): From molecular basis to clinical management. Acta Biomed. 2011, 82, 5–13. [Google Scholar]
- Chavoshzadeh, Z.; Darougar, S.; Momen, T.; Esmaeilzadeh, H.; Abolhassani, H.; Cheraghi, T.; van der Burg, M.; van Zelm, M. Immunodeficiencies affecting cellular and humoral immunity. In Inborn Errors of Immunity—A Practical Guide, 1st ed.; Aghamohammadi, A., Abolhassani, H., Rezaei, N., Yazdani, R., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 9–39, Chapter 2. [Google Scholar]
- Puck, J.M. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol. Rev. 2019, 287, 241–252. [Google Scholar] [CrossRef]
- Dorsey, M.J.; Wright, N.A.M.; Chaimowitz, N.S.; Dávila Saldaña, B.J.; Miller, H.; Keller, M.D.; Thakar, M.S.; Shah, A.J.; Abu-Arja, R.; Andolina, J.; et al. Infections in Infants with SCID: Isolation, Infection Screening, and Prophylaxis in PIDTC Centers. J. Clin. Immunol. 2021, 41, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Bergerson, J.R.E.; Freeman, A.F. An Update on Syndromes with a Hyper-IgE Phenotype. Immunol. Allergy Clin. N. Am. 2019, 39, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, P.; Freeman, A.F. STAT1 and STAT3 mutations: Important lessons for clinical immunologists. Expert Rev. Clin. Immunol. 2018, 14, 1029–1041. [Google Scholar] [CrossRef]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Renner, E.D.; Rylaarsdam, S.; Anover-Sombke, S.; Rack, A.L.; Reichenbach, J.; Carey, J.C.; Zhu, Q.; Jansson, A.F.; Barboza, J.; Schimke, L.F.; et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 2008, 122, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Aggor, F.E.Y.; Break, T.J.; Trevejo-Nuñez, G.; Whibley, N.; Coleman, B.M.; Bailey, R.D.; Kaplan, D.H.; Naglik, J.R.; Shan, W.; Shetty, A.C.; et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci. Immunol. 2020, 5, eaba0570. [Google Scholar] [CrossRef]
- Tsilifis, C.; Freeman, A.F.; Gennery, A.R. STAT3 Hyper-IgE Syndrome-an Update and Unanswered Questions. J. Clin. Immunol. 2021, 41, 864–880. [Google Scholar] [CrossRef]
- Yanagimachi, M.; Ohya, T.; Yokosuka, T.; Kajiwara, R.; Tanaka, F.; Goto, H.; Takashima, T.; Morio, T.; Yokota, S. The Potential and Limits of Hematopoietic Stem Cell Transplantation for the Treatment of Autosomal Dominant Hyper-IgE Syndrome. J. Clin. Immunol. 2016, 36, 511–516. [Google Scholar] [CrossRef]
- Harrison, S.C.; Tsilifis, C.; Slatter, M.A.; Nademi, Z.; Worth, A.; Veys, P.; Ponsford, M.J.; Jolles, S.; Al-Herz, W.; Flood, T.; et al. Hematopoietic Stem Cell Transplantation Resolves the Immune Deficit Associated with STAT3-Dominant-Negative Hyper-IgE Syndrome. J. Clin. Immunol. 2021, 41, 934–943. [Google Scholar] [CrossRef]
- Béziat, V.; Li, J.; Lin, J.X.; Ma, C.S.; Li, P.; Bousfiha, A.; Pellier, I.; Zoghi, S.; Baris, S.; Keles, S.; et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci. Immunol. 2018, 3, eaat4956. [Google Scholar] [CrossRef]
- Frey-Jakobs, S.; Hartberger, J.M.; Fliegauf, M.; Bossen, C.; Wehmeyer, M.L.; Neubauer, J.C.; Bulashevska, A.; Proietti, M.; Fröbel, P.; Nöltner, C.; et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci. Immunol. 2018, 3, eaat4941. [Google Scholar] [CrossRef] [PubMed]
- Sassi, A.; Lazaroski, S.; Wu, G.; Haslam, S.M.; Fliegauf, M.; Mellouli, F.; Patiroglu, T.; Unal, E.; Ozdemir, M.A.; Jouhadi, Z.; et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J. Allergy Clin. Immunol. 2014, 133, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.; Jalazo, E.R.; Evans, A.; Winstead, M.; Moran, T. A De Novo Cause of PGM3 Deficiency Treated with Hematopoietic Stem Cell Transplantation. J. Clin. Immunol. 2022, 42, 691–694. [Google Scholar] [CrossRef]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Constantine, G.M.; Lionakis, M.S. Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 2019, 287, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Pivin, M.; Hangan, T.; Yurkovskaya, O.; Pivina, L. Autoimmune polyendocrine syndrome type 1: Clinical manifestations, pathogenetic features, and management approach. Autoimmun. Rev. 2022, 21, 103135. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, Y.; Givony, T.; Kadouri, N.; Dobeš, J.; Peligero-Cruz, C.; Zalayat, I.; Damari, G.; Dassa, B.; Ben-Dor, S.; Gruper, Y.; et al. Mechanistic dissection of dominant AIRE mutations in mouse models reveals AIRE autoregulation. J. Exp. Med. 2021, 218, e20201076. [Google Scholar] [CrossRef]
- Puel, A.; Döffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachée-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef]
- Kisand, K.; Bøe Wolff, A.S.; Podkrajsek, K.T.; Tserel, L.; Link, M.; Kisand, K.V.; Ersvaer, E.; Perheentupa, J.; Erichsen, M.M.; Bratanic, N.; et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010, 207, 299–308. [Google Scholar] [CrossRef]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Marujo, F.; Pelham, S.J.; Freixo, J.; Cordeiro, A.I.; Martins, C.; Casanova, J.L.; Lei, W.T.; Puel, A.; Neves, J.F. A Novel TRAF3IP2 Mutation Causing Chronic Mucocutaneous Candidiasis. J. Clin. Immunol. 2021, 41, 1376–1379. [Google Scholar] [CrossRef] [PubMed]
- Herjan, T.; Hong, L.; Bubenik, J.; Bulek, K.; Qian, W.; Liu, C.; Li, X.; Chen, X.; Yang, H.; Ouyang, S.; et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat. Immunol. 2018, 19, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Blanco Lobo, P.; Lei, W.T.; Pelham, S.J.; Guisado Hernández, P.; Villaoslada, I.; de Felipe, B.; Lucena, J.M.; Casanova, J.L.; Olbrich, P.; Puel, A.; et al. Biallelic TRAF3IP2 variants causing chronic mucocutaneous candidiasis in a child harboring a STAT1 variant. Pediatr. Allergy Immunol. 2021, 32, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ritelli, M.; Ma, C.S.; Rao, G.; Habib, T.; Corvilain, E.; Bougarn, S.; Cypowyj, S.; Grodecká, L.; Lévy, R.; et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-β. Sci. Immunol. 2019, 4, eaax7965. [Google Scholar] [CrossRef]
- de Beaucoudrey, L.; Samarina, A.; Bustamante, J.; Cobat, A.; Boisson-Dupuis, S.; Feinberg, J.; Al-Muhsen, S.; Jannière, L.; Rose, Y.; de Suremain, M.; et al. Revisiting human IL-12Rβ1 deficiency: A survey of 141 patients from 30 countries. Medicine 2010, 89, 381–402. [Google Scholar] [CrossRef] [PubMed]
- Prando, C.; Samarina, A.; Bustamante, J.; Boisson-Dupuis, S.; Cobat, A.; Picard, C.; AlSum, Z.; Al-Jumaah, S.; Al-Hajjar, S.; Frayha, H.; et al. Inherited IL-12p40 deficiency: Genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine 2013, 92, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Al-Muhsen, S.; Halwani, R.; Ma, C.S.; et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 2015, 349, 606–613. [Google Scholar] [PubMed]
