


Metabolomics Analysis of Sporulation-Associated Metabolites of Metarhizium anisopliae Based on Gas Chromatography–Mass Spectrometry

Abstract
1. Introduction
2. Materials and Methods
2.1. Fungi Materials
2.2. Metabolome Analysis
2.3. Effects of Different Amino Acids on Sporulation
2.4. Data Analysis
3. Results
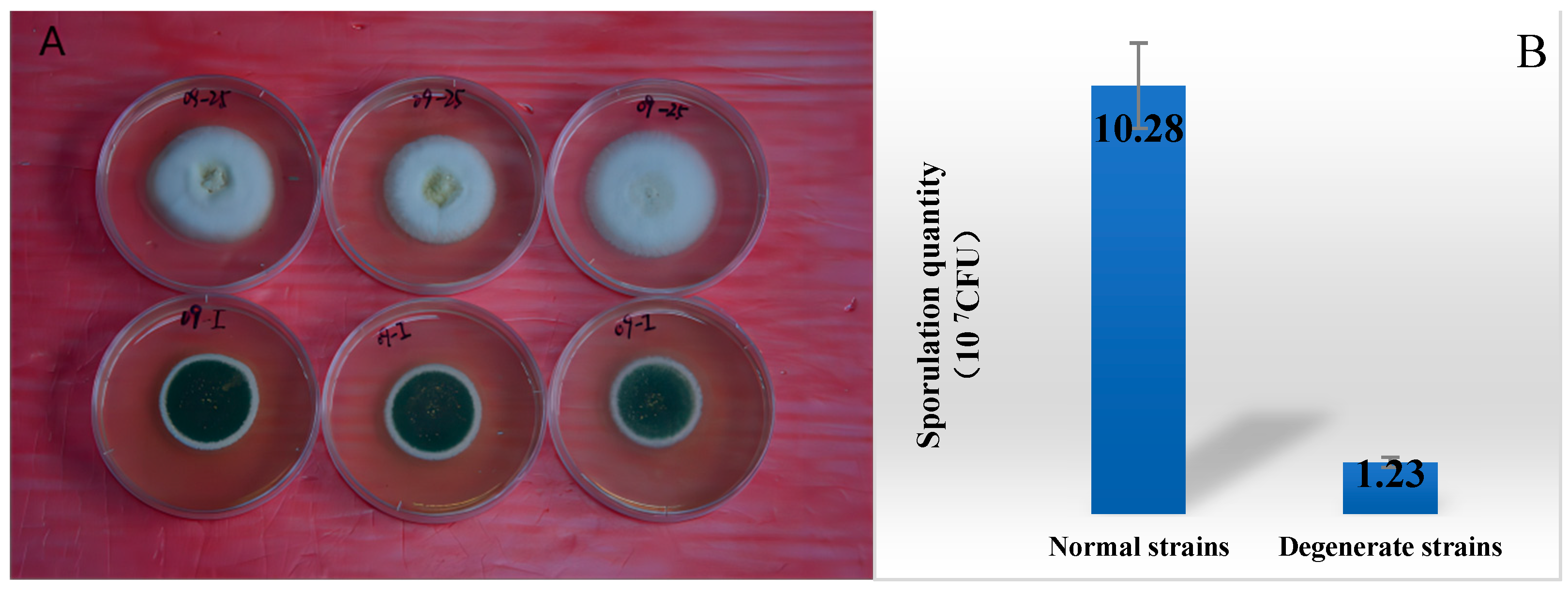
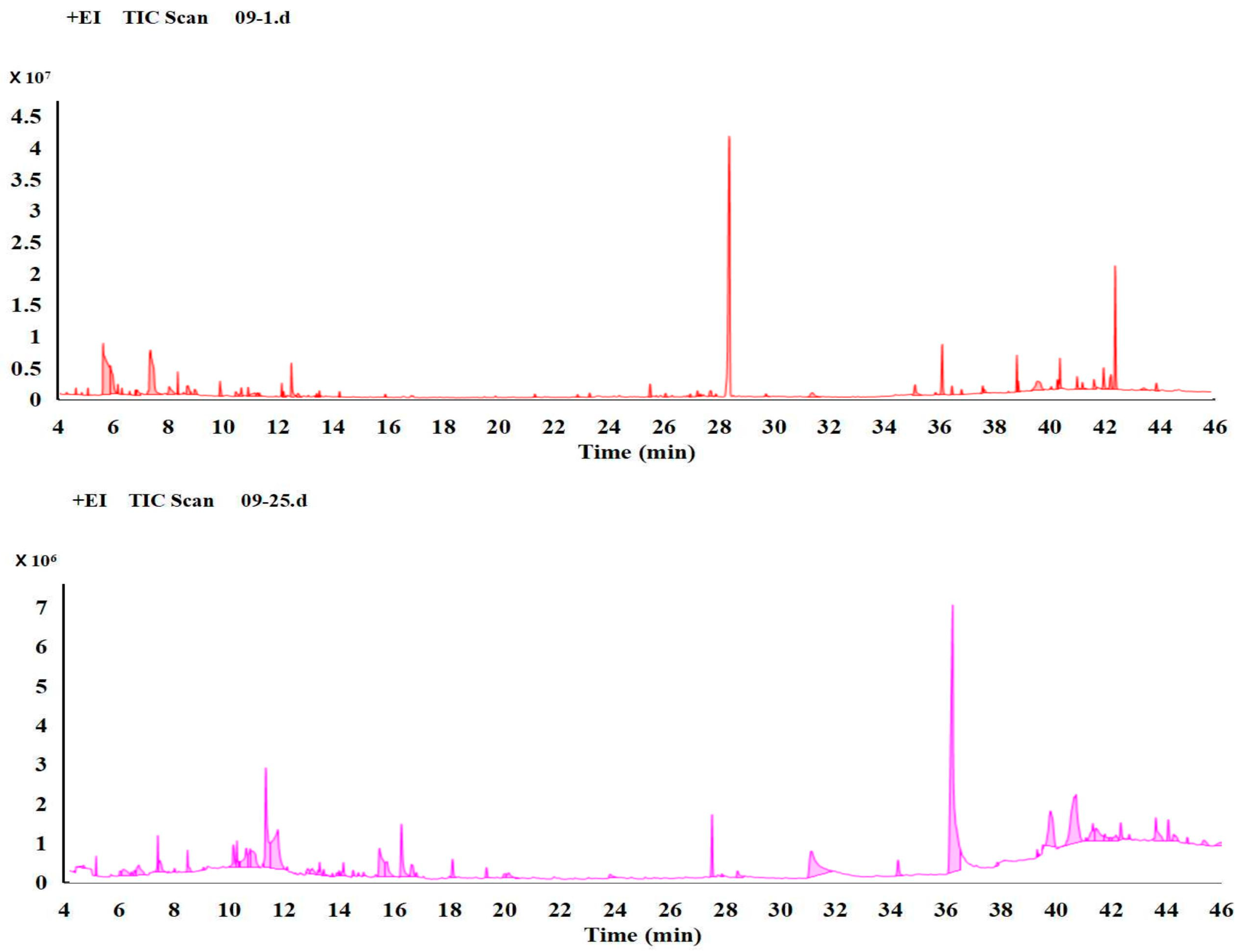
3.1. Comparison of the Metabolism Difference of Ma09
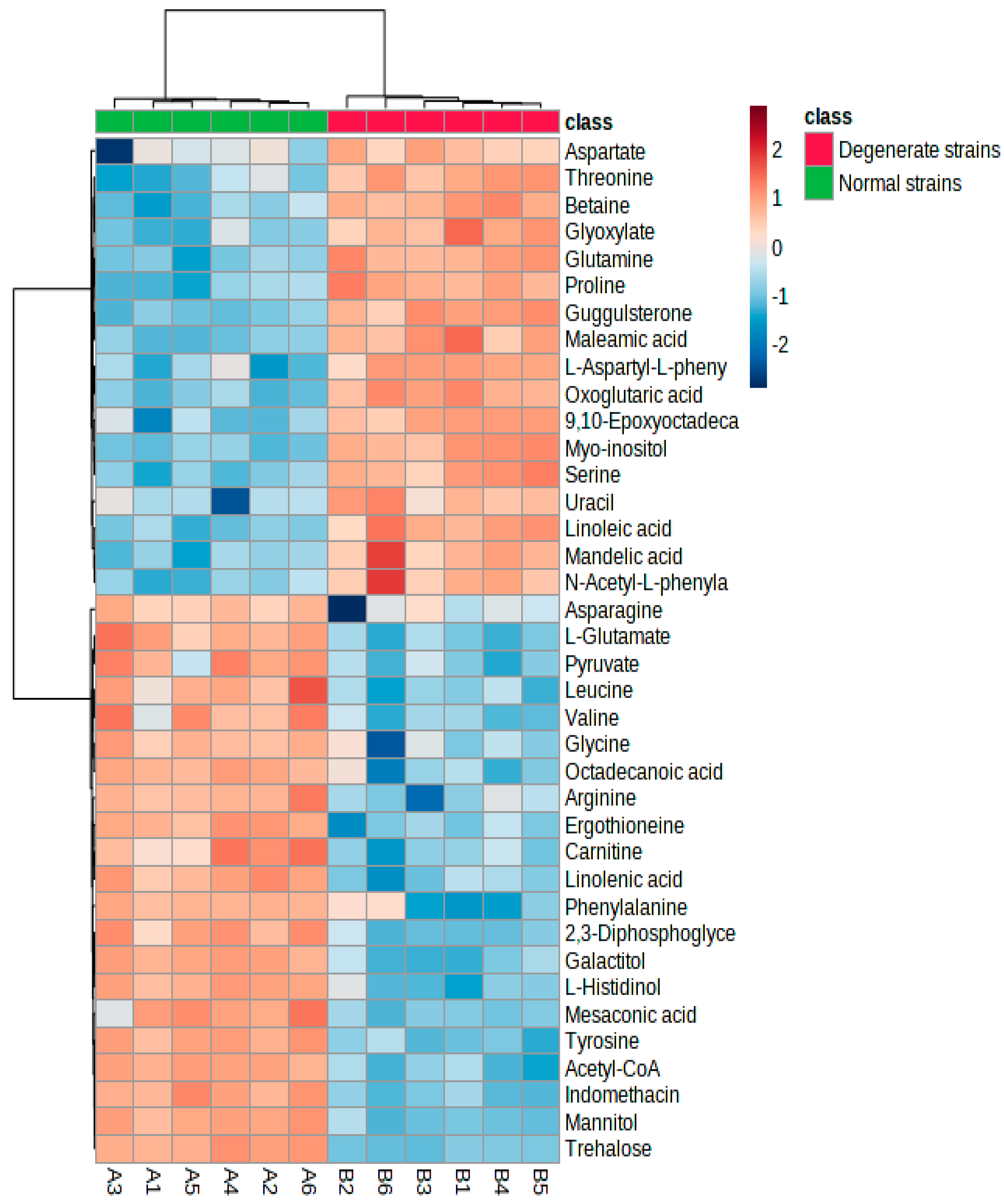
3.2. Multivariate Analysis of the Differential Metabolites
3.3. Pathway Analysis
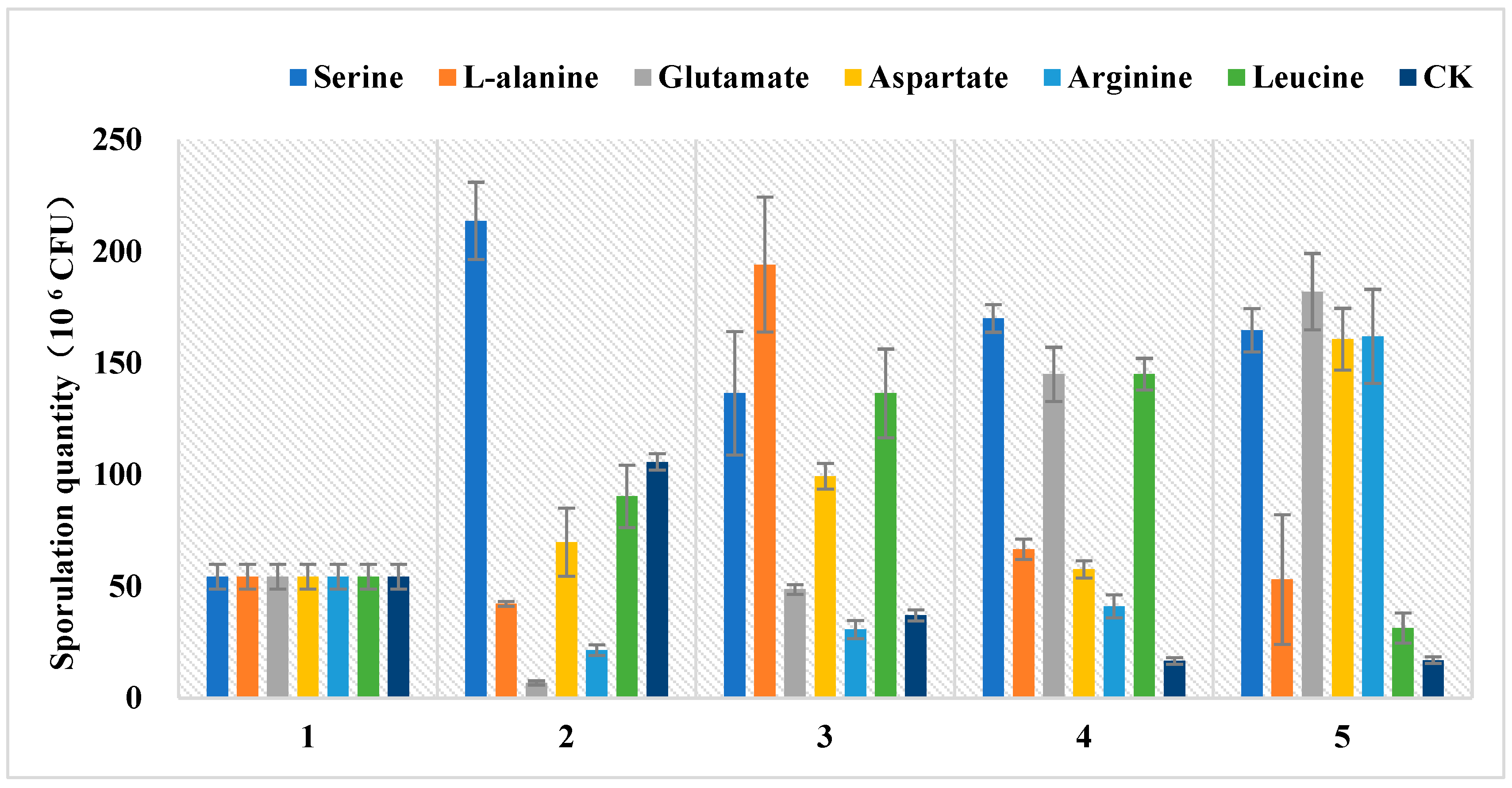
3.4. Amino Acids Have an Effect on Sporulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, H.; Qin, C.S.; Chen, Y.M.; Zhang, G.Y.; Dong, L.H.; Wan, S.Q. Persistence of Metarhizium (Hypocreales: Clavicipitaceae) and Beauveria bassiana (Hypocreales: Clavicipitaceae) in Tobacco Soils and Potential as Biocontrol Agents of Spodoptera litura (Lepidoptera: Noctuidae). Environ. Entomol. 2019, 48, 147–155. [Google Scholar] [CrossRef]
- Pan, X.Y.; Zhang, F. Advances in biological control of the German cockroach, Blattella germanica (L.). Biol. Control 2020, 142, 104104. [Google Scholar]
- Arthurs, S.; Dara, S.K. Microbial biopesticides for invertebrate pests and their markets in the United States. J. Invertebr. Pathol. 2019, 165, 13–21. [Google Scholar] [CrossRef]
- Méndez-González, F.; Loera, O.; Saucedo-Castañeda, G.; Favela-Torres, E. Forced aeration promotes high production and productivity of infective conidia from Metarhizium robertsii in solid-state fermentation. Biochem. Eng. J. 2020, 156, 107492. [Google Scholar] [CrossRef]
