
Ectomycorrhizal Fungi Dominated the Root and Rhizosphere Microbial Communities of Two Willow Cultivars Grown for Six-Years in a Mixed-Contaminated Environment



Abstract
:1. Introduction
2. Materials and Methods
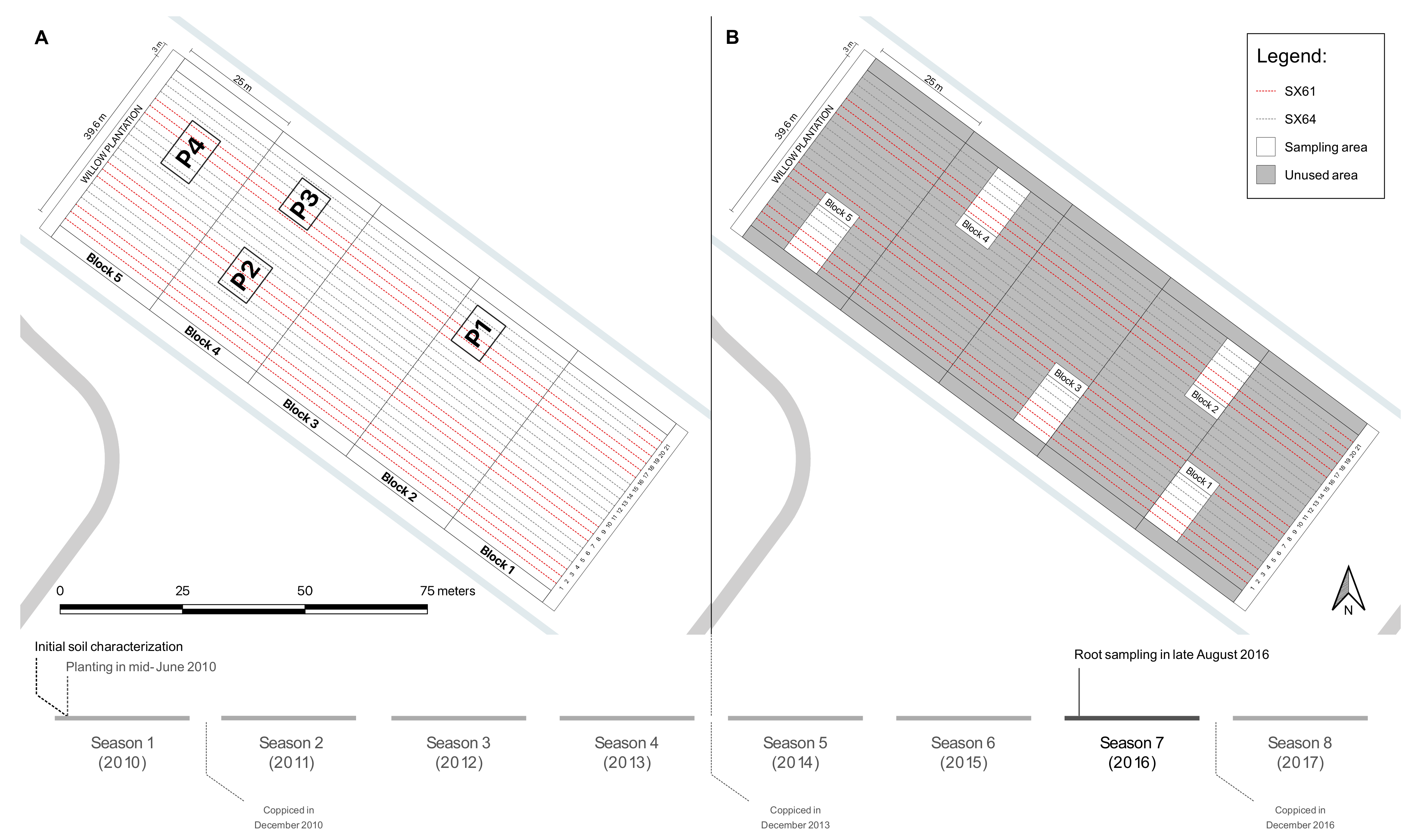
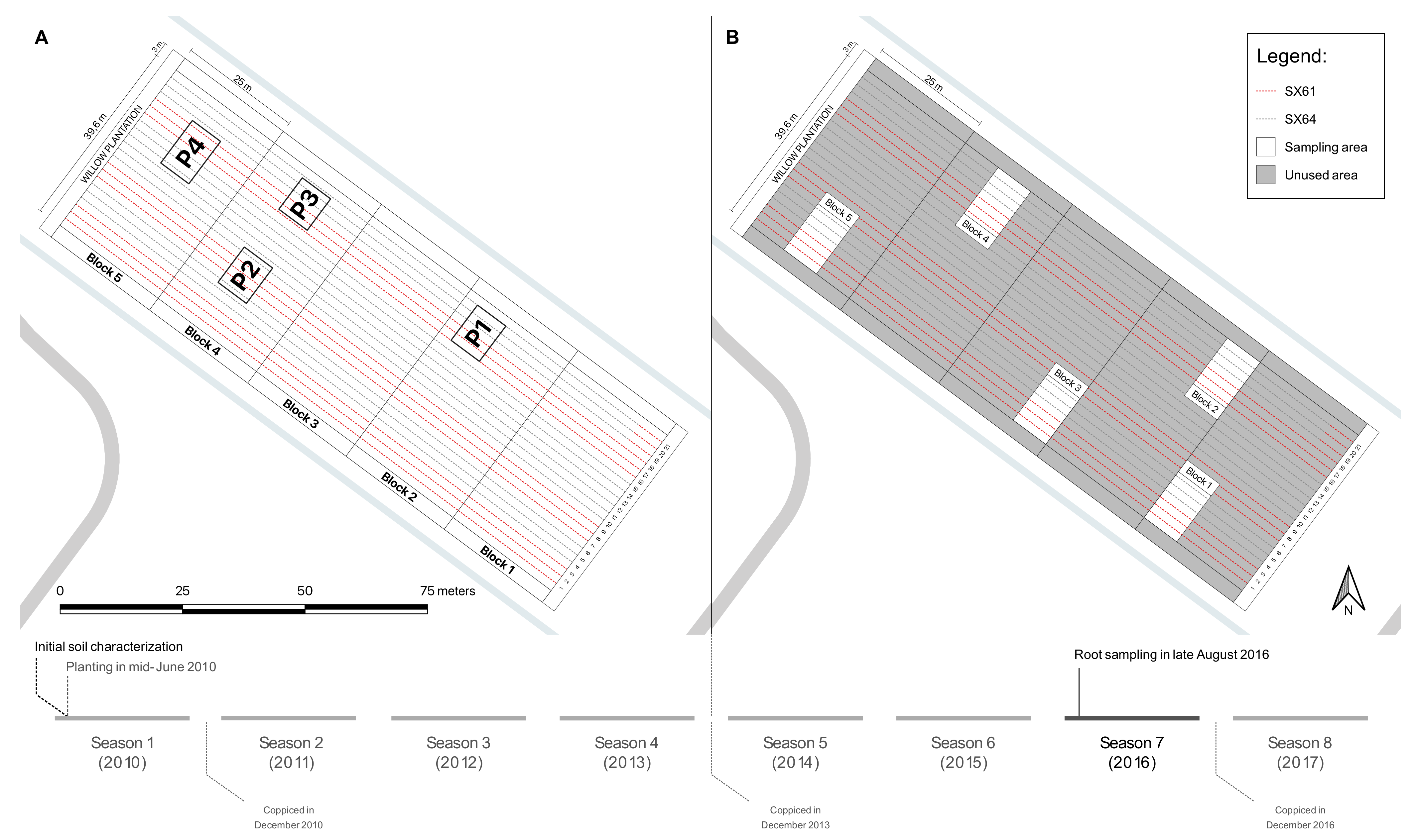
2.1. Experimental Site
2.2. Sample Collection
2.3. DNA Extractions
2.4. PCR Amplifications and Sequencing
2.5. Sequence Processing
2.6. Statistical Analysis
3. Results
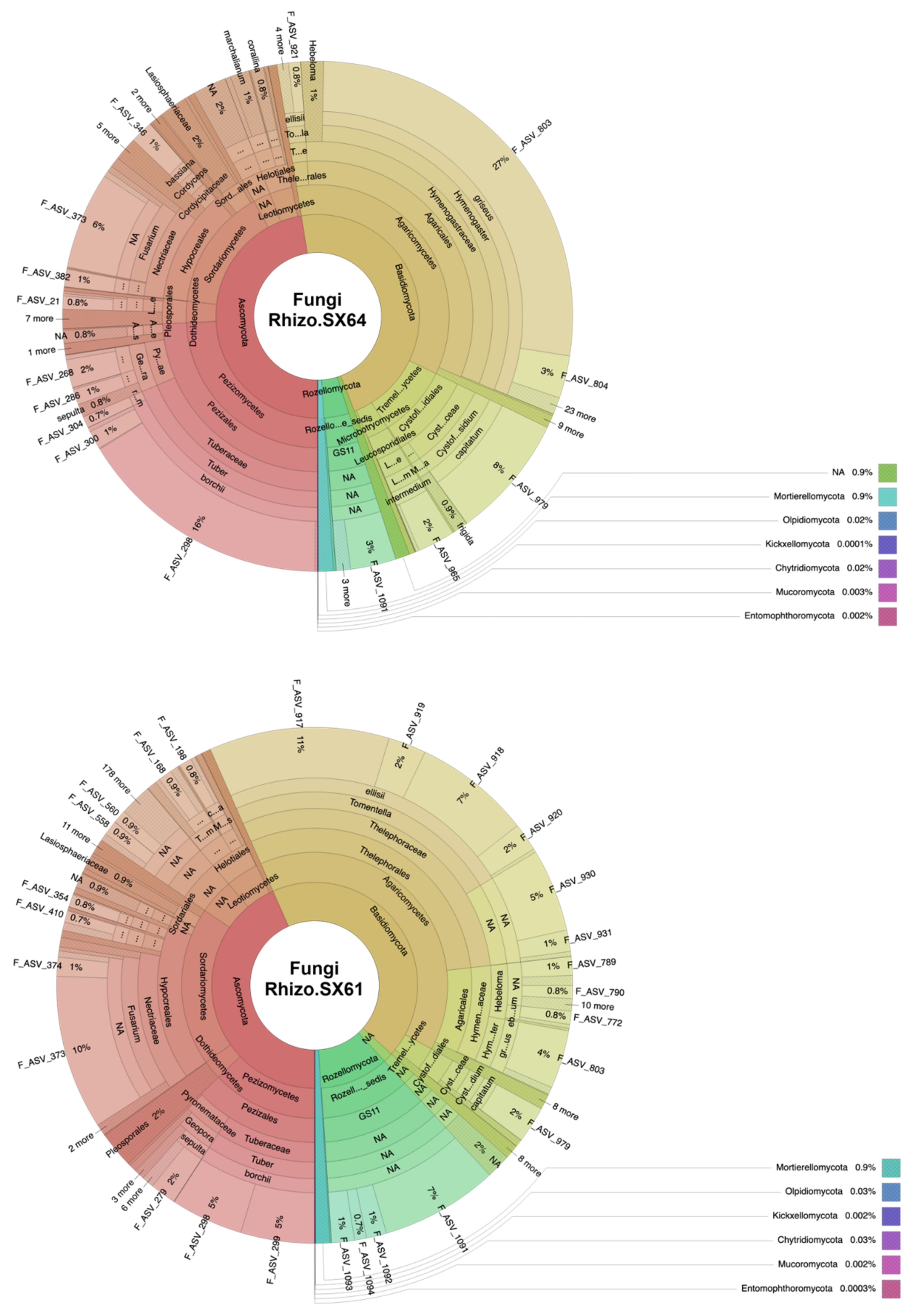
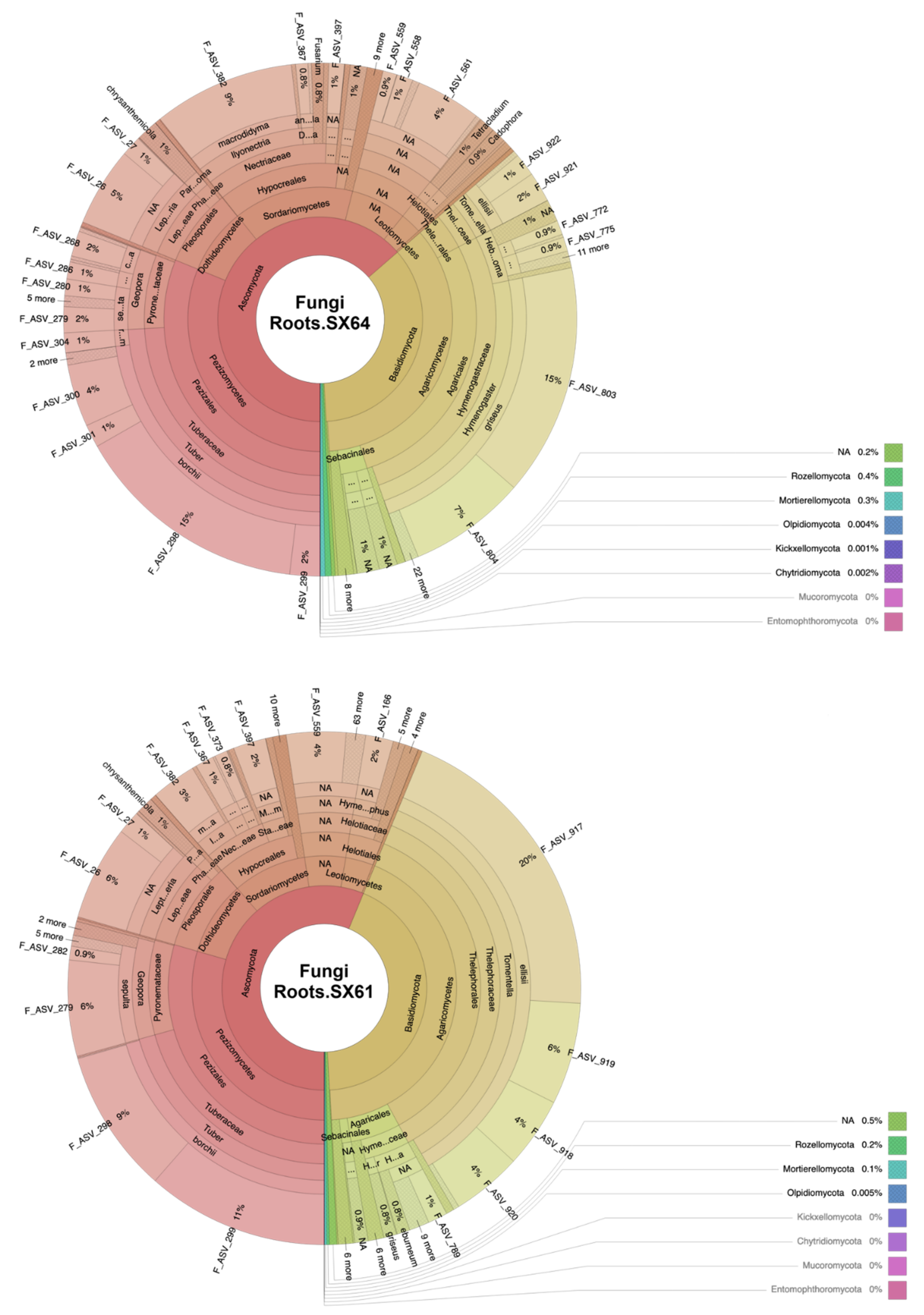
3.1. Fungal Community Structure
3.2. Bacterial Community Structure
3.3. Archaeal Community Structure
3.4. Alpha Diversity
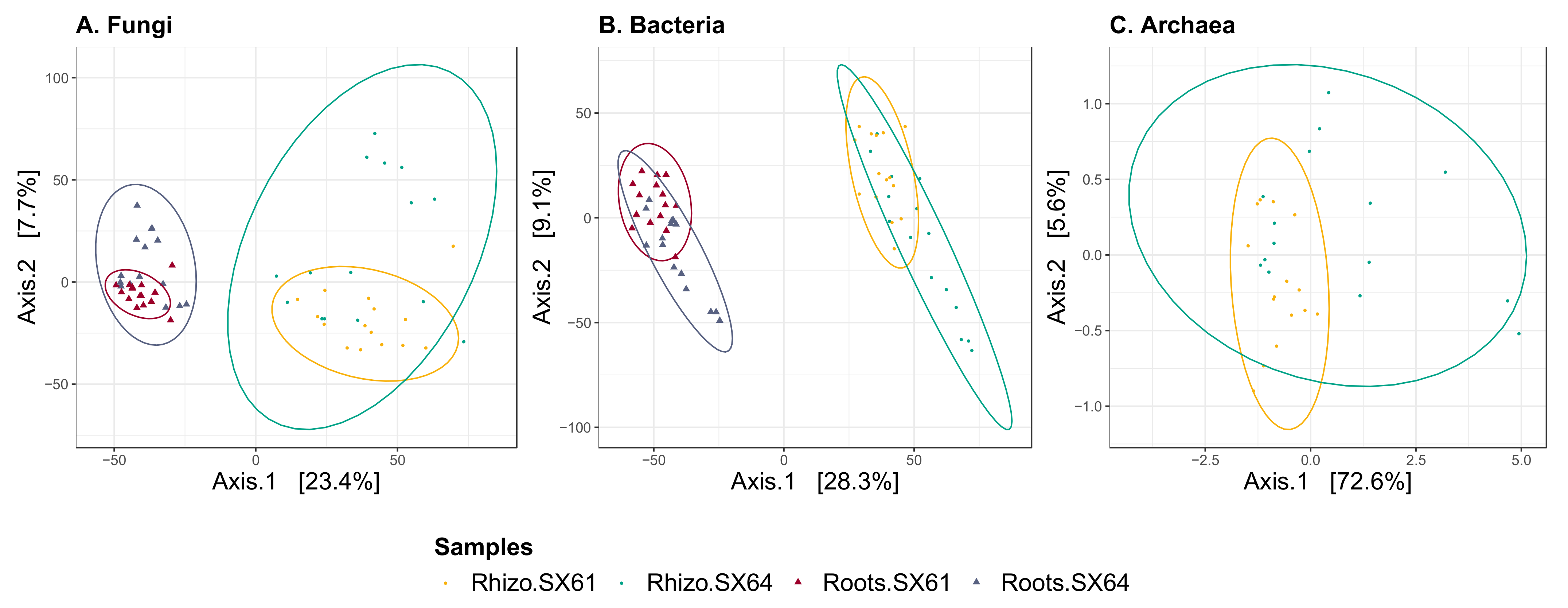
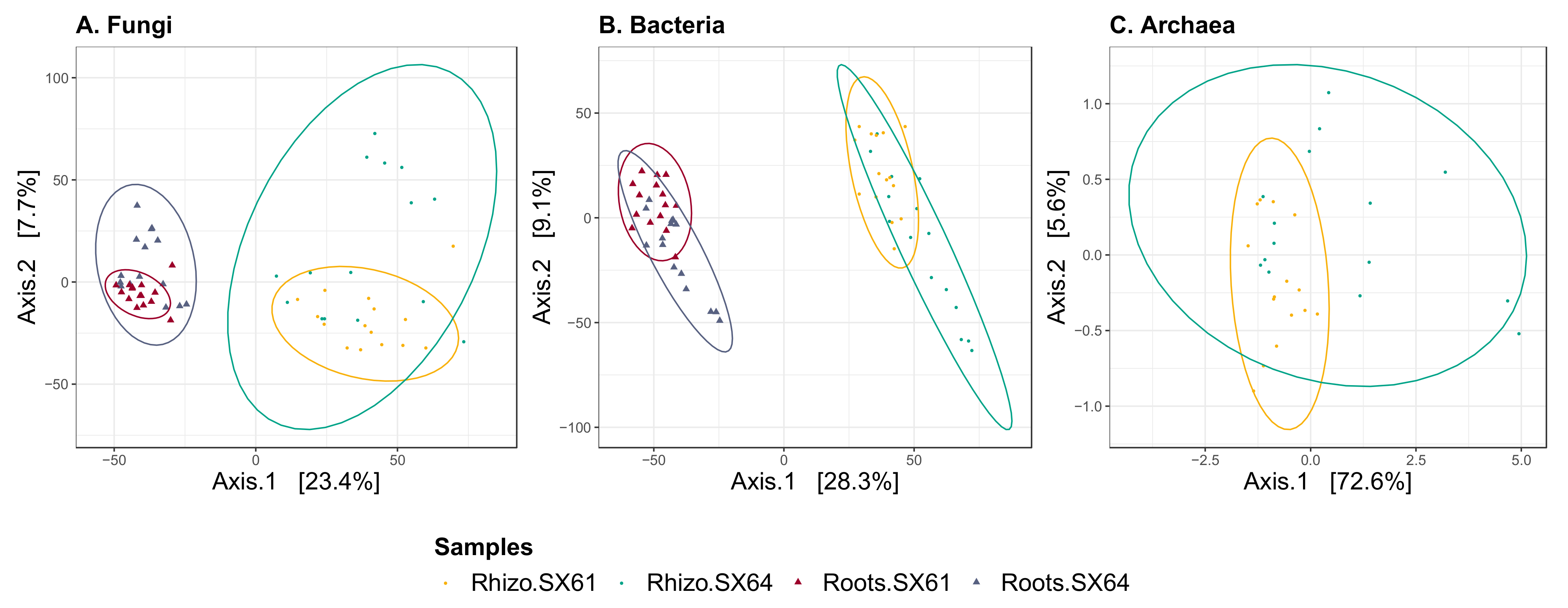
3.5. Beta Diversity
3.6. Differential Abundance of ASVs
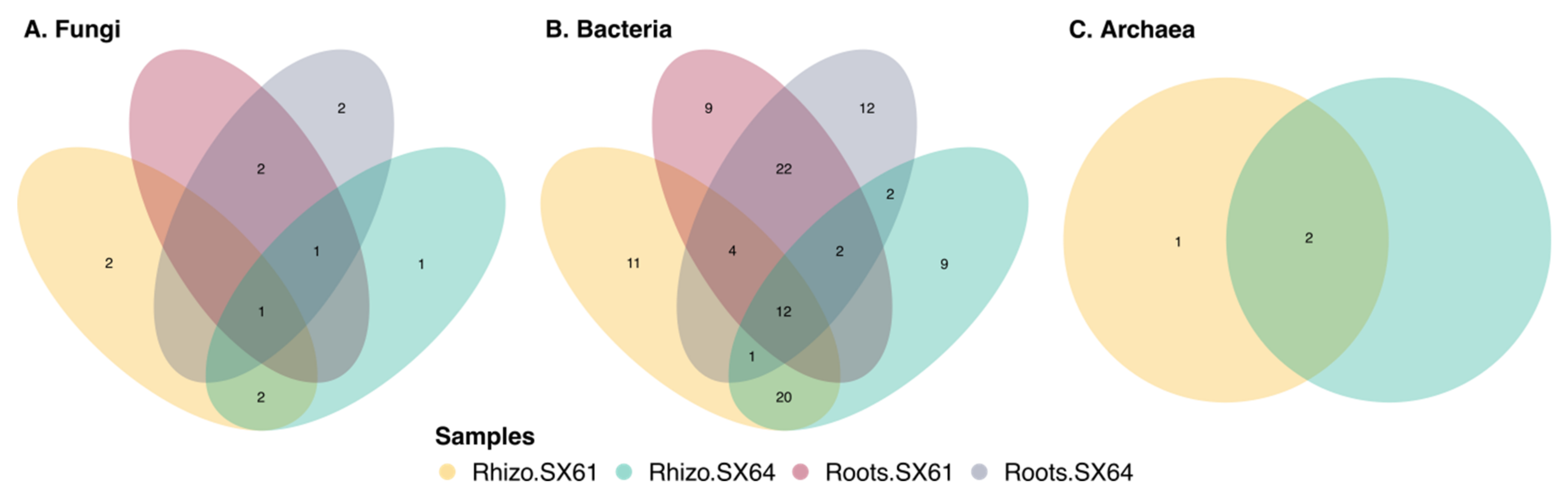
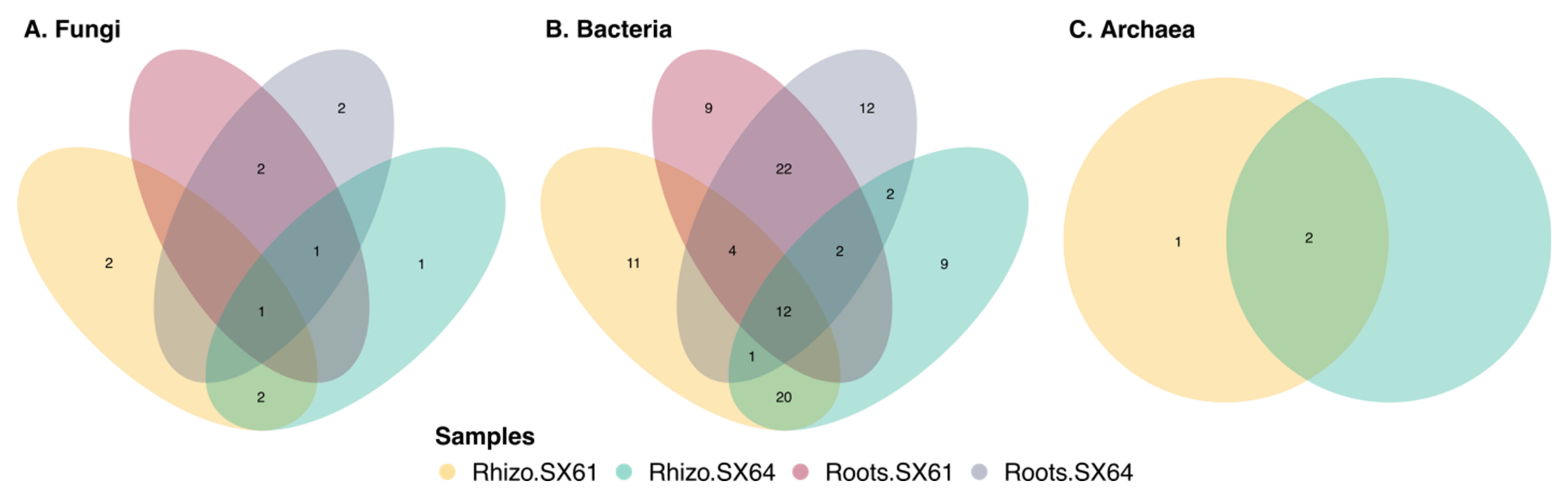
3.7. Common Core Microbiome
4. Discussion
4.1. Beta and Alpha Diversities
4.2. Mycorrhizal Fungi
4.3. Nonmycorrhizal Endophytic Fungi
4.4. Archaeal Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quiza, L.; St-Arnaud, M.; Yergeau, É. Harnessing phytomicrobiome signaling for rhizosphere microbiome engineering. Front. Microbiol. 2015, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Thijs, S.; Sillen, W.; Rineau, F.; Weyens, N.; Vangronsveld, J. Towards an enhanced understanding of plant-microbiome interactions to improve phytoremediation: Engineering the metaorganism. Front. Microbiol. 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Redecker, D.; Kodner, R.; Graham, L.E. Glomalean fungi from the Ordovician. Science 2000, 289, 1920–1921. [Google Scholar] [CrossRef] [Green Version]
- Finlay, R.D. Ecological aspects of mycorrhizal symbiosis: With special emphasis on the functional diversity of interactions involving the extraradical mycelium. J. Exp. Bot. 2008, 59, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.A.; Plenchette, C.; Piché, Y. Les Mycorhizes: L’essor de la Nouvelle Révolution Verte, 2nd ed.; Quae, É., Ed.; Éditions Quae: Paris, France, 2016; ISBN B01DKK6MY4. [Google Scholar]
- Spatafora, J.W.; Chang, Y.; Benny, G.L.; Lazarus, K.; Smith, M.E.; Berbee, M.L.; Bonito, G.; Corradi, N.; Grigoriev, I.; Gryganskyi, A.; et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 2016, 108, 1028–1046. [Google Scholar] [CrossRef] [Green Version]
- Brundrett, M.C.; Tedersoo, L. Evolutionary history of mycorrhizal symbioses and global host plant diversity. New Phytol. 2018, 220, 1108–1115. [Google Scholar] [CrossRef]