- Gross, O.; Gewies, A.; Finger, K.; Schäfer, M.; Sparwasser, T.; Peschel, C.; Förster, I.; Ruland, J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 2006, 442, 651–656. [Google Scholar] [CrossRef]
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638. [Google Scholar] [CrossRef]
- Glocker, E.O.; Hennigs, A.; Nabavi, M.; Schäffer, A.A.; Woellner, C.; Salzer, U.; Pfeifer, D.; Veelken, H.; Warnatz, K.; Tahami, F.; et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009, 361, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Gavino, C.; Cotter, A.; Lichtenstein, D.; Lejtenyi, D.; Fortin, C.; Legault, C.; Alirezaie, N.; Majewski, J.; Sheppard, D.C.; Behr, M.A.; et al. CARD9 deficiency and spontaneous central nervous system candidiasis: Complete clinical remission with GM-CSF therapy. Clin. Infect. Dis. 2014, 59, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Gavino, C.; Hamel, N.; Zeng, J.B.; Legault, C.; Guiot, M.C.; Chankowsky, J.; Lejtenyi, D.; Lemire, M.; Alarie, I.; Dufresne, S.; et al. Impaired RASGRF1/ERK-mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J. Allergy Clin. Immunol. 2016, 137, 1178–1188. [Google Scholar] [CrossRef]
- Li, J.; Vinh, D.C.; Casanova, J.L.; Puel, A. Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr. Opin. Microbiol. 2017, 40, 46–57. [Google Scholar] [CrossRef] [PubMed]
- van de Veerdonk, F.L.; Plantinga, T.S.; Hoischen, A.; Smeekens, S.P.; Joosten, L.A.; Gilissen, C.; Arts, P.; Rosentul, D.C.; Carmichael, A.J.; Smits-van der Graaf, C.A.; et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 2011, 365, 54–61. [Google Scholar] [CrossRef]
- Liu, L.; Okada, S.; Kong, X.F.; Kreins, A.Y.; Cypowyj, S.; Abhyankar, A.; Toubiana, J.; Itan, Y.; Audry, M.; Nitschke, P.; et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011, 208, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Depner, M.; Fuchs, S.; Raabe, J.; Frede, N.; Glocker, C.; Doffinger, R.; Gkrania-Klotsas, E.; Kumararatne, D.; Atkinson, T.P.; Schroeder, H.W., Jr.; et al. The Extended Clinical Phenotype of 26 Patients with Chronic Mucocutaneous Candidiasis due to Gain-of-Function Mutations in STAT1. J. Clin. Immunol. 2016, 36, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, J.; Okada, S.; Hiller, J.; Oleastro, M.; Lagos Gomez, M.; Aldave Becerra, J.C.; Ouachée-Chardin, M.; Fouyssac, F.; Girisha, K.M.; Etzioni, A.; et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016, 127, 3154–3164. [Google Scholar] [CrossRef]
- Frede, N.; Rojas-Restrepo, J.; Caballero Garcia de Oteyza, A.; Buchta, M.; Hübscher, K.; Gámez-Díaz, L.; Proietti, M.; Saghafi, S.; Chavoshzadeh, Z.; Soler-Palacin, P.; et al. Genetic Analysis of a Cohort of 275 Patients with Hyper-IgE Syndromes and/or Chronic Mucocutaneous Candidiasis. J. Clin. Immunol. 2021, 41, 1804–1838. [Google Scholar] [CrossRef]
- Zheng, J.; van de Veerdonk, F.L.; Crossland, K.L.; Smeekens, S.P.; Chan, C.M.; Al Shehri, T.; Abinun, M.; Gennery, A.R.; Mann, J.; Lendrem, D.W.; et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur. J. Immunol. 2015, 45, 2834–2846. [Google Scholar] [CrossRef]
- Hiller, J.; Hagl, B.; Effner, R.; Puel, A.; Schaller, M.; Mascher, B.; Eyerich, S.; Eyerich, K.; Jansson, A.F.; Ring, J.; et al. STAT1 Gain-of-Function and Dominant Negative STAT3 Mutations Impair IL-17 and IL-22 Immunity Associated with CMC. J. Investig. Dermatol. 2018, 138, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, O.; Olbrich, P.; Freeman, A.F.; Rosen, L.B.; Uzel, G.; Zerbe, C.S.; Rosenzweig, S.D.; Kuehn, H.S.; Holmes, K.L.; Stephany, D.; et al. STAT1 Gain-of-Function Mutations Cause High Total STAT1 Levels With Normal Dephosphorylation. Front. Immunol. 2019, 10, 1433. [Google Scholar] [CrossRef] [PubMed]
- Leiding, J.W.; Okada, S.; Hagin, D.; Abinun, M.; Shcherbina, A.; Balashov, D.N.; Kim, V.H.D.; Ovadia, A.; Guthery, S.L.; Pulsipher, M.; et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J. Allergy Clin. Immunol. 2018, 141, 704–717.e5. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, A.; Charbonnier, L.M.; Akcay, A.; Karakoc-Aydiner, E.; Ozen, A.; Ozturk, G.; Chatila, T.A.; Baris, S. Hematopoietic Stem Cell Transplantation in Patients with Heterozygous STAT1 Gain-of-Function Mutation. J. Clin. Immunol. 2019, 39, 37–44. [Google Scholar] [CrossRef]
- Baris, S.; Alroqi, F.; Kiykim, A.; Karakoc-Aydiner, E.; Ogulur, I.; Ozen, A.; Charbonnier, L.M.; Bakır, M.; Boztug, K.; Chatila, T.A.; et al. Severe Early-Onset Combined Immunodeficiency due to Heterozygous Gain-of-Function Mutations in STAT1. J. Clin. Immunol. 2016, 36, 641–648. [Google Scholar] [CrossRef]
- Weinacht, K.G.; Charbonnier, L.M.; Alroqi, F.; Plant, A.; Qiao, Q.; Wu, H.; Ma, C.; Torgerson, T.R.; Rosenzweig, S.D.; Fleisher, T.A.; et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J. Allergy Clin. Immunol. 2017, 139, 1629–1640.e2. [Google Scholar] [CrossRef]
- Bloomfield, M.; Kanderová, V.; Paračková, Z.; Vrabcová, P.; Svatoň, M.; Froňková, E.; Fejtková, M.; Zachová, R.; Rataj, M.; Zentsová, I.; et al. Utility of Ruxolitinib in a Child with Chronic Mucocutaneous Candidiasis Caused by a Novel STAT1 Gain-of-Function Mutation. J. Clin. Immunol. 2018, 38, 589–601. [Google Scholar] [CrossRef]
- Forbes, L.R.; Vogel, T.P.; Cooper, M.A.; Castro-Wagner, J.; Schussler, E.; Weinacht, K.G.; Plant, A.S.; Su, H.C.; Allenspach, E.J.; Slatter, M.; et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J. Allergy Clin. Immunol. 2018, 142, 1665–1669. [Google Scholar] [CrossRef]
- Borgström, E.W.; Edvinsson, M.; Pérez, L.P.; Norlin, A.C.; Enoksson, S.L.; Hansen, S.; Fasth, A.; Friman, V.; Kämpe, O.; Månsson, R.; et al. Three Adult Cases of STAT1 Gain-of-Function with Chronic Mucocutaneous Candidiasis Treated with JAK Inhibitors. J. Clin. Immunol. 2022. [Google Scholar] [CrossRef]
- Deyà-Martínez, A.; Rivière, J.G.; Roxo-Junior, P.; Ramakers, J.; Bloomfield, M.; Guisado Hernandez, P.; Blanco Lobo, P.; Abu Jamra, S.R.; Esteve-Sole, A.; Kanderova, V.; et al. Impact of JAK Inhibitors in Pediatric Patients with STAT1 Gain of Function (GOF) Mutations-10 Children and Review of the Literature. J. Clin. Immunol. 2022, 42, 1071–1082. [Google Scholar] [CrossRef]
- Immunodeficiencies EESf. Multicentric Retrospective Study on JAKinib Treatment of Patients with IEI of the JAK/STAT Pathway. Available online: https://esid.org/Working-Parties/Inborn-Errors-Working-Party-IEWP/Studies/Multicentric-retrospective-study-on-JAKinib-treatment-of-patients-with-IEI-of-the-JAK-STAT-pathway (accessed on 1 January 2020).