- Brownbridge, M.; Costa, S.; Jaronski, S.T. Effects of in Vitro Passage of Beauveria bassiana on Virulence to Bemisia argentifolii. J. Invertebr. Pathol. 2001, 77, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Altre, J.A.; Vandenberg, J.D.; Cantone, F.A. Pathogenicity of Paecilomyces fumosoroseus isolates to diamondback moth, Plutella xylostella: Correlation with spore size, germination speed, and attachment to cuticle. J. Invertebr. Pathol. 1999, 73, 332–338. [Google Scholar] [CrossRef]
- Leland, J.E.; McGuire, M.R.; Grace, J.A.; Jaronski, S.T.; Ulloa, M.; Park, Y.H.; Plattner, R.D. Strain selection of a fungal entomopathogen, Beauveria bassiana, for control of plant bugs (Lugus spp.). Biol. Control 2005, 35, 104–114. [Google Scholar] [CrossRef]
- Safavi, S.A.; Shah, F.A.; Pakdel, A.K.; Reza Rasoulian, G.; Bandani, A.R.; Butt, T.M. Effect of nutrition on growth and virulence of the entomopathogenic fungus Beauveria bassiana. FEMS Microbiol. Lett. 2007, 270, 116–123. [Google Scholar] [CrossRef]
- Xiong, C.; Xia, Y.; Zheng, P.; Wang, C. Increasing oxidative stress tolerance and subculturing stability of Cordyceps militaris by overexpression of a glutathione peroxidase gene. Appl. Microbiol. Biotechnol. 2013, 97, 2009–2015. [Google Scholar] [CrossRef]
- Ryan, M.J.; Bridge, P.D.; Smith, D.; Jeffries, P. Phenotypic degeneration occurs during sector formation in Metarhizium anisopliae. J. Appl. Microbiol. 2002, 93, 163–168. [Google Scholar] [CrossRef]
- Xin, X.; Yin, J.; Zhang, B.; Li, Z.; Zhao, S.; Gui, Z. Genome-wide analysis of DNA methylation in subcultured Cordyceps militaris. Arch. Microbiol. 2019, 201, 369–375. [Google Scholar] [PubMed]
- Zhu, W.; Hu, J.; Chi, J.; Li, Y.; Yang, B.; Hu, W.; Bao, Y. Label-Free Proteomics Reveals the Molecular Mechanism of Subculture Induced Strain Degeneration and Discovery of Indicative Index for Degeneration in Pleurotus ostreatus. Molecules 2020, 25, 4920. [Google Scholar] [PubMed]