- Smith, S.E.; Read, D. Mycorrhizal Symbiosis, 3rd ed.; Academic Press: London, UK, 2008; ISBN 9780123705266. [Google Scholar]
- Ismail, Y.; Mccormick, S.; Hijri, M. The arbuscular mycorrhizal fungus, glomus irregulare, controls the mycotoxin production of fusarium sambucinum in the pathogenesis of potato. FEMS Microbiol. Lett. 2013, 348, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Ismail, Y.; Hijri, M. Arbuscular mycorrhisation with Glomus irregulare induces expression of potato PR homologues genes in response to infection by Fusarium sambucinum. Funct. Plant Biol. 2012, 39, 236–245. [Google Scholar] [CrossRef]
- Ismail, Y.; McCormick, S.; Hijri, M. A fungal symbiont of plant-roots modulates mycotoxin gene expression in the pathogen Fusarium sambucinum. PLoS ONE 2011, 6, e17990. [Google Scholar] [CrossRef] [Green Version]
- Knapp, D.G.; Németh, J.B.; Barry, K.; Hainaut, M.; Henrissat, B.; Johnson, J.; Kuo, A.; Lim, J.H.P.; Lipzen, A.; Nolan, M.; et al. Comparative genomics provides insights into the lifestyle and reveals functional heterogeneity of dark septate endophytic fungi. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barberis, L.; Michalet, S.; Piola, F.; Binet, P. Root fungal endophytes: Identity, phylogeny and roles in plant tolerance to metal stress. Fungal Biol. 2021, 125, 326–345. [Google Scholar] [CrossRef]
- Prakash, J. Chapter 9—Plant growth promoting rhizobacteria in phytoremediation of environmental contaminants: Challenges and future prospects. In Bioremediation for Environmental Sustainability; Elsevier B.V.: Amsterdam, The Netherlands, 2021; pp. 191–218. ISBN 9780128203187. [Google Scholar]
- Taffner, J.; Erlacher, A.; Bragina, A.; Berg, C.; Moissl-Eichinger, C.; Berg, G. What Is the Role of Archaea in Plants? New Insights from the Vegetation of Alpine Bogs. mSphere 2018, 3, e00122-18. [Google Scholar] [CrossRef] [Green Version]
- Akinola, S.A.; Babalola, O.O. The fungal and archaeal community within plant rhizosphere: A review on their contribution to crop safety. J. Plant Nutr. 2021, 44, 600–618. [Google Scholar] [CrossRef]
- Buée, M.; de Boer, W.; Martin, F.; van Overbeek, L.; Jurkevitch, E. The rhizosphere zoo: An overview of plant-associated communities of microorganisms, including phages, bacteria, archaea, and fungi, and of some of their structuring factors. Plant Soil 2009, 321, 189–212. [Google Scholar] [CrossRef]
- Pozo, M.J.; Zabalgogeazcoa, I.; Vazquez de Aldana, B.R.; Martinez-Medina, A. Untapping the potential of plant mycobiomes for applications in agriculture. Curr. Opin. Plant Biol. 2021, 60, 102034. [Google Scholar] [CrossRef] [PubMed]