- Ling, Y.; Cypowyj, S.; Aytekin, C.; Galicchio, M.; Camcioglu, Y.; Nepesov, S.; Ikinciogullari, A.; Dogu, F.; Belkadi, A.; Levy, R.; et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 2015, 212, 619–631. [Google Scholar] [CrossRef]
- Desai, J.V.; Urban, A.; Swaim, D.Z.; Colton, B.; Kibathi, L.W.; Ferrè, E.M.N.; Stratton, P.; Merideth, M.A.; Hunsberger, S.; Matkovits, T.; et al. Efficacy of Cochleated Amphotericin B in Mouse and Human Mucocutaneous Candidiasis. Antimicrob. Agents Chemother. 2022, 66, e0030822. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Oftedal, B.E.; Landegren, N.; Erichsen, M.M.; Bratland, E.; Lima, K.; Jørgensen, A.P.; Myhre, A.G.; Svartberg, J.; Fougner, K.J.; et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J. Clin. Endocrinol. Metab. 2016, 101, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.; Kejariwal, D.; Al-Shehri, T.; Dhar, A.; Lilic, D. Oesophageal candidiasis and squamous cell cancer in patients with gain-of-function STAT1 gene mutation. United Eur. Gastroenterol. J. 2017, 5, 625–631. [Google Scholar] [CrossRef]
- Rotulo, G.A.; Plat, G.; Beaupain, B.; Blanche, S.; Moushous, D.; Sicre de Fontbrune, F.; Leblanc, T.; Renard, C.; Barlogis, V.; Vigue, M.G.; et al. Recurrent bacterial infections, but not fungal infections, characterise patients with ELANE-related neutropenia: A French Severe Chronic Neutropenia Registry study. Br. J. Haematol. 2021, 194, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.E.; Hickstein, D.D.; Bauer, T.R., Jr.; Calder, C.; Manes, B.; Frangoul, H. Matched unrelated bone marrow transplantation with reduced-intensity conditioning for leukocyte adhesion deficiency. Bone Marrow Transpl. 2006, 37, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Cline, M.J. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: The role of myeloperoxidase in resistance to Candida infection. J. Clin. Investig. 1969, 48, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Medicine 2000, 79, 155–169. [Google Scholar]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef]
- Kuhns, D.B.; Alvord, W.G.; Heller, T.; Feld, J.J.; Pike, K.M.; Marciano, B.E.; Uzel, G.; DeRavin, S.S.; Priel, D.A.; Soule, B.P.; et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 2010, 363, 2600–2610. [Google Scholar] [CrossRef]
- Levy, O.; Bourquin, J.P.; McQueen, A.; Cantor, A.B.; Lachenauer, C.; Malley, R. Fatal disseminated Candida lusitaniae infection in an infant with chronic granulomatous disease. Pediatr. Infect. Dis. J. 2002, 21, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Estrada, B.; Mancao, M.Y.; Polski, J.M.; Figarola, M.S. Candida lusitaniae and chronic granulomatous disease. Pediatr. Infect. Dis. J. 2006, 25, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Corvilain, E.; Casanova, J.L.; Puel, A. Inherited CARD9 Deficiency: Invasive Disease Caused by Ascomycete Fungi in Previously Healthy Children and Adults. J. Clin. Immunol. 2018, 38, 656–693. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Lionakis, M.S. Mechanistic Insights into the Role of C-Type Lectin Receptor/CARD9 Signaling in Human Antifungal Immunity. Front. Cell. Infect. Microbiol. 2016, 6, 39. [Google Scholar] [CrossRef]
- Lionakis, M.S. Primary immunodeficiencies and invasive fungal infection: When to suspect and how to diagnose and manage. Curr. Opin. Infect. Dis. 2019, 32, 531–537. [Google Scholar] [CrossRef]
- Drummond, R.A.; Collar, A.L.; Swamydas, M.; Rodriguez, C.A.; Lim, J.K.; Mendez, L.M.; Fink, D.L.; Hsu, A.P.; Zhai, B.; Karauzum, H.; et al. CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog. 2015, 11, e1005293. [Google Scholar] [CrossRef]
- Drummond, R.A.; Swamydas, M.; Oikonomou, V.; Zhai, B.; Dambuza, I.M.; Schaefer, B.C.; Bohrer, A.C.; Mayer-Barber, K.D.; Lira, S.A.; Iwakura, Y.; et al. CARD9(+) microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat. Immunol. 2019, 20, 559–570. [Google Scholar] [CrossRef]
- Drewniak, A.; Gazendam, R.P.; Tool, A.T.; van Houdt, M.; Jansen, M.H.; van Hamme, J.L.; van Leeuwen, E.M.; Roos, D.; Scalais, E.; de Beaufort, C.; et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 2013, 121, 2385–2392. [Google Scholar] [CrossRef]
- Goel, S.; Kuehn, H.S.; Chinen, J.; Niemela, J.; Stoddard, J.; Yamanaka, D.; Garofalo, M.; Samir, S.; Migaud, M.; Oikonomou, V.; et al. CARD9 Expression Pattern, Gene Dosage, and Immunodeficiency Phenotype Revisited. J. Clin. Immunol. 2022, 42, 336–349. [Google Scholar] [CrossRef]
- Queiroz-Telles, F.; Mercier, T.; Maertens, J.; Sola, C.B.S.; Bonfim, C.; Lortholary, O.; Constantino-Silva, R.M.N.; Schrijvers, R.; Hagen, F.; Meis, J.F.; et al. Successful Allogenic Stem Cell Transplantation in Patients with Inherited CARD9 Deficiency. J. Clin. Immunol. 2019, 39, 462–469. [Google Scholar] [CrossRef]
- Pana, Z.D.; Roilides, E.; Warris, A.; Groll, A.H.; Zaoutis, T. Epidemiology of Invasive Fungal Disease in Children. J. Pediatric. Infect. Dis. Soc. 2017, 6 (Suppl. 1), S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C.; Shea, Y.R.; Sugui, J.A.; Parrilla-Castellar, E.R.; Freeman, A.F.; Campbell, J.W.; Pittaluga, S.; Jones, P.A.; Zelazny, A.; Kleiner, D.; et al. Invasive aspergillosis due to Neosartorya udagawae. Clin. Infect. Dis. 2009, 49, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Seyedmousavi, S.; Lionakis, M.S.; Parta, M.; Peterson, S.W.; Kwon-Chung, K.J. Emerging Aspergillus Species Almost Exclusively Associated With Primary Immunodeficiencies. Open Forum Infect. Dis. 2018, 5, ofy213. [Google Scholar] [CrossRef]
- Rieber, N.; Gazendam, R.P.; Freeman, A.F.; Hsu, A.P.; Collar, A.L.; Sugui, J.A.; Drummond, R.A.; Rongkavilit, C.; Hoffman, K.; Henderson, C.; et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight 2016, 1, e89890. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, C.; Song, Y.; Ma, Y.; Wan, Z.; Zhu, X.; Wang, X.; Li, R. Primary Cutaneous Aspergillosis in a Patient with CARD9 Deficiency and Aspergillus Susceptibility of Card9 Knockout Mice. J. Clin. Immunol. 2021, 41, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.H.; DeCarlo, E.S.; Kwon-Chung, K.J.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Aspergillus nidulans infection in chronic granulomatous disease. Medicine 1998, 77, 345–354. [Google Scholar] [CrossRef]
- Lee, M.J.; Liu, H.; Barker, B.M.; Snarr, B.D.; Gravelat, F.N.; Al Abdallah, Q.; Gavino, C.; Baistrocchi, S.R.; Ostapska, H.; Xiao, T.; et al. The Fungal Exopolysaccharide Galactosaminogalactan Mediates Virulence by Enhancing Resistance to Neutrophil Extracellular Traps. PLoS Pathog. 2015, 11, e1005187. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Henriet, S.S.V.; Warris, A. Aspergillosis in Chronic Granulomatous Disease. J. Fungi 2016, 2, 15. [Google Scholar] [CrossRef]