- Mashego, M.R.; Jansen, M.L.; Vinke, J.L.; van Gulik, W.M.; Heijnen, J.J. Changes in the metabolome of Saccharomyces cerevisiae associated with evolution in aerobic glucose-limited chemostats. FEMS Yeast Res. 2005, 5, 419–430. [Google Scholar] [PubMed]
- Karpe, A.V.; Beale, D.J.; Morrison, P.D.; Harding, I.H.; Palombo, E.A. Untargeted metabolic profiling of Vitis vinifera during fungal degradation. FEMS Microbiol. Lett. 2015, 362, fnv060. [Google Scholar]
- Eshelli, M.; Harvey, L.; Edrada-Ebel, R.; McNeil, B. Metabolomics of the bio-degradation process of aflatoxin B1 by actinomycetes at an initial pH of 6.0. Toxins 2015, 7, 439–456. [Google Scholar]
- De Bekker, C.; Smith, P.B.; Patterson, A.D.; Hughes, D.P. Metabolomics reveals the heterogeneous secretome of two entomopathogenic fungi to ex vivo cultured insect tissues. PLoS ONE 2013, 8, e70609. [Google Scholar]
- Park, S.J.; Hyun, S.; Suh, H.W.; Lee, S.; Sung, G.; Kim, S.H.; Choi, H. Biochemical characterization of cultivated Cordyceps bassiana mycelia and fruiting bodies by 1H nuclear magnetic resonance spectroscopy. Metabolomics 2013, 9, 236–246. [Google Scholar]
- Mi, Y.L.; Li, Y.; Qian, G.T.; Vanhaelewyn, L.; Meng, X.X.; Liu, T.X.; Yang, W.; Shi, Y.H.; Ma, P.D.; Tul, W.A.; et al. A transcriptional complex of FtMYB102 and FtbHLH4 coordinately regulates the accumulation of rutin in Fagopyrum tataricum. Plant Physiol. Biochem. 2023, 194, 696–707. [Google Scholar]
- Zeng, L.; Fu, Y.Q.; Yu, Y.L.; Jin, S.H.; Jian, X.C.; Jun, F.Y.; Shan, J.; Wei, J.S.; Yong, Q.X. Comparative analysis of different grades of Tieguanyin oolong tea based on metabolomics and sensory evaluation. LWT 2023, 174, 114423. [Google Scholar]
