
Development of a Method for Detecting and Estimating Moniliophthora roreri Spore Loads Based on Spore Traps and qPCR

 , and
, and
Abstract
1. Introduction
2. Material and Methods
2.1. Fungal Strains and Cultures
2.2. DNA Extractions
2.3. PCR Amplification
2.4. qPCR Optimization
2.5. qPCR Characterization and Validation
2.6. Spore Trap Device
3. Results
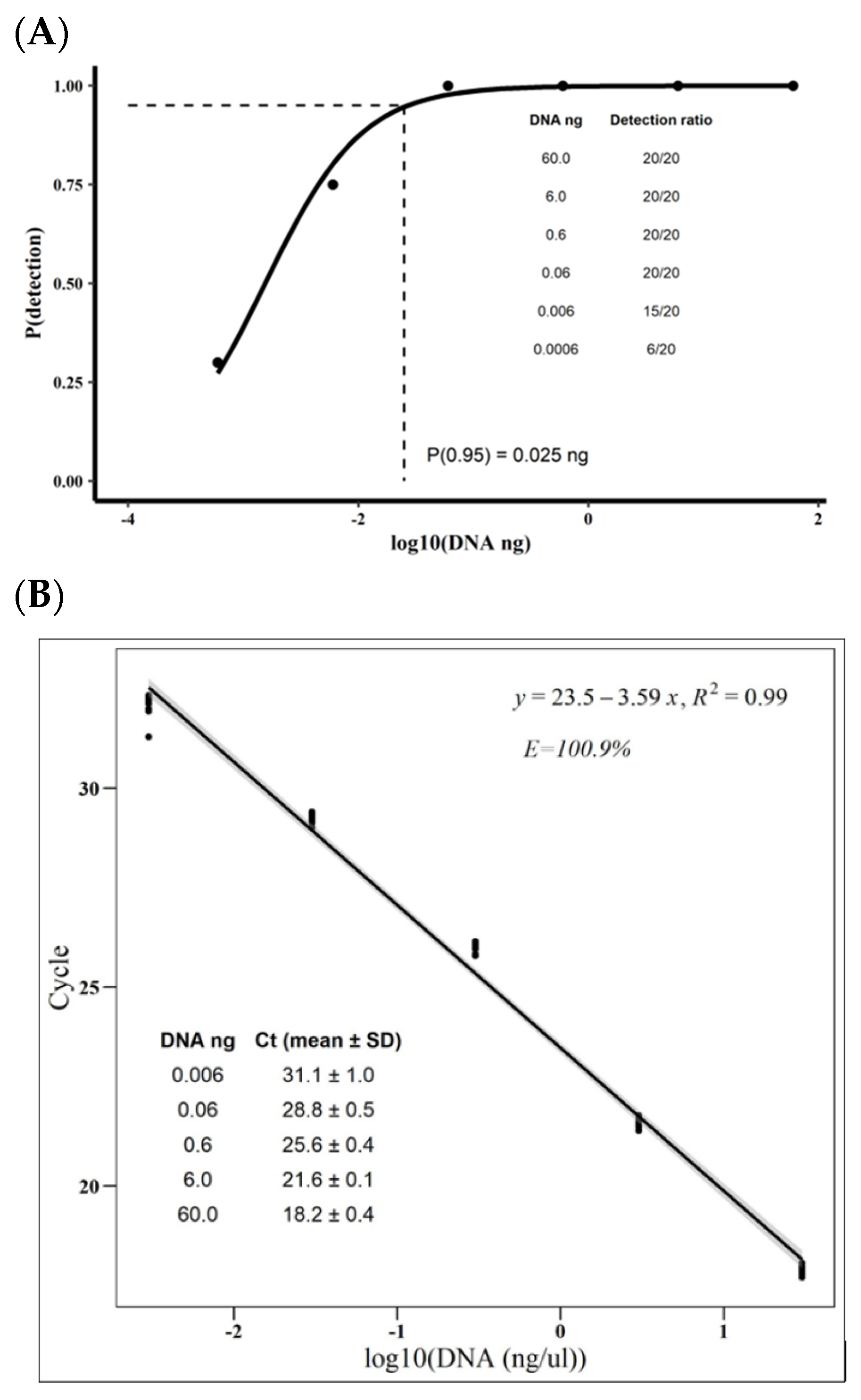
3.1. Moniliophthora roreri qPCR’s Specificity, Efficiency, Limit of Detection, and Presicion
3.2. Moniliophthora roreri Spores Quantification
3.3. Spore Trap Devices
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, H.C.; Holmes, K.A.; Phillips, W.; Wilkinson, M.J. What’s in a Name: Crinipellis, the Final Resting Place for the Frosty Pod Rot Pathogen of Cocoa? Mycologist 2002, 16, 148–152. [Google Scholar] [CrossRef]
- Leandro-Muñoz, M.E.; Tixier, P.; Germon, A.; Rakotobe, V.; Phillips-Mora, W.; Maximova, S.; Avelino, J. Effects of Microclimatic Variables on the Symptoms and Signs Onset of Moniliophthora roreri, Causal Agent of Moniliophthora Pod Rot in Cacao. PLoS ONE 2017, 12, e0184638. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.L.; Alvarez, J.C.; Mosquera, S. Frosty Pod Rot: A Major Threat to Cacao Plantations on the Move. Trop. Plant Pathol. 2021, 47, 187–200. [Google Scholar] [CrossRef]
- Jaimes, Y.; Aranzazu, F. Manejo De Las Enfermedades Del Cacao (Theobroma cacao L.) En Colombia, Con Énfasis En Monilia (Moniliophthora roreri); Corporacion Colombiana de Investigacion Agropecuaria—Corpoica: Bogotá, Colombia, 2010; ISBN 978-958-740-034-2. [Google Scholar]
- Ram, A. Biology, Epidemiology and Control of Moniliasis (Moniliophthora Roreri) of Cacao. Ph.D. Thesis, Imperial College of Science and Technology, University of London, London, UK, 1989. [Google Scholar]
- Torres de la Cruz, H. Temporal Progress and Integrated Management of Frosty Pod Rot (Moniliophthora Roreri) of Cocoa in Tabasco, Mexico. J. Plant Pathol. 2011, 93, 31–36. [Google Scholar]
- Merchán Vargas, V.M. Avances de la Investigación de la Moniliasis del Cacao en Colombia; El Cacaotero Colombiano; Federación Nacional de Cacaoteros—FEDECACAO: Bogotá, Colombia, 1981; pp. 26–41. [Google Scholar]
- Meléndez, L. Microambiente, Cantidad de Esporas en el Aire e Incidencia del Hongo Miniliopthora roreri (Cif & Par). Evans et al. Bajo Tres Sistemas de Manejo de Sombra Leguminosas en Cacao (Theobroma cacao); Tesis de Maestria; CATIE: Turrialba, Costa Rica, 1993. [Google Scholar]