- Gallin, J.I.; Alling, D.W.; Malech, H.L.; Wesley, R.; Koziol, D.; Marciano, B.; Eisenstein, E.M.; Turner, M.L.; DeCarlo, E.S.; Starling, J.M.; et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N. Engl. J. Med. 2003, 348, 2416–2422. [Google Scholar] [CrossRef]
- Lugo Reyes, S.O.; González Garay, A.; González Bobadilla, N.Y.; Rivera Lizárraga, D.A.; Madrigal Paz, A.C.; Medina-Torres, E.A.; Álvarez Cardona, A.; Galindo Ortega, J.L.; Solís Galicia, C.; Espinosa-Padilla, S.E.; et al. Efficacy and Safety of Interferon-Gamma in Chronic Granulomatous Disease: A Systematic Review and Meta-analysis. J. Clin. Immunol. 2022. [Google Scholar] [CrossRef]
- Cordero, E.; Goycochea-Valdivia, W.; Mendez-Echevarria, A.; Allende, L.M.; Alsina, L.; Bravo García-Morato, M.; Gil-Herrera, J.; Gudiol, C.; Len-Abad, O.; López-Medrano, F.; et al. Executive Summary of the Consensus Document on the Diagnosis and Management of Patients with Primary Immunodeficiencies. J. Allergy Clin. Immunol. Pract. 2020, 8, 3342–3347. [Google Scholar] [CrossRef] [PubMed]
- Verweij, P.E.; Weemaes, C.M.; Curfs, J.H.; Bretagne, S.; Meis, J.F. Failure to detect circulating Aspergillus markers in a patient with chronic granulomatous disease and invasive aspergillosis. J. Clin. Microbiol. 2000, 38, 3900–3901. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.L.; Knudsen, J.B.; Jensen-Fangel, S.; Stausbøl-Grøn, B.; Arendrup, M.C.; Petersen, E. Successful management of invasive aspergillosis presenting as pericarditis in an adult patient with chronic granulomatous disease. Mycoses 2011, 54, e233–e236. [Google Scholar] [CrossRef]
- Patterson, T.F.; Thompson, G.R., III; Denning, D.W.; Fishman, J.A.; Hadley, S.; Herbrecht, R.; Kontoyiannis, D.P.; Marr, K.A.; Morrison, V.A.; Nguyen, M.H.; et al. Practice Guidelines for the Diagnosis and Management of Aspergillosis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 63, e1–e60. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C.; Sugui, J.A.; Hsu, A.P.; Freeman, A.F.; Holland, S.M. Invasive fungal disease in autosomal-dominant hyper-IgE syndrome. J. Allergy Clin. Immunol. 2010, 125, 1389–1390. [Google Scholar] [CrossRef]
- Duréault, A.; Tcherakian, C.; Poiree, S.; Catherinot, E.; Danion, F.; Jouvion, G.; Bougnoux, M.E.; Mahlaoui, N.; Givel, C.; Castelle, M.; et al. Spectrum of Pulmonary Aspergillosis in Hyper-IgE Syndrome with Autosomal-Dominant STAT3 Deficiency. J. Allergy Clin. Immunol. Pract. 2019, 7, 1986–1995.e3. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, X.; Gao, G.; Xing, S.; Zhou, L.; Tang, X.; Zhao, X.; An, Y. Clinical Relevance of Gain- and Loss-of-Function Germline Mutations in STAT1: A Systematic Review. Front. Immunol. 2021, 12, 654406. [Google Scholar] [CrossRef]
- Vinh, D.C.; Patel, S.Y.; Uzel, G.; Anderson, V.L.; Freeman, A.F.; Olivier, K.N.; Spalding, C.; Hughes, S.; Pittaluga, S.; Raffeld, M.; et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 2010, 115, 1519–1529. [Google Scholar] [CrossRef]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Dickinson, R.E.; Griffin, H.; Bigley, V.; Reynard, L.N.; Hussain, R.; Haniffa, M.; Lakey, J.H.; Rahman, T.; Wang, X.N.; McGovern, N.; et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 2011, 118, 2656–2658. [Google Scholar] [CrossRef]
- Dickinson, R.E.; Milne, P.; Jardine, L.; Zandi, S.; Swierczek, S.I.; McGovern, N.; Cookson, S.; Ferozepurwalla, Z.; Langridge, A.; Pagan, S.; et al. The evolution of cellular deficiency in GATA2 mutation. Blood 2014, 123, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Dorn, J.M.; Patnaik, M.S.; Van Hee, M.; Smith, M.J.; Lagerstedt, S.A.; Newman, C.C.; Boyce, T.G.; Abraham, R.S. WILD syndrome is GATA2 deficiency: A novel deletion in the GATA2 gene. J. Allergy Clin. Immunol. Pract. 2017, 5, 1149–1152.e1. [Google Scholar] [CrossRef]
- Oleaga-Quintas, C.; de Oliveira-Júnior, E.B.; Rosain, J.; Rapaport, F.; Deswarte, C.; Guérin, A.; Sajjath, S.M.; Zhou, Y.J.; Marot, S.; Lozano, C.; et al. Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J. Clin. Immunol. 2021, 41, 639–657. [Google Scholar] [CrossRef] [PubMed]
- Hickstein, D.D.; Calvo, K.R. The Spectrum of GATA2 Deficiency Syndrome. Blood 2022. [Google Scholar] [CrossRef]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Olivier, K.N.; Folio, L.R.; Zerbe, C.S.; Hsu, A.P.; Freeman, A.F.; Filie, A.C.; Spinner, M.A.; Sanchez, L.A.; Lovell, J.P.; et al. Pulmonary Manifestations of GATA2 Deficiency. Chest 2021, 160, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Kuang, L.; Lv, G.; Wang, B.; Lian, Z. Vertebral aspergillosis in a patient with autosomal-dominant hyper-IgE syndrome. Clin. Vaccine Immunol. 2014, 21, 107–109. [Google Scholar] [CrossRef]
- Natarajan, M.; Hsu, A.P.; Weinreich, M.A.; Zhang, Y.; Niemela, J.E.; Butman, J.A.; Pittaluga, S.; Sugui, J.; Collar, A.L.; Lim, J.K.; et al. Aspergillosis, eosinophilic esophagitis, and allergic rhinitis in signal transducer and activator of transcription 3 haploinsufficiency. J. Allergy Clin. Immunol. 2018, 142, 993–997.e3. [Google Scholar] [CrossRef]
- Rosain, J.; Kong, X.F.; Martinez-Barricarte, R.; Oleaga-Quintas, C.; Ramirez-Alejo, N.; Markle, J.; Okada, S.; Boisson-Dupuis, S.; Casanova, J.L.; Bustamante, J. Mendelian susceptibility to mycobacterial disease: 2014–2018 update. Immunol. Cell. Biol. 2019, 97, 360–367. [Google Scholar] [CrossRef]
- Kerner, G.; Rosain, J.; Guérin, A.; Al-Khabaz, A.; Oleaga-Quintas, C.; Rapaport, F.; Massaad, M.J.; Ding, J.Y.; Khan, T.; Ali, F.A.; et al. Inherited human IFN-γ deficiency underlies mycobacterial disease. J. Clin. Investig. 2020, 130, 3158–3171. [Google Scholar] [CrossRef]
- Knight, V.; Heimall, J.R.; Chong, H.; Nandiwada, S.L.; Chen, K.; Lawrence, M.G.; Sadighi Akha, A.A.; Kumánovics, A.; Jyonouchi, S.; Ngo, S.Y.; et al. A Toolkit and Framework for Optimal Laboratory Evaluation of Individuals with Suspected Primary Immunodeficiency. J. Allergy Clin. Immunol. Pract. 2021, 9, 3293–3307.e6. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.; Boisson-Dupuis, S.; Abel, L.; Casanova, J.L. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 2014, 26, 454–470. [Google Scholar] [CrossRef] [PubMed]
- Zerbe, C.S.; Holland, S.M. Disseminated histoplasmosis in persons with interferon-gamma receptor 1 deficiency. Clin. Infect. Dis. 2005, 41, e38–e41. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C.; Masannat, F.; Dzioba, R.B.; Galgiani, J.N.; Holland, S.M. Refractory disseminated coccidioidomycosis and mycobacteriosis in interferon-gamma receptor 1 deficiency. Clin. Infect. Dis. 2009, 49, e62–e65. [Google Scholar] [CrossRef]