- Yu, Q.Q.; Xiong, Y.; Su, X.L.; Xiong, Y.L.; Dong, Z.X.; Zhao, J.M.; Shu, X.; Bai, S.Q.; Lei, X.; Yan, L.J.; et al. Comparative Mmetabolomic studies of siberian wildrye (Elymus sibiricus L.): A new look at the mechanism of plant drought resistance. Int. J. Mol. Sci. 2022, 24, 452. [Google Scholar]
- Li, Z.; Wang, H.; Feng, L.; Song, L.; Lu, Y.; Li, H.; Li, Y.; Tian, G.; Yang, Y.; Li, H.; et al. Comparative metabolomics provides novel insights into correlation between dominant habitat factors and constituents of Stellaria Radix (Stellaria dichotoma L. var. lanceolata Bge.). Front. Plant Sci. 2022, 13, 1035712. [Google Scholar]
- Yang, H.; Xu, J.Z.; Zhao, D.Y.; Qiu, H.L.; Tian, L.Y.; Qin, C.S. Study on sample preparation for the metabolomics of Metarhizium based on GC-MS. J. South China Agric. Univ. 2021, 42, 69–79. (In Chinese) [Google Scholar]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; Auria, J.D.; Ewald, J.; Ewald, C.J.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Xia, J. MetaboAnalyst 4.0_towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [PubMed]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [PubMed]
- Li, P.; Yin, Y.L.; Li, D.; Kim, S.W.; Wu, G. Amino acids and immune function. Br. J. Nutr. 2007, 98, 237–252. [Google Scholar]
- Gong, X.; Fu, Y.; Jiang, D.; Li, G.; Yi, X.; Peng, Y. L-arginine is essential for conidiation in the filamentous fungus Coniothyrium minitans. Fungal Genet. Biol. 2007, 44, 1368–1379. [Google Scholar]
- Faiza, B.; Halima, Z.; Eddine, K.N. Physiological responses of salt stress and osmoprotection with proline in two strains of lactococci isolated from camel s milk in Southern Algeria. Afr. J. Biotechnol. 2011, 83, 19429–19435. [Google Scholar]
- Liu, Y.; Wang, L.; Chen, H.Z. Two-steps gas double-dynamic solid-state fermentation enhances growth and spore production of Conithyrium minitans. Bioresour. Technol. 2018, 262, 235–241. [Google Scholar]