- Ortega Andrade, S.; Páez, G.T.; Feria, T.P.; Muñoz, J. Climate change and the risk of spread of the fungus from the high mortality of Theobroma cocoa in Latin America. Neotrop. Biodivers. 2017, 3, 30–40. [Google Scholar] [CrossRef]
- Sánchez Mora, F.D.; Garcés Fiallos, F.R. Moniliophthora roreri (Cif y Par) Evans et al. in the crop of cocoa. Sci. Agropecu. 2012, 3, 249–258. [Google Scholar] [CrossRef]
- Leandro-Muñoz, M. Efecto de los Factores Macro y Microclimáticas y las Características Productivas del Cacao Sobre la Epidemiología de la Moniliasis; CATIE: Turrialba, Costa Rica, 2011. [Google Scholar]
- Zhang, Z.; Zhu, Z.; Ma, Z.; Li, H. A Molecular Mechanism of Azoxystrobin Resistance in Penicillium digitatum UV Mutants and a PCR-Based Assay for Detection of Azoxystrobin-Resistant Strains in Packing- or Store-House Isolates. Int. J. Food Microbiol. 2009, 131, 157–161. [Google Scholar] [CrossRef]
- Díaz-Valderrama, J.R.; Aime, M.C. The Cacao Pathogen Moniliophthora roreri (Marasmiaceae) Produces Rhexolytic Thallic Conidia and Their Size Is Influenced by Nuclear Condition. Mycoscience 2016, 57, 208–216. [Google Scholar] [CrossRef]
- Úrbez-Torres, J.R.; Battany, M.; Bettiga, L.J.; Gispert, C.; McGourty, G.; Roncoroni, J.; Smith, R.J.; Verdegaal, P.; Gubler, W.D. Botryosphaeriaceae Species Spore-Trapping Studies in California Vineyards. Plant Dis. 2010, 94, 717–724. [Google Scholar] [CrossRef]
- Bailey, B.A.; Ali, S.S.; Strem, M.D.; Meinhardt, L.W. Morphological Variants of Moniliophthora roreri on Artificial Media and the Biotroph/Necrotroph Shift. Fungal Biol. 2018, 122, 701–716. [Google Scholar] [CrossRef]
- Schweigkofler, W.; O’Donnell, K.; Garbelotto, M. Detection and Quantification of Airborne Conidia of Fusarium circinatum, the Causal Agent of Pine Pitch Canker, from Two California Sites by Using a Real-Time PCR Approach Combined with a Simple Spore Trapping Method. Appl. Environ. Microbiol. 2004, 70, 3512–3520. [Google Scholar] [CrossRef]
- Quesada, T.; Hughes, J.; Smith, K.; Shin, K.; James, P.; Smith, J. A Low-Cost Spore Trap Allows Collection and Real-Time PCR Quantification of Airborne Fusarium circinatum Spores. Forests 2018, 9, 586. [Google Scholar] [CrossRef]
- Dvořák, M.; Janoš, P.; Botella, L.; Rotková, G.; Zas, R. Spore Dispersal Patterns of Fusarium circinatum on an Infested Monterey Pine Forest in North-Western Spain. Forests 2017, 8, 432. [Google Scholar] [CrossRef]