- Vinh, D.C.; Schwartz, B.; Hsu, A.P.; Miranda, D.J.; Valdez, P.A.; Fink, D.; Lau, K.P.; Long-Priel, D.; Kuhns, D.B.; Uzel, G.; et al. Interleukin-12 receptor β1 deficiency predisposing to disseminated Coccidioidomycosis. Clin. Infect. Dis. 2011, 52, e99–e102. [Google Scholar] [CrossRef]
- Moraes-Vasconcelos, D.; Grumach, A.S.; Yamaguti, A.; Andrade, M.E.; Fieschi, C.; de Beaucoudrey, L.; Casanova, J.L.; Duarte, A.J. Paracoccidioides brasiliensis disseminated disease in a patient with inherited deficiency in the beta1 subunit of the interleukin (IL)-12/IL-23 receptor. Clin. Infect. Dis. 2005, 41, e31–e37. [Google Scholar] [CrossRef]
- Rezai, M.S.; Khotael, G.; Kheirkhah, M.; Hedayat, T.; Geramishoar, M.; Mahjoub, F. Cryptococcosis and deficiency of interleukin12r. Pediatr. Infect. Dis. J. 2008, 27, 673. [Google Scholar] [CrossRef]
- Jirapongsananuruk, O.; Luangwedchakarn, V.; Niemela, J.E.; Pacharn, P.; Visitsunthorn, N.; Thepthai, C.; Vichyanond, P.; Piboonpocanun, S.; Fleisher, T.A. Cryptococcal osteomyelitis in a child with a novel compound mutation of the IL12RB1 gene. Asian Pac. J. Allergy Immunol. 2012, 30, 79–82. [Google Scholar]
- Dupuis, S.; Dargemont, C.; Fieschi, C.; Thomassin, N.; Rosenzweig, S.; Harris, J.; Holland, S.M.; Schreiber, R.D.; Casanova, J.L. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 2001, 293, 300–303. [Google Scholar] [CrossRef]
- Chapgier, A.; Boisson-Dupuis, S.; Jouanguy, E.; Vogt, G.; Feinberg, J.; Prochnicka-Chalufour, A.; Casrouge, A.; Yang, K.; Soudais, C.; Fieschi, C.; et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006, 2, e131. [Google Scholar] [CrossRef]
- Tsumura, M.; Okada, S.; Sakai, H.; Yasunaga, S.; Ohtsubo, M.; Murata, T.; Obata, H.; Yasumi, T.; Kong, X.F.; Abhyankar, A.; et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum. Mutat. 2012, 33, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Hsu, A.P.; Pechacek, J.; Bax, H.I.; Dias, D.L.; Paulson, M.L.; Chandrasekaran, P.; Rosen, L.B.; Carvalho, D.S.; Ding, L.; et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J. Allergy Clin. Immunol. 2013, 131, 1624–1634. [Google Scholar] [CrossRef]
- Nemoto, K.; Kawanami, T.; Hoshina, T.; Ishimura, M.; Yamasaki, K.; Okada, S.; Kanegane, H.; Yatera, K.; Kusuhara, K. Impaired B-Cell Differentiation in a Patient With STAT1 Gain-of-Function Mutation. Front. Immunol. 2020, 11, 557521. [Google Scholar] [CrossRef] [PubMed]
- Rottman, M.; Soudais, C.; Vogt, G.; Renia, L.; Emile, J.F.; Decaluwe, H.; Gaillard, J.L.; Casanova, J.L. IFN-gamma mediates the rejection of haematopoietic stem cells in IFN-gammaR1-deficient hosts. PLoS Med. 2008, 5, e26. [Google Scholar] [CrossRef] [PubMed]
- Roesler, J.; Horwitz, M.E.; Picard, C.; Bordigoni, P.; Davies, G.; Koscielniak, E.; Levin, M.; Veys, P.; Reuter, U.; Schulz, A.; et al. Hematopoietic stem cell transplantation for complete IFN-gamma receptor 1 deficiency: A multi-institutional survey. J. Pediatr. 2004, 145, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Donadieu, J.; Lamant, M.; Fieschi, C.; de Fontbrune, F.S.; Caye, A.; Ouachee, M.; Beaupain, B.; Bustamante, J.; Poirel, H.A.; Isidor, B.; et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 2018, 103, 1278–1287. [Google Scholar] [CrossRef]
- Hahn, C.N.; Chong, C.E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef]
- Ostergaard, P.; Simpson, M.A.; Connell, F.C.; Steward, C.G.; Brice, G.; Woollard, W.J.; Dafou, D.; Kilo, T.; Smithson, S.; Lunt, P.; et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 2011, 43, 929–931. [Google Scholar] [CrossRef]
- Pasquet, M.; Bellanné-Chantelot, C.; Tavitian, S.; Prade, N.; Beaupain, B.; Larochelle, O.; Petit, A.; Rohrlich, P.; Ferrand, C.; Van Den Neste, E.; et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood 2013, 121, 822–829. [Google Scholar] [CrossRef]
- Orange, J.S. How I Manage Natural Killer Cell Deficiency. J. Clin. Immunol. 2020, 40, 13–23. [Google Scholar] [CrossRef]
- Imran, T.; Cui, C. GATA2 transcription factor deficiency predisposing to severe disseminated coccidioidomycosis. In Proceedings of the Frontiers in Immunology Conference: 15th International Congress of Immunology, Milan, Italy, 22–27 August 2013. [Google Scholar]
- Spinner, M.A.; Ker, J.P.; Stoudenmire, C.J.; Fadare, O.; Mace, E.M.; Orange, J.S.; Hsu, A.P.; Holland, S.M. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J. Allergy Clin. Immunol. 2016, 137, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Cuellar-Rodriguez, J.; Gea-Banacloche, J.; Freeman, A.F.; Hsu, A.P.; Zerbe, C.S.; Calvo, K.R.; Wilder, J.; Kurlander, R.; Olivier, K.N.; Holland, S.M.; et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood 2011, 118, 3715–3720. [Google Scholar] [CrossRef] [PubMed]
- Grossman, J.; Cuellar-Rodriguez, J.; Gea-Banacloche, J.; Zerbe, C.; Calvo, K.; Hughes, T.; Hakim, F.; Cole, K.; Parta, M.; Freeman, A.; et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol. Blood Marrow Transpl. 2014, 20, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Hickstein, D. HSCT for GATA2 deficiency across the pond. Blood 2018, 131, 1272–1274. [Google Scholar] [CrossRef]
- Nichols-Vinueza, D.X.; Parta, M.; Shah, N.N.; Cuellar-Rodriguez, J.M.; Bauer, T.R.; West, R.R., Jr.; Hsu, A.P.; Calvo, K.R.; Steinberg, S.M.; Notarangelo, L.D.; et al. Donor source and post-transplantation cyclophosphamide influence outcome in allogeneic stem cell transplantation for GATA2 deficiency. Br. J. Haematol. 2022, 196, 169–178. [Google Scholar] [CrossRef]
- França, T.T.; Barreiros, L.A.; Al-Ramadi, B.K.; Ochs, H.D.; Cabral-Marques, O.; Condino-Neto, A. CD40 ligand deficiency: Treatment strategies and novel therapeutic perspectives. Expert Rev. Clin. Immunol. 2019, 15, 529–540. [Google Scholar] [CrossRef]
- Etzioni, A.; Ochs, H.D. The hyper IgM syndrome—An evolving story. Pediatr. Res. 2004, 56, 519–525. [Google Scholar] [CrossRef]
- Leven, E.A.; Maffucci, P.; Ochs, H.D.; Scholl, P.R.; Buckley, R.H.; Fuleihan, R.L.; Geha, R.S.; Cunningham, C.K.; Bonilla, F.A.; Conley, M.E.; et al. Hyper IgM Syndrome: A Report from the USIDNET Registry. J. Clin. Immunol. 2016, 36, 490–501. [Google Scholar] [CrossRef]
- Du, X.; Tang, W.; Chen, X.; Zeng, T.; Wang, Y.; Chen, Z.; Xu, T.; Zhou, L.; Tang, X.; An, Y.; et al. Clinical, genetic and immunological characteristics of 40 Chinese patients with CD40 ligand deficiency. Scand. J. Immunol. 2019, 90, e12798. [Google Scholar] [CrossRef]