- Erb-Downward, J.R.; Huffnagle, G.B. Role of oxylipins and other lipid mediators in fungal pathogenesis. Future Microbiol. 2006, 1, 219–227. [Google Scholar]
- Shea, J.M.; Del Poeta, M. Lipid signaling in pathogenic fungi. Curr. Opin. Microbiol. 2006, 9, 352–358. [Google Scholar] [PubMed]
- Tsitsigiannis, D.I.; Keller, N.P. Oxylipins as developmental and host-fungal communication signals. Trends Microbiol. 2007, 15, 109–118. [Google Scholar] [PubMed]
- Reverberi, M.; Gazzetti, K.; Punelli, F.; Scarpari, M.; Zjalic, S.; Ricelli, A.; Fanelli, C. Aoyap1 regulates OTA synthesis by controlling cell redox balance in Aspergillus ochraceus. Appl. Microbiol. Biotechnol. 2012, 95, 1293–1304. [Google Scholar]
- Mehta, R.J.; Fare, L.R.; Shearer, M.E.; Nash, C.H. Mannitol oxidation in two Micromonospora isolates and in representative species of other actinomycetes. Appl. Environ. Microbiol. 1977, 33, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Gummer, P.A.; Trengove, R.D.; Oliver, R.P.; Solomon, P.S. Dissecting the role of G-protein signalling in primary metabolism in the wheat pathogen Stagonospora nodorum. Microbiology 2013, 159, 1972–1985. [Google Scholar] [CrossRef] [PubMed]
- Krahulec, S.; Armao, G.C.; Klimacek, M.; Nidetzky, B. Enzymes of mannitol metabolism in the human pathogenic fungus Aspergillus fumigatus--kinetic properties of mannitol-1-phosphate 5-dehydrogenase and mannitol 2-dehydrogenase, and their physiological implications. FEBS J. 2011, 278, 1264–1276. [Google Scholar] [PubMed]
- Zhao, X.; Yu, C.; Zhao, Y.; Liu, S.; Wang, H.; Wang, C.; Guo, L.; Chen, M. Changes in Mannitol Content, Regulation of Genes Involved in Mannitol Metabolism, and the Protective Effect of Mannitol on Volvariella volvacea at Low Temperaturs. Biomed. Res. Int. 2019, 2019, 1493721. [Google Scholar]