- Chandelier, A.; Helson, M.; Dvorak, M.; Gischer, F. Detection and Quantification of Airborne Inoculum of Hymenoscyphus pseudoalbidus Using Real-Time PCR Assays. Plant Pathol. 2014, 63, 1296–1305. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- White, T.J. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols, a Guide to Methods and Applications; Academic Press: San Francisco, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Inglis, P.W.; Pappas, M.d.C.R.; Resende, L.V.; Grattapaglia, D. Fast and Inexpensive Protocols for Consistent Extraction of High Quality DNA and RNA from Challenging Plant and Fungal Samples for High-Throughput SNP Genotyping and Sequencing Applications. PLoS ONE 2018, 13, e0206085. [Google Scholar] [CrossRef]
- Raja, H.A.; Miller, A.N.; Pearce, C.J.; Oberlies, N.H. Fungal Identification Using Molecular Tools: A Primer for the Natural Products Research Community. J. Nat. Prod. 2017, 80, 756–770. [Google Scholar] [CrossRef]
- R Development Core Team. Language and Environment for Statistical Computing: Reference Index; R Foundation for Statistical Computing: Vienna, Austria, 2021; ISBN 978-3-900051-07-5. [Google Scholar]
- Lenth, R.V. Emmeans: Estimated Marginal Means, Aka Least-Squares Means. R Package Version 2022, 1, 3. [Google Scholar]
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Ruijter, J.M.; Barnewall, R.J.; Marsh, I.B.; Szentirmay, A.N.; Quinn, J.C.; van Houdt, R.; Gunst, Q.D.; van den Hoff, M.J.B. Efficiency Correction Is Required for Accurate Quantitative PCR Analysis and Reporting. Clin. Chem. 2021, 67, 829–842. [Google Scholar] [CrossRef]
- Ali, S.S.; Shao, J.; Strem, M.D.; Phillips-Mora, W.; Zhang, D.; Meinhardt, L.W.; Bailey, B.A. Combination of RNAseq and SNP Nanofluidic Array Reveals the Center of Genetic Diversity of Cacao Pathogen Moniliophthora roreri in the Upper Magdalena Valley of Colombia and Its Clonality. Front. Microbiol. 2015, 6, 850. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Mora, W.; Aime, M.C.; Wilkinson, M.J. Biodiversity and Biogeography of the Cacao (Theobroma Cacao) Pathogen Moniliophthora roreri in Tropical America. Plant Pathol. 2007, 56, 911–922. [Google Scholar] [CrossRef]
- Nicolaisen, M.; West, J.S.; Sapkota, R.; Canning, G.G.M.; Schoen, C.; Justesen, A.F. Fungal Communities Including Plant Pathogens in Near Surface Air Are Similar across Northwestern Europe. Front. Microbiol. 2017, 8, 1729. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.S.; Turo, C.; Moffat, C.S.; Lopez-Ruiz, F.J.; Gibberd, M.R.; Hamblin, J.; Zerihun, A. Real-Time PCR for Diagnosing and Quantifying Co-Infection by Two Globally Distributed Fungal Pathogens of Wheat. Front. Plant Sci. 2018, 9, 1086. [Google Scholar] [CrossRef]