- Winkelstein, J.A.; Marino, M.C.; Ochs, H.; Fuleihan, R.; Scholl, P.R.; Geha, R.; Stiehm, E.R.; Conley, M.E. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine 2003, 82, 373–384. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Arslanian, C.; Ramos, R.N.; Morato, M.; Schimke, L.; Soeiro Pereira, P.V.; Jancar, S.; Ferreira, J.F.; Weber, C.W.; Kuntze, G.; et al. Dendritic cells from X-linked hyper-IgM patients present impaired responses to Candida albicans and Paracoccidioides brasiliensis. J. Allergy Clin. Immunol. 2012, 129, 778–786. [Google Scholar] [CrossRef]
- Pedroza, L.A.; Guerrero, N.; Stray-Pedersen, A.; Tafur, C.; Macias, R.; Muñoz, G.; Akdemir, Z.C.; Jhangiani, S.N.; Watkin, L.B.; Chinn, I.K.; et al. First Case of CD40LG Deficiency in Ecuador, Diagnosed after Whole Exome Sequencing in a Patient with Severe Cutaneous Histoplasmosis. Front. Pediatr. 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.P.; Lao-Araya, M.; Yang, J.; Chan, K.W.; Ma, H.; Pei, L.C.; Kui, L.; Mao, H.; Yang, W.; Zhao, X.; et al. Application of Flow Cytometry in the Diagnostics Pipeline of Primary Immunodeficiencies Underlying Disseminated Talaromyces marneffei Infection in HIV-Negative Children. Front. Immunol. 2019, 10, 2189. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, H.S.; Lee, M.Y.; Iseki, M.; Lee, J.H.; Song, C.H.; Park, J.K.; Hwang, T.J.; Kook, H. X-linked hyper-IgM syndrome associated with Cryptosporidium parvum and Cryptococcus neoformans infections: The first case with molecular diagnosis in Korea. J. Korean. Med. Sci. 2002, 17, 116–120. [Google Scholar] [CrossRef]
- Suzuki, S.M.L.; Morelli, F.; Negri, M.; Bonfim-Mendonça, P.; Kioshima, É.S.; Salci, T.; Voidaleski, M.F.; Vicente, V.A.; Svidzinski, T. FATAL cryptococcal meningitis in a child with hyper-immunoglobulin M syndrome, with an emphasis on the agent. J. Mycol. Med. 2019, 29, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Athipongarporn, A.; Ittiwut, C.; Manuyakorn, W.; Assawawiroonhakarn, S.; Larbcharoensub, N.; Shotelersuk, V. Diagnosis of Hyper IgM syndrome in a Previously Healthy Adolescent Boy Presented with Cutaneous and Cerebral Cryptococcosis. Pediatr. Infect. Dis. J. 2021, 40, e18–e20. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Williamson, P.R.; Di Cesare, S.; Di Matteo, G.; De Luca, M.; Carsetti, R.; Figà-Talamanca, L.; Cancrini, C.; Rossi, P.; Finocchi, A. Cryptococcal Meningitis and Post-Infectious Inflammatory Response Syndrome in a Patient With X-Linked Hyper IgM Syndrome: A Case Report and Review of the Literature. Front. Immunol. 2021, 12, 708837. [Google Scholar] [CrossRef] [PubMed]
- Françoise, U.; Lafont, E.; Suarez, F.; Lanternier, F.; Lortholary, O. Disseminated Cryptococcosis in a Patient with CD40 Ligand Deficiency. J. Clin. Immunol. 2022, 42, 1622–1625. [Google Scholar] [CrossRef]
- Iseki, M.; Anzo, M.; Yamashita, N.; Matsuo, N. Hyper-IgM immunodeficiency with disseminated cryptococcosis. Acta. Paediatr. 1994, 83, 780–782. [Google Scholar] [CrossRef]
- Tabone, M.D.; Leverger, G.; Landman, J.; Aznar, C.; Boccon-Gibod, L.; Lasfargues, G. Disseminated lymphonodular cryptococcosis in a child with X-linked hyper-IgM immunodeficiency. Pediatr. Infect. Dis. J. 1994, 13, 77–79. [Google Scholar] [CrossRef]
- Odio, C.D.; Milligan, K.L.; McGowan, K.; Rudman Spergel, A.K.; Bishop, R.; Boris, L.; Urban, A.; Welch, P.; Heller, T.; Kleiner, D.; et al. Endemic mycoses in patients with STAT3-mutated hyper-IgE (Job) syndrome. J. Allergy Clin. Immunol. 2015, 136, 1411–1413.e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.P.; Chan, K.W.; Lee, T.L.; Ho, M.H.; Chen, X.Y.; Li, C.H.; Chu, K.M.; Zeng, H.S.; Lau, Y.L. Penicilliosis in children without HIV infection—Are they immunodeficient? Clin. Infect. Dis. 2012, 54, e8–e19. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.P.; Foruraghi, L.; Freeman, A.F.; Uzel, G.; Zerbe, C.S.; Su, H.; Hsu, A.P.; Holland, S.M. Persistent nodal histoplasmosis in nuclear factor kappa B essential modulator deficiency: Report of a case and review of infection in primary immunodeficiencies. J. Allergy Clin. Immunol. 2016, 138, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Krase, I.Z.; Woodward, J.; Bauer, C.S.; Miller, H.; Sacco, K. Seronegative Mediastinal Coccidioidomycosis as a Novel Presentation of CTPS1 Combined Immunodeficiency. Open Forum Infect. Dis. 2022, 9, ofac403. [Google Scholar] [CrossRef]
- Zanelli, G.; Sansoni, A.; Ricciardi, B.; Ciacci, C.; Cellesi, C. Muscular-skeletal cryptococcosis in a patient with idiopathic CD4+ lymphopenia. Mycopathologia 2001, 149, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Kortsik, C.; Elmer, A.; Tamm, I. Pleural effusion due to Histoplasma capsulatum and idiopathic CD4 lymphocytopenia. Respiration 2003, 70, 118–122. [Google Scholar] [CrossRef]
- Zonios, D.I.; Falloon, J.; Huang, C.Y.; Chaitt, D.; Bennett, J.E. Cryptococcosis and idiopathic CD4 lymphocytopenia. Medicine 2007, 86, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Yuanjie, Z.; Julin, G.; Fubing, C.; Jianghan, C. Recurrent pulmonary cryptococcosis in a patient with idiopathic CD4 lymphocytopenia. Med. Mycol. 2008, 46, 729–734. [Google Scholar] [CrossRef]
- Tsalik, E.L.; Jaggers, L.B. Life-threatening asymptomatic incidentaloma: A case report of idiopathic CD4 lymphocytopenia and opportunistic infections. Am. J. Med. Sci. 2010, 340, 158–159. [Google Scholar] [CrossRef]
- Sancesario, G.; Palmieri, G.; Viola, G.; Fontana, C.; Perfetti, A.; Anemona, L.; Floris, R.; Marziali, S.; Bernardi, G.; Spagnoli, L.G. Difficulty diagnosing chronic cryptococcal meningitis in idiopathic CD4+ lymphocytopenia. Neurol. Sci. 2011, 32, 519–524. [Google Scholar] [CrossRef]
- Dalal, P.; Chernin, L.; Swender, D.; Tcheurekdjian, H.; Hostoffer, R. Histoplasmosis in the olecranon bursa of a patient with idiopathic CD4 lymphocytopenia. Ann. Allergy Asthma Immunol. 2011, 107, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Marak, R.S.; Jain, S.; Dhole, T.N. Posterior fossa midline cryptococcoma in a patient with idiopathic CD4 lymphocytopenia. Indian, J. Med. Microbiol. 2012, 30, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.J.; Shen, H.; Xu, A.E. Cutaneous Penicillium marneffei infection in a patient with idiopathic CD4(+) lymphocytopenia. J. Dermatol. 2015, 42, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Hara, S.; Nishioka, H. Disseminated cryptococcosis with granuloma formation in idiopathic CD4 lymphocytopenia. J. Infect. Chemother. 2020, 26, 257–260. [Google Scholar] [CrossRef]
- Thornton, C.S.; Larios, O.; Grossman, J.; Griener, T.P.; Vaughan, S. Pulmonary Cryptococcus infections as a manifestation of idiopathic CD4 lymphocytopenia: Case report and literature review. BMC Infect. Dis. 2019, 19, 862. [Google Scholar] [CrossRef]