- Che-Othman, M.H.; Jacoby, R.P.; Millar, A.H.; Taylor, N.L. Wheat mitochondrial respiration shifts from the tricarboxylic acidcycle to the GABA shunt under salt stress. New Phytologist. 2020, 225, 1166–1180. [Google Scholar]
- Nicholson, J.K.; Lindon, J.C. Metabonomics. Nature 2008, 445, 1054–1056. [Google Scholar]
- Xu, Y.J.; Wang, C.S.; Ho, W.E.; Ong, C.N. Recent developments and applications of metabolomics in microbiological investigations. TrAC Trends Anal. Chem. 2014, 56, 37–48. [Google Scholar]
- Xu, F.; Zou, L.; Ong, C.N. Experiment-originated variations, and multi-peak and multi-origination phenomena in derivatization-based GC-MS metabolomics. TrAC Trends Anal. Chem. 2010, 29, 269–280. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | p-Value | VIP | Trend |
|---|---|---|---|
| Acetyl-CoA | 0 | 1.29195 | down |
| Aflatoxin G2 | 0 | 1.17275 | down |
| Ammonia | 0 | 1.31058 | down |
| Arginine | 0 | 1.19034 | down |
| Asparagine | 0.005 | 1.01498 | down |
| Aspartate | 0.001 | 1.10915 | up |
| Betaine | 0 | 1.28929 | up |
| Carnitine | 0 | 1.16839 | down |
| Ergothioneine | 0 | 1.28954 | down |
| Glutamate | 0 | 1.24028 | down |
| Glutamine | 0 | 1.28597 | up |
| 1-Acylglycerophosphoinositol | 0 | 1.24205 | up |
| Glycine | 0 | 1.20305 | down |
| Glyoxylate | 0 | 1.20335 | up |
| Guggulsterone | 0 | 1.24587 | up |
| Histidinol | 0 | 1.29683 | down |
| Leucine | 0.001 | 1.09971 | down |
| Linoleic acid | 0.002 | 1.07298 | up |
| Linolenic acid | 0 | 1.13966 | down |
| Maleamic acid | 0 | 1.18627 | up |
| Mandelic acid | 0.003 | 1.05333 | up |
| Mannitol | 0 | 1.28916 | down |
| Mesaconic acid | 0.001 | 1.09989 | down |