- Basiru, S.; Mwanza, H.P.; Hijri, M. Analysis of arbuscular mycorrhizal fungal inoculant benchmarks. Microorganisms 2021, 9, 81. [Google Scholar] [CrossRef]
- Dessaux, Y.; Grandclément, C.; Faure, D. Engineering the Rhizosphere. Trends Plant Sci. 2016, 21, 266–278. [Google Scholar] [CrossRef]
- Guidi Nissim, W.; Pitre, F.E.; Teodorescu, T.I.; Labrecque, M. Long-term biomass productivity of willow bioenergy plantations maintained in southern Quebec, Canada. Biomass Bioenergy 2013, 56, 361–369. [Google Scholar] [CrossRef]
- Padoan, E.; Passarella, I.; Prati, M.; Bergante, S.; Facciotto, G.; Ajmone-Marsan, F. The suitability of short rotation coppice crops for phytoremediation of Urban soils. Appl. Sci. 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Hénault-Ethier, L.; Lucotte, M.; Moingt, M.; Paquet, S.; Maccario, S.; Smedbol, É.; Gomes, M.P.; Lepage, L.; Juneau, P.; Labrecque, M. Herbaceous or Salix miyabeana ‘SX64’ narrow buffer strips as a means to minimize glyphosate and aminomethylphosphonic acid leaching from row crop fields. Sci. Total Environ. 2017, 598, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Lafleur, B.; Sauvé, S.; Duy, S.V.; Labrecque, M. Phytoremediation of groundwater contaminated with pesticides using short-rotation willow crops: A case study of an apple orchard. Int. J. Phytoremediation 2016, 18, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Lachapelle, T.X.; Labrecque, M.; Comeau, Y. Treatment and valorization of a primary municipal wastewater by a short rotation willow coppice vegetation filter. Ecol. Eng. 2019, 130, 32–44. [Google Scholar] [CrossRef]
- Lévesque, S.; Demers, E.; Brisson, J.; Comeau, Y. Treatment of a mixed wood preservative leachate by a hybrid constructed wetland and a willow planted filter. Water Sci. Technol. 2017, 76, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Kuzovkina, Y.A.; Quigley, M.F. Willows beyond wetlands: Uses of Salix L. species for environmental projects. Water Air Soil Pollut. 2005, 162, 183–204. [Google Scholar] [CrossRef]
- Fortin Faubert, M.; Hijri, M.; Labrecque, M. Short Rotation Intensive Culture of Willow, Spent Mushroom Substrate and Ramial Chipped Wood for Bioremediation of a Contaminated Site Used for Land Farming Activities of a Former Petrochemical Plant. Plants 2021, 10, 520. [Google Scholar] [CrossRef]