- Dewangan, A.; Singh, J.; Kumar, D.; Kumar, N.; Kumar, K.; Dinkar, A. Disseminated cryptococcosis in Idiopathic CD4+ lymphocytopenia. Infect. Disord. Drug Targets 2023, 23, 2–5. [Google Scholar]
- Lin, W.C.; Dai, Y.S.; Tsai, M.J.; Huang, L.M.; Chiang, B.L. Systemic Penicillium marneffei infection in a child with common variable immunodeficiency. J. Formos. Med. Assoc. 1998, 97, 780–783. [Google Scholar]
- Johnson, M.E.; Rojas-Moreno, C.; Salzer, W.; Regunath, H. Disseminated histoplasmosis in a patient with common variable immunodeficiency: A coincidence or the result of T cell defects? IDCases 2017, 10, 105–107. [Google Scholar] [CrossRef]
- Nazarian, R.M.; Lilly, E.; Gavino, C.; Hamilos, D.L.; Felsenstein, D.; Vinh, D.C.; Googe, P.B. Novel CARD9 mutation in a patient with chronic invasive dermatophyte infection (tinea profunda). J. Cutan. Pathol. 2020, 47, 166–170. [Google Scholar] [CrossRef]
- Zhan, P.; Dukik, K.; Li, D.; Sun, J.; Stielow, J.B.; Gerrits van den Ende, B.; Brankovics, B.; Menken, S.B.J.; Mei, H.; Bao, W.; et al. Phylogeny of dermatophytes with genomic character evaluation of clinically distinct Trichophyton rubrum and T. violaceum. Stud. Mycol. 2018, 89, 153–175. [Google Scholar] [CrossRef]
- Gupta, C.; Das, S.; Gaurav, V.; Singh, P.K.; Rai, G.; Datt, S.; Tigga, R.A.; Pandhi, D.; Bhattacharya, S.N.; Ansari, M.A.; et al. Review on host-pathogen interaction in dermatophyte infections. J. Mycol. Med. 2022, 33, 101331. [Google Scholar] [CrossRef] [PubMed]
- Boral, H.; Durdu, M.; Ilkit, M. Majocchi’s granuloma: Current perspectives. Infect. Drug Resist. 2018, 11, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Lanternier, F.; Pathan, S.; Vincent, Q.B.; Liu, L.; Cypowyj, S.; Prando, C.; Migaud, M.; Taibi, L.; Ammar-Khodja, A.; Stambouli, O.B.; et al. Deep dermatophytosis and inherited CARD9 deficiency. N. Engl. J. Med. 2013, 369, 1704–1714. [Google Scholar] [CrossRef]
- Jachiet, M.; Lanternier, F.; Rybojad, M.; Bagot, M.; Ibrahim, L.; Casanova, J.L.; Puel, A.; Bouaziz, J.D. Posaconazole treatment of extensive skin and nail dermatophytosis due to autosomal recessive deficiency of CARD9. JAMA Dermatol. 2015, 151, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Grumach, A.S.; de Queiroz-Telles, F.; Migaud, M.; Lanternier, F.; Filho, N.R.; Palma, S.M.; Constantino-Silva, R.N.; Casanova, J.L.; Puel, A. A homozygous CARD9 mutation in a Brazilian patient with deep dermatophytosis. J. Clin. Immunol. 2015, 35, 486–490. [Google Scholar] [CrossRef]
- Alves de Medeiros, A.K.; Lodewick, E.; Bogaert, D.J.; Haerynck, F.; Van Daele, S.; Lambrecht, B.; Bosma, S.; Vanderdonckt, L.; Lortholary, O.; Migaud, M.; et al. Chronic and Invasive Fungal Infections in a Family with CARD9 Deficiency. J. Clin. Immunol. 2016, 36, 204–209. [Google Scholar] [CrossRef]
- Boudghene Stambouli, O.; Amrani, N.; Boudghéne Stambouli, K.; Bouali, F. Dermatophytic disease with deficit in CARD9: A new case with a brain impairment. J. Mycol. Med. 2017, 27, 250–253. [Google Scholar] [CrossRef]
- Vaezi, A.; Fakhim, H.; Abtahian, Z.; Khodavaisy, S.; Geramishoar, M.; Alizadeh, A.; Meis, J.F.; Badali, H. Frequency and Geographic Distribution of CARD9 Mutations in Patients With Severe Fungal Infections. Front. Microbiol. 2018, 9, 2434. [Google Scholar] [CrossRef]
- Zhang, Y.; Mijiti, J.; Huang, C.; Song, Y.; Wan, Z.; Li, R.; Kang, X.; Wang, X. Deep dermatophytosis caused by Microsporum ferrugineum in a patient with CARD9 mutations. Br. J. Dermatol. 2019, 181, 1093–1095. [Google Scholar] [CrossRef]
- Huang, C.; Peng, Y.; Zhang, Y.; Li, R.; Wan, Z.; Wang, X. Deep dermatophytosis caused by Trichophyton rubrum. Lancet Infect. Dis. 2019, 19, 1380. [Google Scholar] [CrossRef]
- Wang, X.; Ding, H.; Chen, Z.; Zeng, X.; Sun, J.; Chen, H.; Fu, M. CARD9 Deficiency in a Chinese Man with Cutaneous Mucormycosis, Recurrent Deep Dermatophytosis and a Review of the Literature. Mycopathologia 2020, 185, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yu, Q.; Gao, Z.; Yang, H.; Chen, Q.; Yang, L. Case report: Severe deep ulcer on the left abdomen mimicking mycosis fungoides caused by Trichophyton tonsurans in a patient with novel CARD9 mutation. Front. Immunol. 2022, 13, 1015000. [Google Scholar] [CrossRef]
- Liang, P.; Wang, X.; Wang, R.; Wan, Z.; Han, W.; Li, R. CARD9 deficiencies linked to impaired neutrophil functions against Phialophora verrucosa. Mycopathologia 2015, 179, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Lin, Z.; Wang, X.; Li, T.; Yu, J.; Liu, W.; Tong, Z.; Xu, Y.; Zhang, J.; et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J. Allergy Clin. Immunol. 2014, 133, 905–908.e3. [Google Scholar] [CrossRef]
- Vinh, D.C.; Freeman, A.F.; Shea, Y.R.; Malech, H.L.; Abinun, M.; Weinberg, G.A.; Holland, S.M. Mucormycosis in chronic granulomatous disease: Association with iatrogenic immunosuppression. J. Allergy Clin. Immunol. 2009, 123, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Hanks, M.E.; Chandrasekaran, P.; Davis, B.C.; Hsu, A.P.; Van Wagoner, N.J.; Merlin, J.S.; Spalding, C.; La Hoz, R.M.; Holland, S.M.; et al. Gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation-related primary immunodeficiency is associated with disseminated mucormycosis. J. Allergy Clin. Immunol. 2014, 134, 236–239. [Google Scholar] [CrossRef]
- Wang, X.; Wang, A.; Wang, X.; Li, R.; Yu, J. Cutaneous mucormycosis caused by Mucor irregularis in a patient with CARD9 deficiency. Br. J. Dermatol. 2019, 180, 213–214. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, J.; Ma, Y.; Wan, Z.; Dai, H.; Li, R.; Gu, H.; Wang, X. Primary Cutaneous Mucormycosis, Candida Onychomycosis and Endophthalmitis in a Patient with CARD9 Mutation. Mycopathologia 2022, 187, 305–308. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Inborn Error of Immunity | Gene | Inheritance | Associated Fungal Diseases | Associated Clinical Features | Relevant Immunological Features | Comments |
|---|---|---|---|---|---|---|
| ACT1 deficiency | TRAF3IP2 | AR | CMC | May also get S. aureus skin and soft tissue infections | Impaired signaling of IL-17 receptor | |
| APECED | AIRE | AR | CMC | Endocrinopathies (typically, parathyroid, adrenal, and gonadal insufficiencies) | Auto-antibodies to cytokines, including IL-17 and IL-22 | Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; aka autoimmune polyglandular syndrome type 1 (APS1) |
| Autosomal dominant—Hyper-IgE syndrome | STAT3/Dominant-negative mutations | AD | CMC | Characteristic facial features. Musculoskeletal abnormalities (hyperextensibility; scoliosis). Decreased bone density/pathological fractures. Abnormal retention of primary teeth. Vascular anomalies. Ocular complications. Elevated serum IgE. “Cold” cutaneous S. aureus skin abscesses. | Impaired Th17 development | |