| Myo-inositol | 0 | 1.13666 | up |
| N-Acetyl-L-phenylalanine | 0.002 | 1.06872 | up |
| Octadecanoic acid | 0 | 1.27733 | down |
| 9,10-Epoxyoctadecatrienoic acid | 0 | 1.24609 | up |
| Indomethacin | 0.001 | 1.12263 | down |
| Oxoglutaric acid | 0 | 1.23002 | up |
| Phenylalanine | 0 | 1.26797 | down |
| 2,3-Diphosphoglyceric acid | 0 | 1.22615 | down |
| Proline | 0 | 1.28514 | up |
| Proline betaine | 0.001 | 1.13507 | down |
| Pyruvate | 0 | 1.14563 | down |
| Serine | 0 | 1.25741 | up |
| Threonine | 0 | 1.24688 | up |
| Trehalose | 0 | 1.30756 | down |
| Tyrosine | 0 | 1.27859 | down |
| Uracil | 0.001 | 1.13457 | up |
| Valine | 0 | 1.14533 | down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Tian, L.; Qiu, H.; Qin, C.; Ling, S.; Xu, J. Metabolomics Analysis of Sporulation-Associated Metabolites of Metarhizium anisopliae Based on Gas Chromatography–Mass Spectrometry. J. Fungi 2023, 9, 1011. https://doi.org/10.3390/jof9101011
Yang H, Tian L, Qiu H, Qin C, Ling S, Xu J. Metabolomics Analysis of Sporulation-Associated Metabolites of Metarhizium anisopliae Based on Gas Chromatography–Mass Spectrometry. Journal of Fungi. 2023; 9(10):1011. https://doi.org/10.3390/jof9101011
Chicago/Turabian StyleYang, Hua, Longyan Tian, Hualong Qiu, Changsheng Qin, Siquan Ling, and Jinzhu Xu. 2023. "Metabolomics Analysis of Sporulation-Associated Metabolites of Metarhizium anisopliae Based on Gas Chromatography–Mass Spectrometry" Journal of Fungi 9, no. 10: 1011. https://doi.org/10.3390/jof9101011
APA StyleYang, H., Tian, L., Qiu, H., Qin, C., Ling, S., & Xu, J. (2023). Metabolomics Analysis of Sporulation-Associated Metabolites of Metarhizium anisopliae Based on Gas Chromatography–Mass Spectrometry. Journal of Fungi, 9(10), 1011. https://doi.org/10.3390/jof9101011