- Bailey, B.A.; Meinhardt, L.W. (Eds.) Cacao Diseases: A History of Old Enemies and New Encounters; Springer: Cham, Switzerland; Heidelberg, Germany; New York, NY, USA, 2016; ISBN 978-3-319-24787-8. [Google Scholar]
- Lofgren, L.A.; Uehling, J.K.; Branco, S.; Bruns, T.D.; Martin, F.; Kennedy, P.G. Genome-Based Estimates of Fungal RDNA Copy Number Variation across Phylogenetic Scales and Ecological Lifestyles. Mol. Ecol. 2019, 28, 721–730. [Google Scholar] [CrossRef]
- Klosterman, S.J.; Anchieta, A.; McRoberts, N.; Koike, S.T.; Subbarao, K.V.; Voglmayr, H.; Choi, Y.-J.; Thines, M.; Martin, F.N. Coupling Spore Traps and Quantitative PCR Assays for Detection of the Downy Mildew Pathogens of Spinach (Peronospora Effusa) and Beet (P. Schachtii). Phytopathology 2014, 104, 1349–1359. [Google Scholar] [CrossRef]
- Kennedy, R.; Wakeham, A.J.; Byrne, K.G.; Meyer, U.M.; Dewey, F.M. A New Method To Monitor Airborne Inoculum of the Fungal Plant Pathogens Mycosphaerella brassicicola and Botrytis cinerea. Appl. Environ. Microbiol. 2000, 66, 2996–3003. [Google Scholar] [CrossRef]


{kind=link}
| Strain a | Fungi | Ct (Mean ± SD) b | Tukey Grouping c | Isolation Site |
|---|---|---|---|---|
| MR 124 | Moniliophthora roreri | 15.9 ± 0.3 | i | Necoclí, Antioquia |
| MR126 | 16.6 ± 0.5 | hi | Necoclí, Antioquia | |
| MR68 | 16.8 ± 0.1 | hi | Palestina, Caldas | |
| MR1 | 16.8 ± 0.3 | hi | Barrancabermeja, Santander | |
| MR134 | 17.0 ± 0.1 | h | Orito, Putumayo | |
| MR98 | 18.2 ± 0.1 | g | Villanueva, Casanare | |
| MR38 | 18.8 ± 0.2 | fg | Barrancabermeja, Santander | |
| MR136 | 19.8 ± 0.2 | ef | Orito, Putumayo | |
| MR33 | 19.8 ± 0.1 | ef | San José del Nus, Antioquia | |
| MR108 | 19.8 ± 0.3 | ef | Rivera, Huila | |
| MR30 | 19.9 ± 0.3 | e | San José del Nus, Antioquia | |
| MR28 | 21.1 ± 0.2 | d | San José del Nus, Antioquia | |
| MR2 | 21.1 ± 0.5 | d | Barrancabermeja, Santander | |
| MR48 | 23.1 ± 0.1 | c | Paz de Ariporo, Casanare | |
| EAFIT-F0059 | Alternaria argyroxiphii | 32.3 ± 0.8 | b | -- |
| -- | Pleurotus sp. | 32.8 ± 0.4 | ab | -- |
| N.A. | Moniliophthora perniciosa | 33 ± 0.3 | ab | Barrancabermeja, Santander |
| -- | Ganoderma sp. | 33.3 ± 0.2 | ab | -- |
| EAFIT-F0056 | Diaporthe phaseolorum | 33.3 ± 0.5 | ab | -- |
| EAFIT-F0066 | Colletotrichum siamense | 33.8 ± 0.5 | a | -- |
| N.A. | H2O | 33.8 ± 0.2 | a | -- |