- Fortin Faubert, M.; Desjardins, D.; Hijri, M.; Labrecque, M. Willows used for phytoremediation increased organic contaminant concentrations in soil surface. Appl. Sci. 2021, 11, 2979. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Higuchi, R. Ectomycorrhizal and arbuscular mycorrhizal colonization of two species of floodplain willows. Mycoscience 2003, 44, 339–343. [Google Scholar] [CrossRef]
- Hrynkiewicz, K.; Toljander, Y.K.; Baum, C.; Fransson, P.M.A.; Taylor, A.F.S.; Weih, M. Correspondence of ectomycorrhizal diversity and colonisation of willows (Salix spp.) grown in short rotation coppice on arable sites and adjacent natural stands. Mycorrhiza 2012, 22, 603–613. [Google Scholar] [CrossRef]
- Dagher, D.J.; De La Providencia, I.E.; Pitre, F.E.; St-Arnaud, M.; Hijri, M. Arbuscular mycorrhizal fungal assemblages significantly shifted upon bacterial inoculation in non-contaminated and petroleum-contaminated environments. Microorganisms 2020, 8, 602. [Google Scholar] [CrossRef] [Green Version]
- Dagher, D.J.; de la Providencia, I.E.; Pitre, F.E.; St-Arnaud, M.; Hijri, M. Plant Identity Shaped Rhizospheric Microbial Communities More Strongly Than Bacterial Bioaugmentation in Petroleum Hydrocarbon-Polluted Sediments. Front. Microbiol. 2019, 10, 2144. [Google Scholar] [CrossRef] [Green Version]
- Yergeau, E.; Sanschagrin, S.; Maynard, C.; St-Arnaud, M.; Greer, C.W. Microbial expression profiles in the rhizosphere of willows depend on soil contamination. ISME J. 2014, 8, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Yergeau, E.; Bell, T.H.; Champagne, J.; Maynard, C.; Tardif, S.; Tremblay, J.; Greer, C.W. Transplanting soil microbiomes leads to lasting effects on willow growth, but not on the rhizosphere microbiome. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tardif, S.; Yergeau, É.; Tremblay, J.; Legendre, P.; Whyte, L.G.; Greer, C.W. The willow microbiome is influenced by soil petroleum-hydrocarbon concentration with plant compartment-specific effects. Front. Microbiol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.H.; Cloutier-Hurteau, B.; Al-Otaibi, F.; Turmel, M.C.; Yergeau, E.; Courchesne, F.; St-Arnaud, M. Early rhizosphere microbiome composition is related to the growth and Zn uptake of willows introduced to a former landfill. Environ. Microbiol. 2015, 17, 3025–3038. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.E.D.; Bell, T.H.; Stefani, F.O.P.; Denis, D.; Hijri, M.; St-Arnaud, M. Contrasting the community structure of arbuscular mycorrhizal fungi from hydrocarbon-contaminated and uncontaminated soils following willow (Salix spp. L.) planting. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.H.; El-Din Hassan, S.; Lauron-Moreau, A.; Al-Otaibi, F.; Hijri, M.; Yergeau, E.; St-Arnaud, M. Linkage between bacterial and fungal rhizosphere communities in hydrocarbon-contaminated soils is related to plant phylogeny. ISME J. 2014, 8, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MELCC Normales Climatiques du Québec 1981–2010. Available online: http://www.environnement.gouv.qc.ca/climat/normales/index.asp (accessed on 28 February 2021).
- Turcotte, C. Pétromont ferme ses portes. LEDEVOIR. Available online: https://www.ledevoir.com/economie/227909/petromont-ferme-ses-portes (accessed on 17 January 2009).
- Guidi, W.; Kadri, H.; Labrecque, M. Establishment techniques to using willow for phytoremediation on a former oil refinery in southern Quebec: Achievements and constraints. Chem. Ecol. 2012, 28, 37–41. [Google Scholar] [CrossRef]
- Labrecque, M.; Teodorescu, T.I. La Culture Intensive de Saules en Courtes Rotations (CICR); Institut de recherche en biologie végétale et Jardin botanique de Montréal: Montréal, QC, Canada, 2006. [Google Scholar]
- CEAEQ. MA. 400—BPC 1.0—Détermination des Biphényles Polychlorés: Dosage par Chromatographie en Phase Gazeuse Couplée à un Spectromètre de Masse ou à un Détecteur à Capture D’électrons—Méthode par Congénère et Groupe Homologue, Rév. 5; CEAEQ: Quebec City, QC, Canada, 2014. [Google Scholar]
- CEAEQ. MA. 400—HAP 1.1—Détermination des Hydrocarbures Aromatiques Polycycliques: Dosage par Chromatographie en Phase Gazeuse Couplée à un Spectromètre de Masse, Rév. 5; CEAEQ: Quebec City, QC, Canada, 2016. [Google Scholar]
- CEAEQ. MA. 400—HYD. 1.0—Méthode D’analyse—Dosage des Hydrocarbures Pétroliers (C10 à C50) Dans L’eau; CEAEQ: Quebec City, QC, Canada, 2004. [Google Scholar]
- CEAEQ. MA. 200—Mét. 1.2—Détermination des Métaux: Méthode par Spectrométrie de Masse à Source Ionisante au Plasma D’argon, Rév. 5; CEAEQ: Quebec City, QC, Canada, 2014. [Google Scholar]
- CEAEQ. MA. 203—Mét. 3.2—Méthode D’analyse—Détermination des Métaux Dans L’eau: Méthode par Spectrométrie D’émission au Plasma D’argon, Rév. 2; CEAEQ: Quebec City, QC, Canada, 2008. [Google Scholar]
- Martin, K.J.; Rygiewicz, P.T. Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol. 2005, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- R Core Development Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; pp. 1–2673. [Google Scholar]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pauvert, C. Psadd: Additions to Phyloseq Package for Microbiome Analysis. R Packag, Version 0.1.2. 2020. Available online: https://rdrr.io/github/cpauvert/psadd/ (accessed on 28 February 2021).