| Pulmonary aspergillosis | Pulmonary aspergillosis & aspergillomas typically occurs in established structural lung damage (e.g., pneumatoceles, cysts, cavities) | |||||
| Aspergillomas, ABPA | ||||||
| Extra-pulmonary aspergillosis | ||||||
| TDEF | ||||||
| Cryptococcus | ||||||
| CARD9 deficiency | CARD9 | AR | CMC | Fungal disease may be adult in onset | ||
| Invasive Candidiasis | Especially, spontaneous CNS candidiasis | |||||
| Extrapulmonary aspergillosis | ||||||
| Deep Dermatophytosis | aka Tinea Profunda | |||||
| Mucormycosis | ||||||
| Chronic Granulomatous Disease (CGD) | CYBB | XL | Invasive aspergillosis. May also get: Invasive candidiasis. Mucormycosis | Spontaneously occurring severe or recurrent bacterial infections of lung, liver, bone, skin, and/or lymph nodes. Granulomatous inflammation in gastrointestinal and/or genitourinary organs. Abnormal wound healing. Colitis, especially with fistulae and fissures. | Impaired phagocyte oxidative burst | Aspergillosis is the main fungal disease in CGD, primarily A. fumigatus complex and A. nidulans complex |
| CYBA | AR | |||||
| CYBC1 | AR | Mucormycosis especially in the context of recent steroid use | ||||
| NCF1 | AR | |||||
| NCF2 | AR | |||||
| NCF4 | AR | |||||
| CD40LG deficiency | CD40LG | XL | TDEF | Elevated or normal serum IgM. Decreased IgG, IgA, and IgE. Combined T and B immunodeficiency. | Impaired CD40L-CD40 interactions, leading to impaired costimulatory signal for T activation | X-linked Hyper-IgM syndrome (X-HIGM) |
| Cryptococcus | ||||||
| GATA2 deficiency | GATA2 | AD | TDEF | Some features may be adult-onset. Susceptibility to mycobacterial and viral (esp. HPV) infections. May develop Pulmonary alveolar proteinosis. | Monocytopenia, B lymphopenia, NK lymphopenia. Variable T cell dysfunction. | Causes: MonoMAC, DCML, familial acute myeloid leukemia, Emberger syndrome, WILD syndrome, chronic neutropenia, and/or classical NK cell deficiency |
| Pulmonary aspergillosis | Pulmonar aspergillosis especially in context of pulmonary alveolar proteinosis | |||||
| Cryptococcus | ||||||
| Mucormycosis | ||||||
| IL17F deficiency | IL17F | AD | CMC | May also get S. aureus skin and soft tissue infections | Impaired IL-17 function | |
| IL17RA deficiency | IL17RA | AR | CMC | May also get S. aureus skin and soft tissue infections | Impaired IL-17 function | |
| IL17RC deficiency | IL17RC | AR | CMC | Impaired IL-17 function | Isolated CMC | |
| JNK1 deficiency | JNK1 | AD | May also get S. aureus skin and soft tissue infections | Impaired IL-17 and TGFβ1 signaling pathway | Features of Ehlers-Danlos like connective tissue disorder | |
| MSMD | Mendelian Susceptibility to Mycobacterial Disease | |||||
| IL12B | AR | CMC | Susceptibility to Salmonella, Mycobacteria | Defect in production or response to IFN-γ | ||
| IL12RB1 | AR | CMC | Susceptibility to Salmonella, Mycobacteria | Defect in production or response to IFN-γ | ||
| TDEF | Defect in production or response to IFN-γ | |||||
| Cryptococcus | Defect in production or response to IFN-γ | |||||
| IL12RB2 | AR | Susceptibility to Salmonella, Mycobacteria | Defect in production or response to IFN-γ | |||
| IL23R | AR | Susceptibility to Salmonella, Mycobacteria | Defect in production or response to IFN-γ | |||
| TYK2 | AR | Susceptibility to Mycobacteria.Isolated and syndromic forms exist. Syndromic form may have elevated IgE and susceptibility to viral diseases | Defect in production or response to IFN-γ | |||
| RORC | AR | CMC | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | ||
| IFNG | AR | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | |||
| IFNGR1 | AD or AR | TDEF | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | ||
| IFNGR2 | AD or AR | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | |||
| JAK1 | AR | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | Impaired response to type I IFN | ||
| STAT1/LOF/Negative dominance | AD | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | Impaired response to type I IFN | ||
| STAT1/LOF | AR | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | Impaired response to type I IFN | ||
| IRF8 | AD | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | |||
| SPPL2a | AR | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | |||
| ISG15 | AR | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | |||
| CYBB | XL | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | Defect in macrophage oxidative burst | ||
| IKBKG | XL | Susceptibility to Mycobacteria | Defect in production or response to IFN-γ | |||
| (Severe) Combined Immunodeficiency | IL2RG | XL | CMC | T- B+ NK- SCID | ||
| JAK3 | AR | CMC | ||||
| PTPRC | AR | CMC | aka CD45 deficiency | |||
| CD3D | AR | CMC | ||||
| CD3E | AR | CMC | ||||
| CD3Z | AR | CMC | ||||
| LAT | AR | CMC | ||||
| IL7RA | AR | CMC | T- B+ NK+ SCID | |||
| CORO1A | AR | CMC | ||||
| RMRP | AR | CMC | SCID with skeletal dysplasia, short stature, thin/sparse hair growth, neuronal colonic dysplasia (Hirschsprung-like anomaly), increased risk of malignancy | |||
| ADA | AR | CMC | T- B- NK- | |||
| AK2 | CMC | |||||
| RAG1/2 | AR | CMC | T- B- NK+ | |||
| DCLRE1C | AR | CMC | ||||
| RAC2 | AD/GOF | CMC | ||||
| NHEJ1 | AR | CMC | SCID with microcephaly, growth retardation | T- B- NK+ | aka Cernunnos-XLF deficiency | |
| LIG4 | AR | CMC | ||||
| PRKDC | AR | CMC | ||||
| STAT1 GOF | STAT1/GOF | AD | CMC | Molecular gain of phosphorylation, with cellular loss of response to IFN restimulation due to prolonged phosphorylation. Impaired Th17 response | CMC may improve with JAK inhibitors | |
| TDEF | ||||||
| Cryptococcus | ||||||
| Mucomycosis | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olbrich, P.; Vinh, D.C. Inborn Errors of Immunity Causing Pediatric Susceptibility to Fungal Diseases. J. Fungi 2023, 9, 149. https://doi.org/10.3390/jof9020149
Olbrich P, Vinh DC. Inborn Errors of Immunity Causing Pediatric Susceptibility to Fungal Diseases. Journal of Fungi. 2023; 9(2):149. https://doi.org/10.3390/jof9020149
Chicago/Turabian StyleOlbrich, Peter, and Donald C. Vinh. 2023. "Inborn Errors of Immunity Causing Pediatric Susceptibility to Fungal Diseases" Journal of Fungi 9, no. 2: 149. https://doi.org/10.3390/jof9020149
APA StyleOlbrich, P., & Vinh, D. C. (2023). Inborn Errors of Immunity Causing Pediatric Susceptibility to Fungal Diseases. Journal of Fungi, 9(2), 149. https://doi.org/10.3390/jof9020149