| qPCR Run | Load | Within qPCR Run | Between qPCR Runs | ||
|---|---|---|---|---|---|
| Estimated DNA ng (Mean ± SD) a | CV b | Estimated DNA ng (Mean ± SD) c | CV d | ||
| DNA suspension | |||||
| 1 | 6 ng | 6.32 ± 0.59 | 0.09 | 6.51 ±0.27 | 0.04 |
| 2 | 6.7 ± 0.84 | 0.13 | |||
| 1 | 0.6 ng | 0.72 ± 0.08 | 0.11 | 0.80 ± 0.11 | 0.14 |
| 2 | 0.87 ± 0.02 | 0.02 | |||
| Spore suspensions | |||||
| 1 | 2.9 × 106 spores | 99.7 ± 11.68 | 0.12 | 81.83 ± 25.27 | 0.31 |
| 2 | 63.96 ± 28.62 | 0.45 | |||
| 1 | 2.9 × 105 spores | 13.56 ± 3.62 | 0.27 | 10.9 ± 3.76 | 0.34 |
| 2 | 8.25 ± 7.92 | 0.96 | |||
| 1 | 2.9 × 104 spores | 0.52 ± 0.78 | 1.5 | 0.56 ± 0.06 | 0.1 |
| 2 | 0.6 ± 0.81 | 1.35 | |||
| Spore traps | |||||
| 1 | 2.9 × 106 spores | 10.87 ± 6.51 | 0.6 | 8.36 ± 3.55 | 0.42 |
| 2 | 5.86 ± 6.64 | 1.13 | |||
| 1 | 2.9 × 105 spores | 0.49 ± 0.62 | 1.26 | 0.35 ± 0.20 | 0.57 |
| 2 | 0.21 ± 0.16 | 0.78 | |||
| 1 | 2.9 × 104 spores | 0.1 ± 0.16 | 1.73 | 0.07 ± 0.04 | 0.56 |
| 2 | 0.04 ± 0.01 | 0.14 | |||
| No-template control (H2O) | |||||
| 1 | -- | 0 ± 0 | -- | 0 ± 0 | -- |
| 2 | -- | 0 ± 0 | -- | ||
| Negative Control (M. pernisiosa DNA) | |||||
| 1 | -- | 0 ± 0 | -- | 0 ± 0 | -- |
| 2 | -- | 0 ± 0 | -- | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Zapata, D.L.; Quiroga-Pérez, M.; Quiroz-Yepes, M.; Marulanda-Tobón, A.; Álvarez, J.C.; Mosquera-López, S. Development of a Method for Detecting and Estimating Moniliophthora roreri Spore Loads Based on Spore Traps and qPCR. J. Fungi 2023, 9, 47. https://doi.org/10.3390/jof9010047
Jiménez-Zapata DL, Quiroga-Pérez M, Quiroz-Yepes M, Marulanda-Tobón A, Álvarez JC, Mosquera-López S. Development of a Method for Detecting and Estimating Moniliophthora roreri Spore Loads Based on Spore Traps and qPCR. Journal of Fungi. 2023; 9(1):47. https://doi.org/10.3390/jof9010047
Chicago/Turabian StyleJiménez-Zapata, Diana L., Manuela Quiroga-Pérez, Manuela Quiroz-Yepes, Alejandro Marulanda-Tobón, Javier C. Álvarez, and Sandra Mosquera-López. 2023. "Development of a Method for Detecting and Estimating Moniliophthora roreri Spore Loads Based on Spore Traps and qPCR" Journal of Fungi 9, no. 1: 47. https://doi.org/10.3390/jof9010047
APA StyleJiménez-Zapata, D. L., Quiroga-Pérez, M., Quiroz-Yepes, M., Marulanda-Tobón, A., Álvarez, J. C., & Mosquera-López, S. (2023). Development of a Method for Detecting and Estimating Moniliophthora roreri Spore Loads Based on Spore Traps and qPCR. Journal of Fungi, 9(1), 47. https://doi.org/10.3390/jof9010047