- Kandlikar, G.S.; Gold, Z.J.; Cowen, M.C.; Meyer, R.S.; Freise, A.C.; Kraft, N.J.B.; Moberg-Parker, J.; Sprague, J.; Kushner, D.J.; Curd, E.E. Ranacapa: An R package and shiny web app to explore environmental DNA data with exploratory statistics and interactive visualizations. F1000Research 2018, 7, 1–8. [Google Scholar] [CrossRef]
- Quensen, J. R Functions Useful for Community Ecology. R Packag, Version 0.1.4. 2020. Available online: https://john-quensen.com/wp-content/uploads/2020/12/QsRutils_0.1.4.pdf (accessed on 28 February 2021).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, R.P.; O’Hara, R.B.; Simpson, L.G.; Solymos, P.; et al. vegan: Community Ecology Package. R Packag, Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 28 February 2021).
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. R Packag, Version 0.4.0. 2020. Available online: https://rdrr.io/cran/ggpubr/ (accessed on 28 February 2021).
- Dusa, A. venn: Draw Venn Diagrams. R Packag, Version 1.9. 2020. Available online: https://cran.r-project.org/web/packages/venn/venn.pdf (accessed on 28 February 2021).
- Risely, A. Applying the core microbiome to understand host–microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, D.B.; Vogel, C.; Bai, Y.; Vorholt, J.A. The Plant Microbiota: Systems-Level Insights and Perspectives. Annu. Rev. Genet. 2016, 50, 211–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iffis, B.; St-Arnaud, M.; Hijri, M. Petroleum Contamination and Plant Identity Influence Soil and Root Microbial Communities While AMF Spores Retrieved from the Same Plants Possess Markedly Different Communities. Front. Plant Sci. 2017, 8, 1381. [Google Scholar] [CrossRef] [PubMed]
- Blažková, A.; Jansa, J.; Püschel, D.; Vosátka, M.; Janoušková, M. Is mycorrhiza functioning influenced by the quantitative composition of the mycorrhizal fungal community? Soil Biol. Biochem. 2021, 157, 108249. [Google Scholar] [CrossRef]
- Bell, T.H.; Camillone, N.; Abram, K.; Bruns, M.A.; Yergeau, E.; St-Arnaud, M. Hydrocarbon substrate richness impacts microbial abundance, microbiome composition, and hydrocarbon loss. Appl. Soil Ecol. 2021, 165, 104015. [Google Scholar] [CrossRef]
- Bell, T.H.; Stefani, F.O.P.; Abram, K.; Champagne, J.; Yergeau, E.; Hijri, M.; St-Arnaud, M. A diverse soil microbiome degrades more crude oil than specialized bacterial assemblages obtained in culture. Appl. Environ. Microbiol. 2016, 82, 5530–5541. [Google Scholar] [CrossRef] [Green Version]
- Jansa, J.; Mozafar, A.; Frossard, E. Long-distance transport of P and Zn through the hyphae of an arbuscular mycorrhizal fungus in symbiosis with maize. Agronomie 2003, 23, 481–488. [Google Scholar] [CrossRef]
- Berruti, A.; Borriello, R.; Orgiazzi, A.; Barbera, A.C.; Lumini, E.; Bianciotto, V. Chapter 8—Arbuscular Mycorrhizal Fungi and their Value for Ecosystem Management. In Biodiversity—The Dynamic Balance of the Planet; IntechOpen: Rijeka, Croatia, 2014; pp. 159–192. [Google Scholar]
- Parádi, I.; Baar, J. Mycorrhizal fungal diversity in willow forests of different age along the river Waal, The Netherlands. For. Ecol. Manag. 2006, 237, 366–372. [Google Scholar] [CrossRef]
- Van der Heijden, E.W. Differential benefits of arbuscular mycorrhizal and ectomycorrhizal infection of Salix repens. Mycorrhiza 2001, 10, 185–193. [Google Scholar] [CrossRef]
- Pray, T.J. The Effect of Mycorrhizal Fungi Associated with Willows Growing on Marginal Agricultural Land. Ph.D. Thesis, Université de Montréal, Montréal, QC, Canada, 2017. [Google Scholar]
- Lee, J.; Lee, S.; Young, J.P.W. Improved PCR primers for the detection and identification of arbuscular mycorrhizal fungi. FEMS Microbiol. Ecol. 2008, 65, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Pray, T.J.; Guidi Nissim, W.; St-Arnaud, M.; Labrecque, M. Investigating the effect of a mixed mycorrhizal inoculum on the productivity of biomass plantation willows grown on marginal farm land. Forests 2018, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Bourdel, G.; Roy-Bolduc, A.; St-Arnaud, M.; Hijri, M. Concentration of petroleum-hydrocarbon contamination shapes fungal endophytic community structure in plant roots. Front. Microbiol. 2016, 7, 685. [Google Scholar] [CrossRef] [PubMed]
- Iffis, B.; St-Arnaud, M.; Hijri, M. Petroleum hydrocarbon contamination, plant identity and arbuscular mycorrhizal fungal (AMF) community determine assemblages of the AMF spore-associated microbes. Environ. Microbiol. 2016, 18, 2689–2704. [Google Scholar] [CrossRef] [PubMed]
- Bücking, H.; Mensah, J.A.; Fellbaum, C.R. Common mycorrhizal networks and their effect on the bargaining power of the fungal partner in the arbuscular mycorrhizal symbiosis. Commun. Integr. Biol. 2016, 9, e1107684. [Google Scholar] [CrossRef] [PubMed]
- Egger, K. The surprising diversity of ascomycetous mycorrhizas. New Phytol. 2006, 170, 421–428. [Google Scholar] [CrossRef]
- Tedersoo, L.; Hansen, K.; Perry, B.A.; Kjøller, R. Molecular and morphological diversity of pezizalean ectomycorrhiza. New Phytol. 2006, 170, 581–596. [Google Scholar] [CrossRef]
- Hrynkiewicz, K.; Baum, C.; Leinweber, P.; Weih, M.; Dimitriou, I. The significance of rotation periods for mycorrhiza formation in Short Rotation Coppice. For. Ecol. Manage. 2010, 260, 1943–1949. [Google Scholar] [CrossRef]
- Pierleoni, R.; Buffalini, M.; Vallorani, L.; Guidi, C.; Zeppa, S.; Sacconi, C.; Pucci, P.; Amoresano, A.; Casbarra, A.; Stocchi, V. Tuber borchii fruit body: 2-Dimensional profile and protein identification. Phytochemistry 2004, 65, 813–820. [Google Scholar] [CrossRef]
- Bellion, M.; Courbot, M.; Jacob, C.; Blaudez, D.; Chalot, M. Extracellular and cellular mechanisms sustaining metal tolerance in ectomycorrhizal fungi. FEMS Microbiol. Lett. 2006, 254, 173–181. [Google Scholar] [CrossRef]
- Marjanović, Ž.; Grebenc, T.; Marković, M.; Glišić, A.; Milenković, M. Ecological specificities and molecular diversity of truffles (genus Tuber) originating from mid-west of the Balkan Peninsula. Sydowia 2010, 62, 67–87. [Google Scholar]
- Erlandson, S.R.; Savage, J.A.; Cavender-Bares, J.M.; Peay, K.G. Soil moisture and chemistry influence diversity of ectomycorrhizal fungal communities associating with willow along an hydrologic gradient. FEMS Microbiol. Ecol. 2016, 92, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrynkiewicz, K.; Baum, C.; Niedojadło, J.; Dahm, H. Promotion of mycorrhiza formation and growth of willows by the bacterial strain Sphingomonas sp. 23L on fly ash. Biol. Fertil. Soils 2009, 45, 385–394. [Google Scholar] [CrossRef]
- Tedersoo, L.; May, T.W.; Smith, M.E. Ectomycorrhizal lifestyle in fungi: Global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 2010, 20, 217–263. [Google Scholar] [CrossRef] [PubMed]
- Pruett, G.; Bruhn, J.; Mihail, J. Temporal dynamics of ectomycorrhizal community composition on root systems of oak seedlings infected with Burgundy truffle. Mycol. Res. 2008, 112, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.A.; Wilson, J.; Last, F.T.; Walker, C. The concept of succession in relation to the spread of sheathing mycorrhizal fungi on inoculated tree seedlings growing in unsterile soils. Plant Soil 1983, 71, 247–256. [Google Scholar] [CrossRef]
- Püttsepp, Ü.; Rosling, A.; Taylor, A.F.S. Ectomycorrhizal fungal communities associated with Salix viminalis L. and S. dasyclados Wimm. clones in a short-rotation forestry plantation. For. Ecol. Manag. 2004, 196, 413–424. [Google Scholar] [CrossRef]
- Arraiano-Castilho, R.; Bidartondo, M.I.; Niskanen, T.; Zimmermann, S.; Frey, B.; Brunner, I.; Senn-Irlet, B.; Hörandl, E.; Gramlich, S.; Suz, L.M. Plant-fungal interactions in hybrid zones: Ectomycorrhizal communities of willows (Salix) in an alpine glacier forefield. Fungal Ecol. 2020, 45, 100936. [Google Scholar] [CrossRef]
- Hrynkiewicz, K.; Haug, I.; Baum, C. Ectomycorrhizal community structure under willows at former ore mining sites. Eur. J. Soil Biol. 2008, 44, 37–44. [Google Scholar] [CrossRef]
- Wijesinghe, M. Communities Associated with Soil Aggregates in the Rhizosphere of Willows (Salix spp.) Inoculated with Rhizophagus intraradices and Hebeloma cylindrosporum. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 2013. [Google Scholar]
- Sell, J.; Kayser, A.; Schulin, R.; Brunner, I. Contribution of ectomycorrhizal fungi to cadmium uptake of poplars and willows from a heavily polluted soil. Plant Soil 2005, 277, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Kozdrój, J.; Piotrowska-Seget, Z.; Krupa, P. Mycorrhizal fungi and ectomycorrhiza associated bacteria isolated from an industrial desert soil protect pine seedlings against Cd(II) impact. Ecotoxicology 2007, 16, 449–456. [Google Scholar] [CrossRef]
- Chelius, M.K.; Triplett, E.W. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 2001, 41, 252–263. [Google Scholar] [CrossRef]
- Deng, Z.; Cao, L. Fungal endophytes and their interactions with plants in phytoremediation: A review. Chemosphere 2017, 168, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, X.; Hou, L.; Ren, Y.; Wang, S.; Su, F. Dark septate endophytes isolated from a xerophyte plant promote the growth of Ammopiptanthus mongolicus under drought condition. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Likar, M.; Regvar, M. Isolates of dark septate endophytes reduce metal uptake and improve physiology of Salix caprea L. Plant Soil 2013, 370, 593–604. [Google Scholar] [CrossRef]
- Kaczmarek, J.; Jędryczka, M. Characterization of two coexisting pathogen populations of Leptosphaeria spp., The cause of stem canker of brassicas. Acta Agrobot. 2011, 64, 3–14. [Google Scholar] [CrossRef]
- Spadaro, D.; Pellegrino, C.; Garibaldi, A.; Gullino, M.L. Development of SCAR primers for the detection of Cadophora luteo-olivacea on kiwifruit and pome fruit and of Cadophora malorum on pome fruit. Phytopathol. Mediterr. 2011, 50, 430–441. [Google Scholar] [CrossRef]
- Auger, J.; Pérez, I.; Osorio-Navarro, C.; Esterio, M. First Report of Cadophora luteo-olivacea Causing Side Rot on Kiwifruit in Chile. Plant Dis. 2018, 102, 680. [Google Scholar] [CrossRef]
- Úrbez-Torres, J.R.; Peduto, F.; Gubler, W.D. First Report of Ilyonectria macrodidyma Causing Root Rot of Olive Trees (Olea europaea) in California. Plant Dis. 2012, 96, 1378. [Google Scholar] [CrossRef]
- Farh, M.E.A.; Kim, Y.J.; Kim, Y.J.; Yang, D.C. Cylindrocarpon destructans/Ilyonectria radicicola-species complex: Causative agent of ginseng root-rot disease and rusty symptoms—Review. J. Ginseng Res. 2018, 42, 9–15. [Google Scholar] [CrossRef]
- Corredor, A.H.; Van Rees, K.; Vujanovic, V. Changes in root-associated fungal assemblages within newly established clonal biomass plantations of Salix spp. For. Ecol. Manag. 2012, 282, 105–114. [Google Scholar] [CrossRef]
- Hirose, D.; Tanabe, Y.; Uchida, M.; Kudoh, S.; Osono, T. Microfungi associated with withering willow wood in ground contact near Syowa Station, East Antarctica for 40 years. Polar Biol. 2013, 36, 919–924. [Google Scholar] [CrossRef] [Green Version]
- Hosseini-Nasabnia, Z.; Van Rees, K.; Vujanovic, V. Preventing unwanted spread of invasive fungal species in willow (Salix spp.) plantations. Can. J. Plant Pathol. 2016, 38, 325–337. [Google Scholar] [CrossRef]
- Jumpponen, A.; Trappe, J.M. Dark septate endophytes: A review of facultative biotrophic root-colonizing fungi. New Phytol. 1998, 140, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Zhang, S.; Wang, H.; Li, X.; Li, X.; Zhang, H. Distribution of fungal endophytes in roots of Stipa krylovii across six vegetation types in grassland of northern China. Fungal Ecol. 2018, 31, 47–53. [Google Scholar] [CrossRef]
- Maciá-Vicente, J.G.; Jansson, H.B.; Abdullah, S.K.; Descals, E.; Salinas, J.; Lopez-Llorca, L.V. Fungal root endophytes from natural vegetation in Mediterranean environments with special reference to Fusarium spp. FEMS Microbiol. Ecol. 2008, 64, 90–105. [Google Scholar] [CrossRef] [Green Version]
- Khalmuratova, I.; Kim, H.; Nam, Y.J.; Oh, Y.; Jeong, M.J.; Choi, H.R.; You, Y.H.; Choo, Y.S.; Lee, I.J.; Shin, J.H.; et al. Diversity and plant growth promoting capacity of endophytic fungi associated with halophytic plants from the west coast of Korea. Mycobiology 2015, 43, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Michielse, C.B.; Rep, M. Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 2009, 10, 311–324. [Google Scholar] [CrossRef]
- Poletto, T.; Muniz, M.F.B.; Fantinel, V.S.; Harakava, R.; Rolim, J.M. Characterization and pathogenicity of Fusarium oxysporum associated with Carya illinoinensis seedlings. Floresta Ambiente 2020, 27, 1–9. [Google Scholar] [CrossRef]
- Wei, F.; Zhang, Y.; Shi, Y.; Feng, H.; Zhao, L.; Feng, Z.; Zhu, H. Evaluation of the Biocontrol Potential of Endophytic Fungus Fusarium solani CEF559 against Verticillium dahliae in Cotton Plant. BioMed Res. Int. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Caliz, J.; Montes-Borrego, M.; Triadó-Margarit, X.; Metsis, M.; Landa, B.B.; Casamayor, E.O. Influence of edaphic, climatic, and agronomic factors on the composition and abundance of nitrifying microorganisms in the rhizosphere of commercial olive crops. PLoS ONE 2015, 10, e0125787. [Google Scholar] [CrossRef] [Green Version]
- Taffner, J.; Cernava, T.; Erlacher, A.; Berg, G. Novel insights into plant-associated archaea and their functioning in arugula (Eruca sativa Mill.). J. Adv. Res. 2019, 19, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Taffner, J.; Bergna, A.; Cernava, T.; Berg, G. Tomato-Associated Archaea Show a Cultivar-Specific Rhizosphere Effect but an Unspecific Transmission by Seeds. Phytobiomes J. 2020, 4, 133–141. [Google Scholar] [CrossRef]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Joa, J.H.; Sang, M.K.; Song, J.; Weon, H.Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chai, L.; Huang, M.; Jia, W.; Guo, J.; Huang, Y. Deciphering the archaeal communities in tree rhizosphere of the Qinghai-Tibetan plateau. BMC Microbiol. 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Cann, I.K.O.; Mackie, R.I. Response of archaeal communities in the rhizosphere of maize and soybean to elevated atmospheric CO2 concentrations. PLoS ONE 2010, 5, e15897. [Google Scholar] [CrossRef] [Green Version]
- Breidenbach, B.; Pump, J.; Dumont, M.G. Microbial community structure in the rhizosphere of rice plants. Front. Microbiol. 2016, 6, 1537. [Google Scholar] [CrossRef] [Green Version]
- Bessetti, J. Profiles in DNA—An Introduction to Pcr Inhibitors; Promega. Available online: https://www.promega.es/-/media/files/resources/profiles-in-dna/1001/an-introduction-to-pcr-inhibitors.pdf?la=es-es (accessed on 20 December 2021).
- Oliveira, M.N.V.; Santos, T.M.A.; Vale, H.M.M.; Delvaux, J.C.; Cordero, A.P.; Ferreira, A.B.; Miguel, P.S.B.; Tótola, M.R.; Costa, M.D.; Moraes, C.A.; et al. Endophytic microbial diversity in coffee cherries of Coffea arabica from southeastern Brazil. Can. J. Microbiol. 2013, 59, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Lv, X.; Warren, A.; Gong, J. Shifts in diversity and community structure of endophytic bacteria and archaea across root, stem and leaf tissues in the common reed, Phragmites australis, along a salinity gradient in a marine tidal wetland of northern China. Antonie Van Leeuwenhoek 2013, 104, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Qiu, F.; Zhang, X.; Dai, X.; Dong, X.; Song, W. Endophytic bacterial diversity in rice (Oryza sativa L.) roots estimated by 16S rDNA sequence analysis. Microb. Ecol. 2008, 55, 415–424. [Google Scholar] [CrossRef]
- Müller, H.; Berg, C.; Landa, B.B.; Auerbach, A.; Moissl-Eichinger, C.; Berg, G. Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize Mediterranean olive trees. Front. Microbiol. 2015, 6, 138. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Units | Values | Parameters | Units | Values |
|---|---|---|---|---|---|
| Cation-exchange capacity | meq 100 g−1 | 43.50 | PCBs c | mg kg−1 | 57.58 ± 11.70 |
| pH a | - | 7.70 | Cadmium c | mg kg−1 | 1.75 ± 0.15 |
| pH buffer | - | >7.50 | Chromium c | mg kg−1 | 659.50 ± 127.22 |
| Soil texture | - | Clay | Copper c | mg kg−1 | 1380.00 ± 201.57 |
| Clay | % | 46.00 | Nickel c | mg kg−1 | 42.90 ± 2.22 |
| Silt | % | 33.90 | Lead c | mg kg−1 | 34.00 ± 8.12 |
| Sand | % | 20.10 | Zinc c | mg kg−1 | 386.50 ± 72.13 |
| Organic matter | % | 9.60 | Acenaphthene c | mg kg−1 | 0.56 ± 0.18 |
| K + Mg + Ca saturation | % | 100.00 | Acenaphtylene c | mg kg−1 | 1.98 ± 0.38 |
| P (P/Al) saturation | % | 16.50 | Anthracene c | mg kg−1 | 18.15 ± 4.90 |
| Ca saturation | % | 81.60 | Benz[a]anthracene c | mg kg−1 | 0.43 ± 0.09 |
| K saturation | % | 3.10 | Benzo[a]pyrene c | mg kg−1 | 0.28 ± 0.07 |
| Mg saturation | % | 15.30 | Benzo[ghi]perylene c | mg kg−1 | 0.48 ± 0.12 |
| Parameters | Units | Values | Chrysene c | mg kg−1 | 0.40 ± 0.09 |
| Al b | mg kg−1 | 48.00 | Fluoranthene c | mg kg−1 | 0.54 ± 0.20 |
| B b | mg kg−1 | 1.40 | Fluorene c | mg kg−1 | 0.94 ± 0.21 |
| Ca b | mg kg−1 | 7090.00 | Indeno [1,2,3-cd]pyrene c | mg kg−1 | 0.32 ± 0.09 |
| Cu b | mg kg−1 | 417.00 | Naphthalene c | mg kg−1 | 0.42 ± 0.13 |
| Fe b | mg kg−1 | 178.00 | Phenanthrene c | mg kg−1 | 2.62 ± 0.71 |
| K b | mg kg−1 | 525.00 | Pyrene c | mg kg−1 | 1.34 ± 0.41 |
| Mg b | mg kg−1 | 800.00 | 1-Methylnaphthalene c | mg kg−1 | 0.42 ± 0.13 |
| Mn b | mg kg−1 | 11.00 | 2-Methylnaphthalene c | mg kg−1 | 0.42 ± 0.12 |
| P b | mg kg−1 | 80.00 | 1,3-Dimethylnaphthalene c | mg kg−1 | 0.55 ± 0.18 |
| Zn b | mg kg−1 | 85.60 | 2,3,5-Trimethylnaphthalene c | mg kg−1 | 0.40 ± 0.13 |
| SX61 | SX64 | p-Value | Interpretation | |||||
|---|---|---|---|---|---|---|---|---|
| Roots | Rhizosphere | Roots | Rhizosphere | Cultivar | Compartment | Cultivar × break//Compartment | ||
| Fungi | 1.95 ± 0.84 | 2.87 ± 0.63 | 2.52 ± 0.75 | 2.67 ± 0.89 | 0.385 | 0.016 | 0.035 | Roots.SX61 < Rhizo.SX61 |
| Bacteria | 5.75 ± 0.31 | 6.92 ± 0.17 | 6.07 ± 0.37 | 7.07 ± 0.11 | 0.117 | <0.001 | 0.040 | Roots.SX61 < Rhizo.SX61 Roots.SX64 < Rhizo.SX64 |
| Archaea | - | 0.68 ± 0.07 | - | 1.15 ± 0.59 | 0.158 | - | - | - |
| Factor | Fungi | Bacteria | Archaea | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Df | F.Model | R2 | Pr (>F) | Df | F.Model | R2 | Pr (>F) | Df | F.Model | R2 | Pr (>F) | |
| Cultivar | 1 | 2.7657 | 0.0363 | 0.006 | 1 | 4.1311 | 0.0489 | 0.002 | 1 | 6.0091 | 0.1767 | 0.001 |
| Compartment | 1 | 16.3440 | 0.2145 | 0.001 | 1 | 23.0238 | 0.2725 | 0.001 | - | - | - | - |
| Cultivar × Compartment | 1 | 1.0759 | 0.0141 | 0.209 | 1 | 1.3507 | 0.0160 | 0.125 | - | - | - | - |
| Residuals | 56 | - | 0.7351 | - | 56 | - | 0.6627 | - | 28 | - | 0.8233 | - |
| Total | 59 | - | 1 | - | 59 | - | 1 | - | 29 | - | 1 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortin Faubert, M.; Labrecque, M.; Hijri, M. Ectomycorrhizal Fungi Dominated the Root and Rhizosphere Microbial Communities of Two Willow Cultivars Grown for Six-Years in a Mixed-Contaminated Environment. J. Fungi 2022, 8, 145. https://doi.org/10.3390/jof8020145
Fortin Faubert M, Labrecque M, Hijri M. Ectomycorrhizal Fungi Dominated the Root and Rhizosphere Microbial Communities of Two Willow Cultivars Grown for Six-Years in a Mixed-Contaminated Environment. Journal of Fungi. 2022; 8(2):145. https://doi.org/10.3390/jof8020145
Chicago/Turabian StyleFortin Faubert, Maxime, Michel Labrecque, and Mohamed Hijri. 2022. "Ectomycorrhizal Fungi Dominated the Root and Rhizosphere Microbial Communities of Two Willow Cultivars Grown for Six-Years in a Mixed-Contaminated Environment" Journal of Fungi 8, no. 2: 145. https://doi.org/10.3390/jof8020145
APA StyleFortin Faubert, M., Labrecque, M., & Hijri, M. (2022). Ectomycorrhizal Fungi Dominated the Root and Rhizosphere Microbial Communities of Two Willow Cultivars Grown for Six-Years in a Mixed-Contaminated Environment. Journal of Fungi, 8(2), 145. https://doi.org/10.3390/jof8020145